

Abstract
Approximately 1% of known protein structures display knotted configurations in their native fold, but the function of these configurations is not understood. It has been speculated that the entanglement may inhibit mechanical protein unfolding or transport, e.g., as in cellular threading or translocation processes through narrow biological pores. Protein knot manipulation, e.g., knot tightening and localization, has become possible in single-molecule experiments. Here, we investigate tight peptide knot (TPK) characteristics in detail by pulling selected 31 and 41-knotted peptides using all-atom molecular dynamics computer simulations. We find that the 31- and 41-TPK lengths are typically Δl ≈ 47± 4 Å and 69 ± 4 Å, respectively, for a wide range of tensions (0.1 nN ≲ F ≲ 1.5 nN). The 41-knot length is in agreement with recent atomic force microscopy pulling experiments. Calculated TPK radii of gyration point to a pore diameter of ∼20 Å, below which a translocated knotted protein might get stuck. TPK characteristics, however, may be sequence-specific: we find a different size and structural behavior in polyglycines, and, strikingly, a strong hydrogen bonding and water trapping capability of hydrophobic TPKs. Water capture and release is found to be controllable by the tightening force in a few cases. These mechanisms result in a sequence-specific “locking” and metastability of TPKs, which might lead to a blocking of knotted peptide transport at designated sequence positions. We observe that macroscopic tight 41-knot structures are reproduced microscopically (“figure of eight” versus the “pretzel”) and can be tuned by sequence, in contrast to mathematical predictions. Our findings may explain a function of knots in native proteins, challenge previous studies on macromolecular knots, and prove useful in bio- and nanotechnology.
Introduction
After the discovery of the first knotted structure in the native fold of a protein in 1994 (1), additional studies (2,3) and, in particular, a recent survey, have identified almost 300 more knotted proteins, constituting ∼1% of known structures in the protein database (4). Most of them have the simplest 31 (trefoil) topology; only a few have been found to possess the more complicated 41 (typically called a “figure-of-eight” knot) and 52 types of prime knots (6). (The nomenclature “Xn knot” refers to the number of strand crossings of the knot structure projected onto a two-dimensional plane, i.e, X = 3 for the trefoil and 4 for the figure-of-eight topology. The index n refers to the type of prime knot with the same number of crossings. For the three- and fourfold crossings, only one prime knot exists; thus, for them, always, n = 1.) Fig. 1 provides an illustration of tightened 31 and 41 open knot topologies. (Mathematically these knots are open, i.e., no closed loops, but throughout the work we just use the term “knot” for simplicity.) Whereas the question of the physiological relevance of protein knots is still a matter of debate (3,8,9), it has been proposed that the entangled structure might have a stabilizing effect against thermal or mechanical protein unfolding (4,9–11). An interesting possible consequence is the inhibition of knotted protein translocation and threading through the narrow pores of biological membranes or proteasomes (4). In this respect, it is tempting to speculate that the steric blocking of narrow pathways by a localized or tightly pulled protein knot may have bio(techno)logical significance. Very recently, another complex entanglement referred to as a “slipknot” has been discovered in proteins, the existence of which is also linked to a stabilizing function (12). Also relevant in this respect are cyclotides, a superstable family of proteins that feature a cyclic peptide backbone and a tightly entangled topology in their interior, showing strong biological activity and high pharmaceutical potential (13). In contrast to that of protein knots, however, the tight structure of cyclotides is generated by covalent connections between cysteine side chains. We note that the synthesis and design of artificially interlocked molecules has become possible in supramolecular chemistry with applications in bio- or nanotechnology, e.g., as molecular receptors, locks, or machines (14,15).
Figure 1.

Self-made photographs of open tight-knot configurations in a tensioned computer cable. (a) A trefoil (31)-knot. (b) A “figure-of-eight” configuration in a 41 knot. (c) A “pretzel” configuration in a 41 knot.
The study of tight-knot characteristics in (bio)polymers has been of interest for a long time, as knots easily self-tie and localize in any long chain (16–19). More than 20 years ago, de Gennes argued that knots may self-tie in crystallizing or sheared polymer melts, changing the macroscopic relaxation behavior of the latter (20). Possible self-tying mechanisms may be based on electrostatic repulsion (21), entropic tightening in wormlike chains (22), or localization of polymer knots either in confinement (23–25) or in bad solvent conditions (17). Externally controlled manipulation and characterization of microscopic knots has become accessible experimentally by employing optical tweezer methods (26,27) or atomic force microscopy (AFM) (9,28,29). Molecular knot behavior has been addressed from a theoretical perspective using scaling arguments (22), vacuum quantum calculations (30), or coarse-grained computer simulations (17,31–35). Previous studies focused almost exclusively on homogeneous systems such as polyethylene, DNA, or actin filaments; only recently have tight protein knots (TPKs) with specific, inhomogeneous sequences been investigated by Sułkowska et al. using an implicit-solvent Go model (36). Due to local geometry (side-chain size or kinks in the peptide), knot localization and diffusion was dominated by jumps of the knot's ends to specific peptide locations, suggesting qualitatively different, sequence-dependent fluctuations of TPKs when compared to homopolymers (21,27,34,35,37).
Although highly relevant for transport, translocation, and threading processes of knotted proteins, the size of a TPK has not been determined before in a systematic way to our knowledge. For a first rough guess, consider a rope of contour length lc, tie a knot in it, and tighten it by pulling the rope's ends. The end-to-end distance of the rope, l, will be reduced by the presence of the knot by Δl = lc – l. In the following, we refer to Δl as the “tight knot length”, i.e., the length of the rope involved in the open knot. By dividing Δl by the rope thickness, D, we obtain the characteristic quantity (38)
| (1) |
which has been shown to be minimized by the tight knot conformation, and is Λ = 10.1 and 13.7 for 31 and 41 knots, respectively, for idealized hard-core ropes (38). If we apply this simplified picture to molecular entities and assume a typical peptide thickness of the order of an atomic size D ≃ 3.5 − 5 Å, we would anticipate Δl = Λ D ≃ 35 − 51 Å and 48–69 Å for tight 31- and 41-peptide knots, respectively. This coarse size estimate for idealized, nonelastic hard-core peptides agrees well with recent AFM predictions of the 41-TPK length Δl = 62 ± 10 Å in bacterial phytochrome (29). If the knot is further assumed, naively, to be a circle with circumference Δl = 2πR, we infer that a typical TPK radius would be R ≃ 6 − 11 Å.
In this work, we take a more detailed look at TPKs by performing explicit-water molecular dynamics (MD) computer simulations (39) of 31 and 41 knots in selected polypeptides. The simulated peptides involve up to 30 amino acids and we systematically study knot sizes and their structural behavior. We find TPK lengths of Δl ≃ 47 ± 4 Å (involving 13 ± 1 amino acids) and 69 ± 4 Å (19 ± 1 amino acids) for the 31 and 41 knots, respectively. The knot sizes are found to be surprisingly constant for a wide range of stretching forces (F ≲1.5 nN). Typical tight-knot radii of gyration are Rg ≃ 7 − 8 Å. All sizes are in the range of the macroscopic estimate (1). The 41-TPK length is in agreement with recent AFM pulling experiments on the natively knotted bacterial phytochrome (29). Detailed tight-knot characteristics, however, may be sequence-specific: we find smaller knots, and different structural and stability behavior, in the special case of polyglycines. We find that TPKs have a strong water-capturing and hydrogen-bonding capability within their closely packed interior that is sequence-specific and promoted by nonpolar side chains. Buried water and long-lived intraknot hydrogen bonds lead to surprisingly rigid and stable tight knots in free simulations on a ∼100-ns timescale. An intriguing finding is that macroscopic tight 41-knot structures are reproduced microscopically (in a “figure-of-eight” versus a “pretzel” configuration) but depend on peptide sequence. This stands in contrast to mathematical predictions of the tight 41-knot structure, which is the “figure-of-eight” structure (38). We predict strongly localized tight knots after peptide stretching, and a preferential affinity toward regions with dominantly nonpolar side chains. We demonstrate that the accurate modeling of specific side chains and the aqueous environment is crucial for a full understanding of protein knot characteristics.
Methods and Systems
MD simulations
Our all-atom MD simulations are performed using the software package Amber9.0 with the ff03 force-field and TIP3P solvent (40). Systems are maintained at a fixed pressure, P = 1 bar, and temperature, T = 300 K, by coupling to a Berendsen barostat and Langevin thermostat, respectively. System sizes vary between N ≃ 4000 and N ≃ 8000 atoms. Electrostatic neutrality is assured by additional Na+ counterions compensating the net peptide charge given at pH 7. The rectangular and periodically repeated simulation box has lateral edge lengths Lx ≃ Ly ≃ 30 − 35 Å while in the peptide stretching direction, Lz ≃ 55 − 70 Å, depending on amino acid and peptide size. Given the observed maximum extension in the x- or y-direction of TPKs of ∼15–20 Å, this allows for at least a 15-Å distance between the peptide and its nearest image. Box sizes are based on thorough testing of finite-size effects before the production runs. Electrostatic interactions are calculated by particle mesh Ewald summation and real-space interactions have a cut-off of 9 Å. Polypeptides are generated using the Amber “tleap” tool. Knots are tied into them utilizing interactive MD (IMD) in visual MD (VMD) (41): while a Langevin simulation of the peptide is running and visualized, a force can be applied to selected fragments by using the computer mouse. Thus, the peptide can be dragged by hand into a finally knotted configuration. Thereafter, the system is equilibrated for ≃ 5 ns with Langevin dynamics, solvated with TIP3P water, and further equilibrated by an ≃ 5-ns MD simulation. For peptide stretching and loosening, we utilize the Amber steered MD (SMD) tool: a constant pulling velocity of 0.1 Å/ns (0.01 m/s) drives the first and last atom (in a distance l) of the peptide backbone in opposite directions, and force-extension curves F(l) are calculated. Pulling is terminated after the mean force reaches ∼1.5 nN, a value at which covalent bond breaking can occur experimentally (28). For every investigated system, at least one stretching-loosening (“reverse pulling”) loop is performed to check for reproducibility, a possible hysteresis, and nonequilibrium effects. This leads to a simulation time of ≃ 200–300 ns per peptide and stretching-loosening loop. Simulation snapshots are generated using VMD (41). Hydrogen bonds, radii of gyration, and root mean-square deviations (RMSDs) are analyzed using the Amber “ptraj” tool.
Systems
Knots of types 31 and 41 are investigated. To study the influence of amino acid type on the tight knot structure we opt for three different homopeptides: the hydrophobic polyleucine (sequence ), the partly hydrophilic and charged polyglutamic acid (), and the slim, amphiphilic polyglycine (). The peptides have a total number of amino acids of Naa = 21 and 30 for the 31 and 41 knots, respectively. Furthermore, two randomly picked pieces from the knotted cores of the natively 31-knotted YibK methyltransferase (42) and the 41-knotted Class II ketol-acid reductoisomerase (43) are considered to directly connect to naturally occurring protein knots. In the following, we name the knotted peptides by knot type and sequence, e.g., “31L” for a polyleucine trefoil and “41mix” for the 41 knot in a mixed sequence. The different knotted-peptide systems, their amino acid (aa) sequence and number (Naa) are summarized in Table 1. The nomenclature “Xn”-knot refers to the number of strand crossings of the knot structure projected onto a two-dimensional plane, i.e, X = 3 for the trefoil and X = 4 for the figure-of-eight topology. The index n refers to the type of prime knot with the same number of crossings. For the three- and fourfold crossings only one prime knot exists, thus for them always n = 1.
Table 1.
Simulated knotted peptide systems
| Knot | Amino acid sequence | Naa | lc(F1) (Å) | l(F1) (Å) | Δl(F1) (Å) | naa(F1) | lc(F2) (Å) | l(F2) (Å) | Δl(F2) (Å) | naa(F2) |
|---|---|---|---|---|---|---|---|---|---|---|
| 31L | L21 | 21 | 77.3 | 27.5 | 49.8 | 14 | 79.0 | 32.5 | 46.5 | 12 |
| 31E | E21 | 21 | 77.3 | 31.0 | 46.3 | 12 | 79.0 | 32.5 | 46.5 | 12 |
| 31G | G21 | 21 | 77.3 | 33.0 | 44.3 | 12 | 79.0 | 42.0 | 37.0 | 10 |
| 31mix | AHSQVKFKLG DYLMFGPETRG | 21 | 77.3 | 27.8 | 49.5 | 13 | 79.0 | 32.9 | 46.1 | 12 |
| 41L | L30 | 30 | 110.4 | 40.0 | 70.4 | 19 | 112.8 | 44.0 | 68.8 | 18 |
| 41E | E30 | 30 | 110.4 | 42.0 | 68.4 | 19 | 112.8 | 45.1 | 67.3 | 18 |
| 41G | G30 | 30 | 110.4 | 50.0 | 60.4 | 16 | 112.8 | 63.0 | 49.8 | 13 |
| 41mix | TKGMLALYNS LSEEGKKDFQ AAYSASYYPS | 30 | 110.4 | 38.7 | 71.7 | 19 | 112.8 | 44.3 | 68.5 | 18 |
Systems are named by knot type and sequence, e.g., “31L” for a polyleucine trefoil, “31E” for a polyglutamic acid trefoil, “31G” for a polyglycine trefoil, and “41mix” for the 41-knot in a mixed sequence. The peptides have Naa amino acids with the sequence shown. lc is the estimated contour length of the unknotted peptide, l the measured end-to-end distance of the knotted peptide, and Δl the tight-knot length involving naa amino acids. The lengths are evaluated at a stretching force of F1 = 200 pN and F2 = 1 nN.
A comment is in order here regarding the chirality of the knots. Each crossing in a protein knot can be assigned a “handedness” (6). If the undercrossing strand passes the direction of the overcrossing strand from right to left, then it is righthanded (R); if the reverse is true, it is left-handed (L). A knot nomenclature can be defined by listing the handedness of the crossings according to their sequential occurrence, so that a righthanded trefoil is “RRR” and a left-handed trefoil is “LLL”, whereas the figure of eight is “RLRL” or “LRLR” (8). All types of handedness (or chirality) have been observed in native proteins (8,42,44,45). As our work connects to the natively lefthanded trefoil in the YibK protein (42) and the “LRLR” reductoisomerase (8,43,), all our 31 and 41 knots are lefthanded and of “LRLR” chirality, respectively. For the TPK properties investigated in this work, however, we do not expect any noticeable influence of chirality.
Results and Discussion
Tight-knot size and structure
A typical initial configuration of a 31-knotted peptide in our simulation is shown in Fig. 2 a, where a snapshot of 31G is sketched before pulling it tight. The end-to-end extension here is l ≃ 25 Å. A tight-knot situation for the same peptide is shown in Fig. 2 b for a large stretching force of ∼1.5 nN ( l ≃ 45 Å). For an elastic peptide, as considered in this study, the final “tightness” of the knot will naturally depend on the external stretching force, F. The calculated force-extension curve, F(l), for 31G is shown in Fig. 3 a together with the data for 31E and 41L. We observe an overall monotonic nonlinear increase of the force. Fluctuations are moderate on that scale and have local standard deviations ranging from ∼20 pN to ∼50 pN. We also plot F(l) of knot loosening, i.e., “reverse pulling”, showing complete reversibility of the process and no obvious hysteresis within the statistical fluctuations. This indicates that our systems are close to equilibrium at the chosen pulling rate of 0.1 Å/ns.
Figure 2.
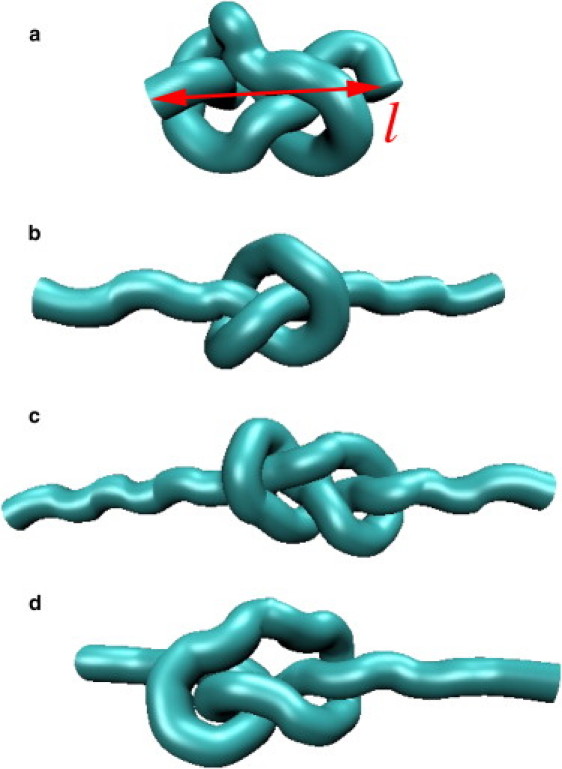
MD simulation snapshots of different protein knots in a “cartoon” representation. (a) Initial configuration of peptide 31G, where l ≃ 25 Å is the end-to-end distance. (b) Tight-knot configuration of peptide 31G. The end-to-end distance is l ≃ 45.0 Å at stretching force F ≃ 1.5 nN. (c) A tight “figure-eight” knot configuration of peptide 41G at F ≃ 1 nN. (d) A tight “pretzel” knot configuration of peptide 41L at F = 1 nN.
Figure 3.
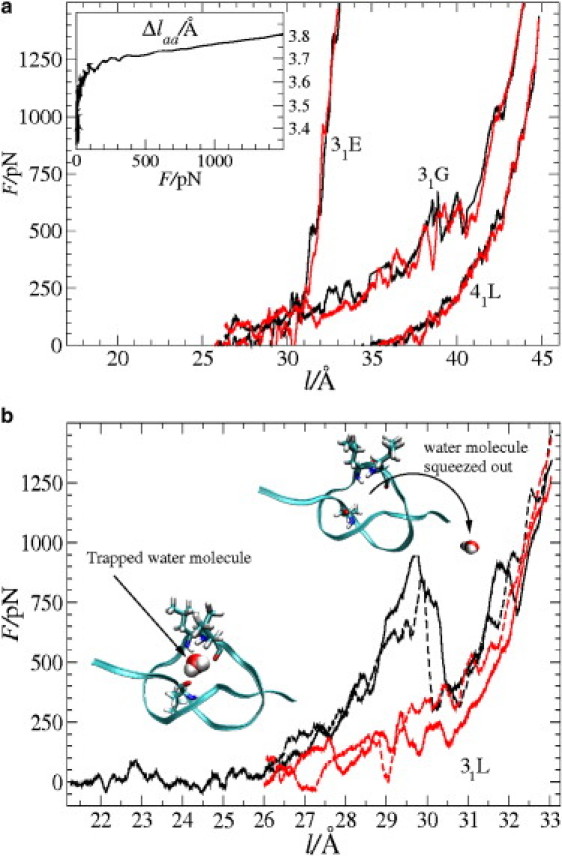
(a) Force (F)-extension (l) curves for the peptides 31E, 31G, and 41L. Stretching curves (black lines) and loosening curves (red lines) lie on top of each other, indicating a small hysteresis. The pulling rate is 0.1 Å/ns. The inset shows the mean distance Δlaa between neighboring backbone nitrogen atoms versus stretching force, F, in an unknotted peptide. (b) Force-extension curves for the polyleucine 31L. Black lines correspond to stretching, whereas red lines correspond to loosening of the knot. While stretching, for extensions l ≲ 30 Å, a single water molecule is permanently trapped by the polar backbone of the peptide knot (left inset). When l ≳ 30 Å, the water molecule is squeezed out (right inset), giving rise to a significant peak in the force-extension stretching curve. This transition leads to a considerable hysteresis when stretching and loosening curves are compared. The effect is reproducible when the stretching-loosening loop is repeated (dashed lines).
To determine the tight knot length, Δl(F) = lc(F) – l(F), an accurate estimate of the force-dependent contour length lc(F) of the unknotted peptide is needed. For this, we calculate the average amino acid length Δlaa(F) by measuring the mean distance between neighboring backbone nitrogen atoms in short (Naa = 8), unknotted peptides. From the resulting force-extension curves F(l) we obtain, by inversion and division by the number of amino acids, Δlaa(F) = l(F)/Naa. The result, which we find to be independent of the choice of amino acid sequence, is presented in the inset to Fig. 3 a: below a stretching force of ∼10 pN the length thermally fluctuates around Δlaa (F) ≃ 3.5 Å, then rises quickly with force in a nonlinear fashion in the low-stretching, thermal regime (F ∼ 10–150 pN) to eventually increase linearly in the high stretching regime, F ≳ 150 pN. At F = 1.5 nN, a value of Δlaa (F) ≃ 3.8 Å is reached. From the slope, b, of the linear part, we estimate the linear elastic modulus Γ = Δlaa (F = 0)/b ≃42 nN, which is in agreement with AFM pulling experiments, where Γ ≃ 50 ± 15 nN (46). This agreement is remarkable, since MD force fields are typically not benchmarked to be accurate at the large tensions considered in this work.
In pulling experiments, rupture of some terminal bonds at the AFM tip can occur at forces of ∼100–200 pN (28), supplying thereby the relevant range for comparing to experimental TPK lengths. At F1 = 200 pN we find Δlaa (F1) ≃ 3.68 Å, leading to contour length estimates lc (F1) = Naa Δlaa ≃ 77.3 Å and lc (F1) ≃ 110.4 Å for the trefoil and 41 peptide, respectively. Consequently, subtracting the calculated end-to-end distances at F1, we find that the tight-knot lengths for the trefoil peptides are between Δl(F1) ≃ 44.3 Å (31G) and 49.8 Å (31L). The number of amino acids involved in the knot is thus naa = Δl/Δlaa ≃ 13 ± 1. For the 41 knots, three of the four values lie between Δl ≃ 68.4 Å and 71.9 Å (naa ≃ 19), whereas for the polyglycine knot (41G), we find Δl (F1) ≃ 60.4 Å (naa ≃ 16), ∼14% smaller. The lengths are summarized in Table 1. A typical error of these values is given by the fluctuations of the F(l) curve and is roughly of amino acid size (±4 Å).
Let us now consider more intense stretching and study the knot lengths at a larger force, F2 = 1 nN. Δlaa(F) increases to ≃ 3.76 Å, giving rise to a slightly larger contour length for the unknotted peptides. In evaluating the particular knot lengths (see Table 1), we observe that the knots shrink in size (while the whole peptide is more stretched), as could have been anticipated. Although the pulling force is substantially increased, typically only one amino acid less is involved in a single knot, so that, surprisingly, the knot sizes vary by only a few percent for a wide range of tensions. Both of the polyglycine peptides are exceptions, however: here, the tightening effect is considerable and the final knot lengths are 20–25% smaller than those of the other studied peptides. All lengths are summarized in Table 1.
The knot lengths of ≃ 37 − 50 Å and ≃ 50 − 72 Å for the 31 and 41 knots, respectively, fall within the predicted range (1). This agreement indicates that TPK lengths are primarily determined by generic packing effects, with an effective excluded volume thickness, D, that is similar for most of the peptides, including the mixed sequences. Furthermore, macroscopic arguments roughly hold on the molecular scale. In contrast, hydrophobicity and hydrophilicity seem to have no direct influence on tight knot size in the considered force regime. A closer examination of the nature of amino acid side chains supports this statement: while glycine has basically no side chain, and packing is performed by its backbone only, a typical-sized residue with a few carbon atoms gives rise to a more difficult molecular arrangement close to or inside the tight knot. This presumably leads to the 20–25% smaller knots in the special case of polyglycine. We thus find a smaller effective thickness, D ≃ 3.7 Å, for polyglycine than for the other peptides, where D ≃ 4.6 − 5.0 Å). It is important to note that, apart from the polyglycine, the 41-TPK lengths are in agreement with recent AFM pulling experiments on the natively knotted bacterial phytochrome, where Δl = 62 ± 10 Å has been measured at an ≃ 70 pN pulling force (29).
Illustrative simulation snapshots are shown in Fig. 4, where we plot tight knot situations for peptides 41G and 41mix, including their side chains. Large side chains obviously impede tight peptide packing. We also calculate the radius of gyration, Rg, of the knots by averaging RMS atomic distances from the geometric center of the atoms involved in the knot. We measure Rg ≃ 7.2 ± 0.2 Å and Rg ≃ 7.8 ± 0.2 Å for the 31 and 41 knots, respectively, with only weak dependence on the stretching force for all considered peptides apart from the polyglycine. For the latter, radii of gyration are found to be close to the values above for moderate stretching (F ≃ 200 pN), but 20% smaller for strong stretching (F ≳ 1 nN). These TPK sizes are larger than the typical size of biological channels, e.g., those found in the protease enzyme (11,47), which is responsible for protein degradation. Thus, a translocation or threading of a protein would indeed be blocked in vivo by the presence of a tightened knot.
Figure 4.
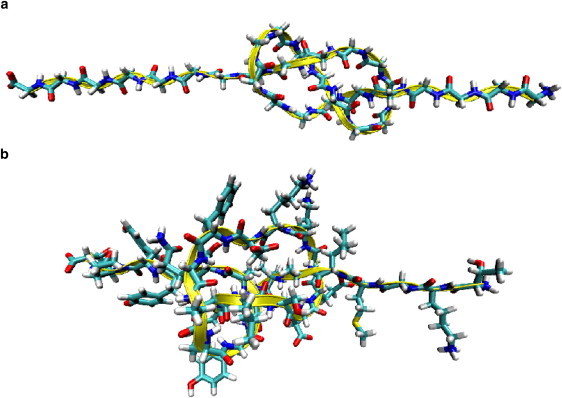
MD simulation snapshots of the tightly knotted 41G (a) and 41mix (b). The backbone is sketched in a yellow ribbon for better identification and all amino acids are resolved in a “licorice” representation (F ≃ 1 nN).
An intriguing topological feature appears when inspecting the overall 41-knot structure without the obscuring side chains, as in Fig. 2, c and d. Although the 41G knot is, figuratively indeed, in a “figure-of-eight” configuration, 41L displays a “pretzel” configuration, as illustrated in Fig. 1 c. Actually, we find that all of the 41 knots considered except 41G reproducibly prefer the pretzel shape when inspected by eye. This comes as a surprise, as the tight 41-knot configuration that minimizes (1) has been shown to be the “figure of eight”, at least using simplifying mathematical assumptions (38). Presumably, the reasons are rather physics-based, i.e., possibly the amino acid packing and their interactions in a pretzel-like configuration minimizes the system free energy. It is interesting to note that the pretzel-like configuration can be a stable 41-configuration in macroscopic knots under tension, e.g., as can be easily self-demonstrated using a simple computer cable (see Fig. 1 c) or as taught in books on cowboy rope tricks (48). This is believed to be the first observation of a tight pretzel-like structure on microscopic scales. Recently, a somewhat looser pretzel configuration was found in collagen by transmission electron microscopy (49).
Water trapping, hysteresis, and hydrogen bonding
A striking structural feature observed in this study is the capability of some peptide knots to capture and strongly bind water molecules in their interior. The simulated peptides show this effect with varying magnitude: we find no bound water in polyglycine (31G and 41G) and the mixed peptide 31mix for any simulated peptide extension, whereas in 31E, a single trapped water molecule is reproducibly found only in the case of very close peptide packing at high forces, F ≳ 1 nN. We find stronger water-binding qualities for the other four peptides, 31L, 41E, 41L, and 41mix, for a wider range of simulated peptide extensions. Here, water was bound for simulation times of the order of ∼10–100 ns per peptide, pointing to a mechanism that is quite stable. It is surprising that, on first glance, both homopeptides with the purely hydrophobic leucine side chains show the strongest water-trapping capability.
Simulation snapshots are shown in Fig. 5 for peptides 41E and 41mix: the water molecule bonds to the backbone amides in the knot interior, involving at least three hydrogen bonds per molecule, and is rotationally immobilized. Apparently, the water binding is made possible by the tight peptide packing in the highly bent knot, allowing for multiple bonds of a water molecule to the polar backbone. A particularly interesting case is the water binding in 31L. Here, the bound water molecule is “squeezed out” of the knot interior for large stretching forces (F ≃ 1 nN). This behavior leads to a high force peak in the force extension curve, as shown in Fig. 3 b: for extensions l ≲ 30 Å, the water molecule is bound as shown in the left snapshot. At l ≃ 30 Å and F ≃ 1 nN, the bound water is “wrung” out and the force drops significantly before further increasing. When the knot is loosened, F(l) shows a considerable hysteresis. However, a water molecule is captured by the knot again during loosening at extensions l ≲ 27 Å and F ≃ 200 pN. Repeating the stretching-loosening loop twice shows quantitative reproducibility of this effect (cf. Fig. 3 b). The occurrence of the hysteresis points to the fact that the water binding-unbinding events fluctuate on large timescales, and this simulation deviates therefore from equilibrium. The magnitude of the hysteresis can be estimated by integrating over the F(l) stretching-loosening cycle, which gives rise to a large dissipation energy of about Δ G ≃ 30 − 35 kBT. This value is indeed comparable to the energy of three to four hydrogen bonds between a water molecule and a peptide environment (8–10 kBT per hydrogen bond) (50).
Figure 5.
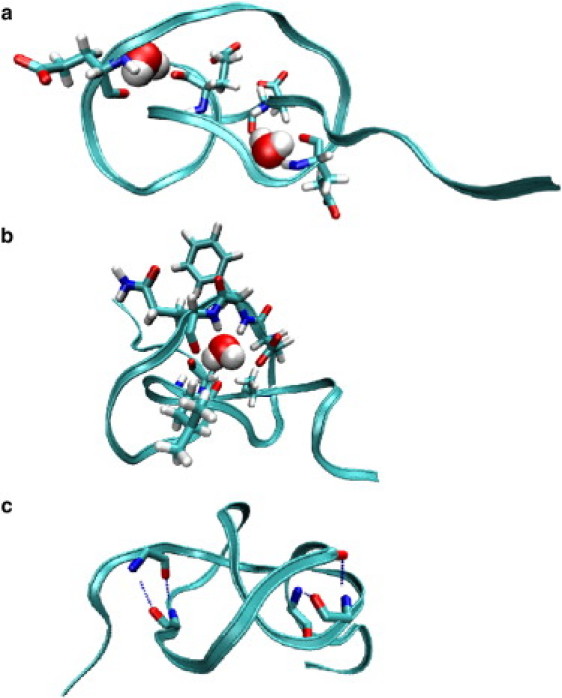
MD simulation snapshots of water trapped in peptide 41E (a) and 41mix (b) at force F ≃ 1 nN. The backbone is shown in ribbon structure and only those residues are sketched (in “licorice” representation) that are actively involved in water binding. Water (red and white spheres) is hydrogen-bonded to the backbone amides. (c) MD snapshot of 41mix in an unconstrained MD simulation. Four long-lived hydrogen bonds between backbone amides at the knot's ends are explicitly drawn (dotted blue lines).
It is a well-known fact that buried water molecules constitute an integral part of many native protein structures, contributing to stability, flexibility, folding, and mechanical and enzymatic function (51–54). It is noteworthy that our measured ΔG is very close to the binding enthalpy of a buried water molecule in the polar pocket of bovine pancreatic trypsin inhibitor (BPTI) (55), where four hydrogen bonds constitute Δ H ≃ 36 kBT. We find a similar large dissipation energy in the peptide 41mix (Δ G ≃ 20 kBT) and less pronounced dissipation energy in 41E (Δ G ≃ 12 kBT) and 31E (Δ G ≃ 5 kBT) due to partial water hydrogen binding events during knot tightening. No hysteresis is found in 41L, as water is bound here during the full stretching-loosening loop without any binding/unbinding transition.
It is interesting to note that in the sequence 31mix, we find no water trapped in the knot interior for the entire peptide extension, in contrast to 41mix, where we observe water trapping on a ∼10-ns timescale, with three binding/unbinding events for tensions F ≲ 500 pN. A closer inspection of the MD trajectory reveals that the immediate surrounding of the buried water molecule consists of six amino acids, ALD FQS, which create a mostly hydrophobic environment (see Fig. 5 b). This observation and the strong water binding capabilities of the polyleucines indicate that a nonpolar-side-chain environment promotes water-hydrogen bonding to the tightly packed polar backbone. We explain this by the textbook fact that hydrogen bonds are generally stronger in a nonpolar and/or desolvated protein environment (50,56), where electrostatic interactions are only weakly screened. We suspect, in addition, that the hydrophobic side chains impose a large energy barrier for possible escape of a water molecule. In the polyglutamic acids, water screening and the (probably lower) barrier is likely to be provided by the methylene groups of the side chains. A nonpolar environment is clearly absent in the polyglycines. However, a strong water trapping capability seems to result from a unique and delicate combination of local backbone structure and a specific, but rather nonpolar, amino acid side chain environment.
Related to this, another consequence of the tight peptide packing, as further revealed by our simulations, is the existence of long-lived hydrogen bonds between particular backbone amide groups. During the ≃ 200-ns stretching and loosening loop of polyleucine 41L, for instance, we find that the backbone of amino acids 10 and 24 hydrogen-bonds for ≃ 80% of the simulation time. Detailed analysis yields similar behavior for the other peptides, yielding stable intrapeptide hydrogen bonds on a long ∼10- to 100-ns timescale. An exception is polyglycine, where the longest hydrogen bond life expectancy is found to be one or two orders of magnitude shorter.
Free simulations and tight knot stability
We also conduct free simulations of the knotted peptides without any constraints to check whether the knots dissolve on a typical simulation timescale. Initial configurations are taken from a stretched situation at F ≃ 200 pN. Dissolution of a knot is defined here by connecting the peptide ends with an imaginary line and observing whether or not we find a knot in the closed loop. Only the two polyglycine knots, 31G and 41G, show strong fluctuations and unknot quickly, on a timescale of ∼10 ns. All other investigated knots do not dissolve in an ≃ 100-ns simulation, pointing to a (meta)stable tight knot situation. To quantify this, we measure the root mean-square deviation (RMSD) from the initial structure of the knotted backbone part only. By our definition, this includes the amino acids 5–17 and 6–25 in the 31 and 41-knotted peptides, respectively. We find values of RMSD ≃ 2 Å, increasing quickly within ≃ 10 ns to 7–8 Å for the polyglycines. For the other peptides, however, the RMSD value stays small, at ≃ 2 Å, for the total simulation time, supporting the observation that, apart from the polyglycines, tight knots stay stable and quite rigid after peptide stretching on relatively long timescales. The RMSD and illustrating snapshots for 41G and 41mix are presented in Fig. 6. The initial knot in 41G quickly dissolves into a random coil structure within 10–20 ns. In contrast, system 41mix displays almost the same knotted core structure after 110 ns when compared to the initial structure. An RMSD value of 1–2 Å is typical for thermal fluctuations of a stable protein structure (57). We note that dissolution of the polyglycine proceeds via a “swelling” rather than a “slithering” mechanism of the knot (where the knot stays tight and diffuses to the end), possibly to relax the highly bent backbone. This needs not to be in contrast to the study of Grosberg and Rabin (22), where an entropic tightening and slithering was predicted, as this might be the dominant mechanism for somewhat looser tight knots.
Figure 6.
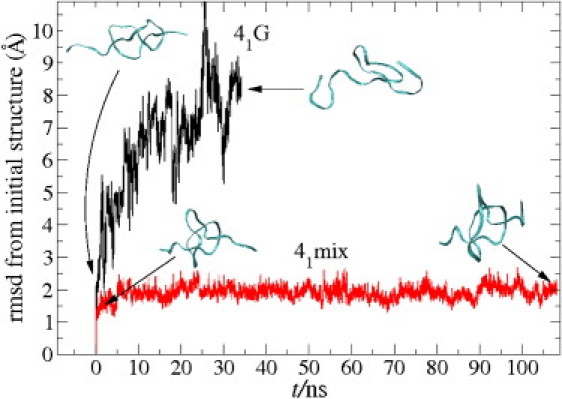
RMSD of the knotted backbone part (amino acids 6–25) from the initial configuration at t = 0 for 41G and 41mix in a free, unconstrained MD simulation run. The MD snapshots correspond to initial and final configurations. Note that the knotted structure of 41mix hardly changes in time.
As in the constrained case, closer inspection of the knot structure reveals a few long-lived hydrogen bonds in all stable knots also in the free simulations. A representative illustration is shown in Fig. 5 c, where we plot an MD snapshot of 41mix after an ≃ 100-ns free simulation. Four hydrogen bonds are found between amide backbone groups right at the knot's ends, clearly inhibiting the opening of the knot. Typically, we find that these hydrogen bonds persist on average for 80–90% of the total free simulation time, even noticeably longer for the polyleucines. In 31mix, the longest hydrogen-bond lifetime is shorter and is 50–60 ns. (Recall that also in 31mix, no buried water could be detected.) It is worthy of note, in addition, that in 31L and 41L, one water molecule was trapped during the full, unconstrained simulation, constituting a total of seven to eight long-lived hydrogen bonds within the knots! Again the high quantity and persistence strength of hydrogen bonds in 31L and 41L must be attributed to the desolvated, strongly nonpolar side-chain environment of the tight knot.
Concluding Remarks
In summary, our MD study has revealed some generic and some specific structural behavior of tight peptide knots and provokes a few interesting conclusions and future prospects: TPKs exhibit an unexpectedly strong stability after stretching, and their radii of gyration are all indeed bigger (∼7–8 Å) at moderate stretching (F ≲ 200 pN) than the radius of the protease pore (∼6.5 Å) (11,47). As a consequence, the steric blocking of narrow pathways by a localized or tightly pulled knot might be possible in vivo. We predict that a translocated knotted protein should get stuck in pores of diameter ≲ 20 Å.
Of interest for future investigation, not only from a topological point of view (57), is the observation that most 41 knots are not in a figure of eight, but rather in a “pretzel” configuration. The pretzel might be a (meta)stable configuration in “physical” open tight knots, in contrast to those underlying simplifying mathematical assumptions (38). The pretzel may be preferred by the lowering of the system free energy due to favorable amino acid arrangements. This seems to be in contrast to the macroscopic pretzel, which is determined by the way the knot is tied and is then stabilized by friction. On microscopic scales, however, the configuration can be tuned by the molecular sequence.
A striking result is our finding that the TPK backbone has a strong water-binding and hydrogen-bonding capability, promoted by rather nonpolar side-chain environments. This mechanism results in “locking” of the tight knot structure and surprisingly stable and rigid TPKs after peptide stretching. The observed quantitative reproducibility of squeezing out and capturing a water molecule at well-defined tensions may allow for an external mechanical control of single (water)-molecule binding. This might be a useful feature in biotechnological applications. It is mportant, in this respect, that buried water is known to be an integral part of native protein structures and can be essential for protein flexibility, folding (52,53,55), and catalytic action (54).
Furthermore, TPKs might resemble structural elements of the cyclotide protein family—constituted of short (≃30 amino acids), tightly entangled cyclic peptides—which has strong potential for drug design (13). In view of their structural complexity and molecule-binding potential, engineered protein knots thus may serve as an important model system for a deeper understanding of protein stability (58) and enzymatic activity, and may be useful for pharmaceutical purposes due to a possible catalytic function.
Finally, we would like to encourage further experimentation in this stimulating field, readily accessible by AFM (29) or optical tweezer methods (26,27). Studies on protein knot size, stability, and diffusion behavior along stretched peptides, as well as some on the refolding of knotted proteins after stretching, are desirable. Of particular interest would be a study focusing on knotted peptide translocation to verify/falsify our prediction about whether and where the protein gets stuck. Knotted protein threading may be possible in vitro using narrow biological or solid-state nanopores (59). Buried water molecules are detectable by nuclear magnetic relaxation dispersion methods (52), allowing for experimental exploration of the existence of bound water in protein knots, and consequently a means to further explore TPK fluctuations and energy landscapes.
Acknowledgments
J.D. is grateful to Thomas Bornschlögl, Katrina Forest, and Matthias Rief for pointing him toward this interesting project, Lyderic Bocquet, Ralf Metzler, Roland Netz, and Joachim Seel for useful comments, and Michael Hinczewski for a critical reading of the manuscript. Computing time on the HLRBII computer cluster of the Leibniz-Rechenzentrum München is acknowledged.
The author acknowledges the support of the Deutsche Forschungsgemeinschaft (DFG) through the Emmy-Noether-Program.
References
- 1.Mansfield M.L. Are there knots in proteins? Nat. Struct. Biol. 1994;1:213–214. doi: 10.1038/nsb0494-213. [DOI] [PubMed] [Google Scholar]
- 2.Taylor W.R., Lin K. A tangled problem. Nature. 2003;421:25. doi: 10.1038/421025a. [DOI] [PubMed] [Google Scholar]
- 3.Lua R.C., Grosberg A.Y. Statistics of knots, geometry of conformations, and evolution of proteins. PLOS Computational Biology. 2006;2:e45. doi: 10.1371/journal.pcbi.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Virnau P., Mirny L.A., Kardar M. Intricate knots in proteins: function and evolution. PLOS Comput. Biol. 2006;2:e122. doi: 10.1371/journal.pcbi.0020122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reference deleted in proof.
- 6.Adams C.C. W.H. Freeman; New York: 1994. The Knot Book: An Elementary Introduction to the Mathematical Theory of Knots. [Google Scholar]
- 7.Reference deleted in proof.
- 8.Taylor W.R. Protein knots and fold complexity: some new twists. Comput. Biol. Chem. 2007;31:151–162. doi: 10.1016/j.compbiolchem.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Alam M.T., Yamada T., Carlsson U., Ikai A. The importance of being knotted: effects of the C-terminal knot structure on enzymatic and mechanical properties of bovine carbonic anhydrase II. FEBS Lett. 2002;519:35–40. doi: 10.1016/s0014-5793(02)02693-5. [DOI] [PubMed] [Google Scholar]
- 10.Wallin S., Zeldovich K.B., Shakhnovich E.I. The folding mechanics of a knotted protein. J. Mol. Biol. 2007;368:884–893. doi: 10.1016/j.jmb.2007.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prakash S., Matouschek S. Protein unfolding in the cell. Trends Biochem. Sci. 2004;29:593–600. doi: 10.1016/j.tibs.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 12.King N.P., Yeates E.O., Yeates T.O. Identification of rare slipknots in proteins and their implications for stability and folding. J. Mol. Biol. 2007;373:153–166. doi: 10.1016/j.jmb.2007.07.042. [DOI] [PubMed] [Google Scholar]
- 13.Rosengren K.J., Daly N.L., Plan M.R., Waine C., Craik D.J. Twists, knots, and rings in proteins: structural definition of the cyclotide framework. J. Biol. Chem. 2002;278:8606–8616. doi: 10.1074/jbc.M211147200. [DOI] [PubMed] [Google Scholar]
- 14.Breault G.A., Hunter C.A., Mayers P.C. Supramolecular topology. Tetrahedron. 1999;55:5265–5293. [Google Scholar]
- 15.Williams A., Northrop B.H., Chang T., Stoddart J.F., White A.J.P. Suitanes. Angew. Chem. Int. Ed. 2006;40:6665–6669. doi: 10.1002/anie.200602173. [DOI] [PubMed] [Google Scholar]
- 16.Katritch V., Olson W.K., Vologodskii A., Dubochet J., Stasiak A. Tightness of random knotting. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 2000;61:5545–5549. doi: 10.1103/physreve.61.5545. [DOI] [PubMed] [Google Scholar]
- 17.Virnau P., Kantor V., Kardar M. Knots in globule and coil phases of a model polyethylene. J. Am. Chem. Soc. 2005;127:15102–15106. doi: 10.1021/ja052438a. [DOI] [PubMed] [Google Scholar]
- 18.Belmonte A. The tangled web of self-tying knots. Proc. Natl. Acad. Sci. USA. 2007;104:17243–17244. doi: 10.1073/pnas.0708150104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kardar M. The elusiveness of polymer knots. Eur. Phys.J. B. 2007;64:519–523. [Google Scholar]
- 20.de Gennes P.-G. Tight knots. Macromolecules. 1984;17:703–704. [Google Scholar]
- 21.Dommersnes P.G., Kantor Y., Kardar M. Knots in charged polymers. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2002;66:031802. doi: 10.1103/PhysRevE.66.031802. [DOI] [PubMed] [Google Scholar]
- 22.Grosberg A.Y., Rabin Y. Metastable tight knots in a worm-like polymer. Phys. Rev. Lett. 2007;99:217801. doi: 10.1103/PhysRevLett.99.217801. [DOI] [PubMed] [Google Scholar]
- 23.Metzler R., Hanke A., Dommersnes P.G., Kantor Y., Kardar M. Equilibrium shapes of flat knots. Phys. Rev. Lett. 2002;88:188101. doi: 10.1103/PhysRevLett.88.188101. [DOI] [PubMed] [Google Scholar]
- 24.Ercolini E., Valle F., Adamcik J., Witz G., Metzler R. Fractal dimension and localization of DNA knots. Phys. Rev. Lett. 2007;98:058102. doi: 10.1103/PhysRevLett.98.058102. [DOI] [PubMed] [Google Scholar]
- 25.Marcone B., Orlandini E., Stella A.L. Knot localization in adsorbing polymer rings. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2007;76:051804. doi: 10.1103/PhysRevE.76.051804. [DOI] [PubMed] [Google Scholar]
- 26.Arai Y. Tying a molecular knot with optical tweezers. Nature. 1999;399:446–448. doi: 10.1038/20894. [DOI] [PubMed] [Google Scholar]
- 27.Bao X.R., Lee H.J., Quake S.R. Behavior of complex knots in single DNA molecules. Phys. Rev. Lett. 2003;91:265506. doi: 10.1103/PhysRevLett.91.265506. [DOI] [PubMed] [Google Scholar]
- 28.Hugel T., Seitz M. The study of molecular interactions by AFM force spectroscopy. Macromol. Rapid Commun. 2001;22:989–1016. [Google Scholar]
- 29.Bornschlögl T., Anstrom D., Dzubiella J., Rief M., Forest K.T. Tightening the knot in phytochrome by single molecule atomic force microscopy. Biophys. J. 2009 doi: 10.1016/j.bpj.2008.11.012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saitta A.M., Soper P.D., Wasserman E., Klein M.L. Influence of a knot on the strength of a polymer strand. Nature. 1999;399:46–48. doi: 10.1038/19935. [DOI] [PubMed] [Google Scholar]
- 31.Mansfield M.L. Tight knots in polymers. Macromolecules. 1997;31:4030–4032. [Google Scholar]
- 32.Farago O., Kantor Y., Kardar M. Pulling knotted polymers. Europhys. Lett. 2002;60:53–59. [Google Scholar]
- 33.Arteca G.A. Externally steered relaxation of tight polyethylene tangles with different initial knot topologies. Theor. Chem. Acc. 2007;118:549–556. [Google Scholar]
- 34.Vologodskii A. Brownian dynamics simulation of knot diffusion along a stretched DNA molecule. Biophys. J. 2006;90:1594–1597. doi: 10.1529/biophysj.105.074682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang L., Makarov D.E. Langevin dynamics simulations of the diffusion of molecular knots in tensioned polymer chains. J. Phys. Chem. A. 2007;111:10338–10344. doi: 10.1021/jp071940+. [DOI] [PubMed] [Google Scholar]
- 36.Sułkowska J.I., Sułkowski P., Szymczak P., Cieplak M. Tightening of knots in proteins. Phys. Rev. Lett. 2008;100:058106. doi: 10.1103/PhysRevLett.100.058106. [DOI] [PubMed] [Google Scholar]
- 37.Metzler R., Reisner W., Riehn R., Austin R., Tegenfeldt J.O. Diffusion mechanisms of localised knots along a polymer. Europhys. Lett. 2006;76:696–702. [Google Scholar]
- 38.Pierański P., Przybył S., Stasiak A. Tight open knots. Eur. Phys. J. E. 2001;6:123–128. [Google Scholar]
- 39.Karplus M., McCammon J.A. Molecular dynamics simulations of macromolecules: a perspective. Nat. Struct. Mol. Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 40.Case D.A., Darden T., Cheatham T.E., III, Simmerling C., Wang J. University of California; San Francisco: 2006. AMBER9.0. [Google Scholar]
- 41.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 42.Lim K., Zhang H., Tempczyk A., Krajewski W., Bonander N. Structure of the YibK methyltransferase from Haemophilus influenzae (HI0766): a cofactor bound at a site formed by a knot. Proteins Struct. Funct. Genet. 2003;51:56–67. doi: 10.1002/prot.10323. [DOI] [PubMed] [Google Scholar]
- 43.Biou V., Dumas R., Cohen-Addad C., Douce R., Job D. The crystal structure of plant acetohydroxy acid isomeroreductase complexed with NADPH, two magnesium ions and a herbicidal transition state analog determined at 1.65 Å resolution. EMBO J. 1997;16:3405–3415. doi: 10.1093/emboj/16.12.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pleshe E., Truesdell J., Batey R.T. Structure of a class II TrmH tRNA-modifying enzyme from Aquifex aeolicus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2005;61:722–728. doi: 10.1107/S1744309105022980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Roon A.-M.M., Loening N.M., Obayashi E., Yang J.-C., Newman A.J. Solution structure of the U2 snRNP protein Rds3p reveals a knotted zinc-finger motif. Proc. Natl. Acad. Sci. USA. 2008;105:9621–9626. doi: 10.1073/pnas.0802494105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ptak A., Takeda S., Nakamura C., Kageshima J.M.M., Jarvis S.P. Modified atomic force microscope applied to the measurement of elastic modulus for a single peptide molecule. J. Appl. Phys. 2001;90:3095–3099. [Google Scholar]
- 47.Pickart C.M., VanDemark A.P. Opening doors into the proteasome. Nat. Struct. Mol. Biol. 2000;7:999–1001. doi: 10.1038/81018. [DOI] [PubMed] [Google Scholar]
- 48.Mason, B.S. 1928. How to Spin a Rope: Lariat Throwing, Rope Spinning and Trick Cowboy Knots. Self-published, Columbus, OH. http://www.inquiry.net/outdoor/spinrope/trickknots.htm.
- 49.Myers J.C., Amenta P.S., Dion A.S., Sciancalepore J.P., Nagaswami C. The molecular structure of human tissue type XV presents a unique conformation among the collagens. Biochem. J. 2007;404:535–544. doi: 10.1042/BJ20070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jackson M.B. Cambridge University Press; Cambridge, UK: 2006. Molecular and Cellular Biophysics. [Google Scholar]
- 51.Baker E.N. Solvent interactions with proteins as revealed by X-ray crystallographic studies. In: Gregroy R.B., editor. Protein-Solvent Interactions. Marcel Dekker; New York: 1995. pp. 143–189. [Google Scholar]
- 52.Denisov V.P., Peters J., Hörlein H.D., Halle B. Using buried water molecules to explore the energy landscape of proteins. Nat. Struct. Biol. 1996;3:505–509. doi: 10.1038/nsb0696-505. [DOI] [PubMed] [Google Scholar]
- 53.Dougan L., Lu G.F.H., Fernandez J.M. Solvent molecules bridge the mechanical unfolding transition state of a protein. Proc. Natl. Acad. Sci. USA. 2008;105:3185–3190. doi: 10.1073/pnas.0706075105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Ball P. Water as an active constituent in cell biology. Chem. Rev. 2008;108:74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- 55.Fischer S., Verma C.S. Binding of buried structural water increases the flexibility of proteins. Proc. Natl. Acad. Sci. USA. 1999;96:9613–9615. doi: 10.1073/pnas.96.17.9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roseman M.A. Hydrophobicity of the peptide C=OH—N hydrogen-bonded group. J. Mol. Biol. 1988;201:621–623. doi: 10.1016/0022-2836(88)90642-0. [DOI] [PubMed] [Google Scholar]
- 57.Katritch V., Bednar J., Michoud D., Scharein R.G., Dubochet J. Geometry and physics of knots. Nature. 1996;384:142–145. doi: 10.1038/384122a0. [DOI] [PubMed] [Google Scholar]
- 58.Yeates T., Norcross S., King N.P. Knotted and topologically complex proteins as models for studying folding and stability. Curr. Opin. Chem. Biol. 2007;11:595–603. doi: 10.1016/j.cbpa.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dekker C. Solid-state nanopores. Nat. Nanotechnol. 2007;2:209–215. doi: 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]