

Abstract
Synaptotagmin 1 (Syt1) is the Ca+2 receptor for fast, synchronous vesicle fusion in neurons. Because membrane fusion is an inherently mechanical, force-driven event, Syt1 must be able to adapt to the energetics of the fusion apparatus. Syt1 contains two C2 domains (C2A and C2B) that are homologous in sequence and three-dimensional in structure; yet, a number of observations have suggested that they have distinct biochemical and biological properties. In this study, we analyzed the mechanical stability of the C2A and C2B domains of human Syt1 using single-molecule atomic force microscopy. We found that stretching the C2AB domains of Syt1 resulted in two distinct unfolding force peaks. The larger force peak of ∼100 pN was identified as C2B and the second peak of ∼50 pN as C2A. Furthermore, a significant fraction of C2A domains unfolded through a low force intermediate that was not observed in C2B. We conclude that these domains have different mechanical properties. We hypothesize that a relatively small stretching force may be sufficient to deform the effector-binding regions of the C2A domain and modulate the affinity for soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptors (SNAREs), phospholipids, and Ca+2.
Introduction
Biological systems have evolved a relatively small set of proteins known as soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptors (SNAREs) to catalyze the fusion of cargo-containing phospholipid vesicles with a target membrane. The assembly of the SNARE proteins (synaptobrevin, syntaxin, and SNAP-25) into a parallel four-helix bundle at the vesicle docking interface provides the free energy required in its role as the fusion engine for exocytosis (1–3). Synaptotagmin 1 (Syt1) is a vesicle-associated protein that interacts with the SNARE complex (4,5), and is thought to fine-tune the probability of calcium-ion dependence of release (6).
The very nature of exocytosis, that is, the fusion of juxtaposed phospholipid membranes, implies a great deal of mechanical force. For example, the interaction of forces among the three protein components of the SNARE complex have been measured by single-molecule atomic force microscopy (AFM) to be in excess of 285 pN (7,8). Because the Syt1 protein directly interacts with the SNARE complex, it must be able to adjust its structure to this highly mechanical, force-driven framework. In this work, we used single-molecule AFM to study the mechanical properties of the tandem C2 domains (C2A and C2B) of human Syt1, because these domains are the Ca+2/phospholipid and SNARE interacting portion of the Syt1 protein (Fig. 1 A).
Figure 1.
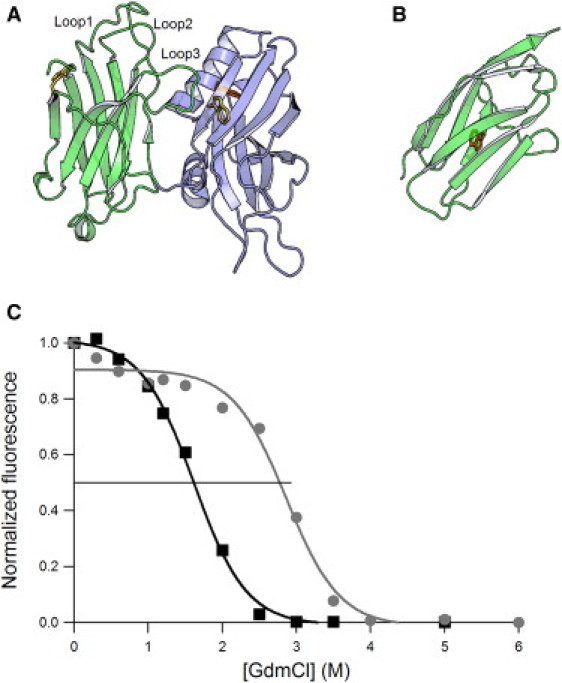
Equilibrium denaturation for Syt1 C2AB and titin I27 domains. (A) X-ray structure of human Syt1 C2AB showing the position of solvent-accessible (yellow) and buried tryptophan residues (orange). The left domain (green) is C2A. Ca+2/phospholipid binding loops are labeled in C2A as loop 1, 2, and 3. The blue domain is C2B. (B) NMR structure of I27; the buried Trp is shown in orange. (C) Chemical denaturation curves for human Syt1 C2AB domains (black squares) and titin I27 domain (gray circles). The data were fit by a simple sigmoid. The black horizontal line demarcates the point at which 50% of the normalized fluorescent signal [D]50%. The estimated [D]50% are ∼1.6 M for C2AB and 2.7 M for I27.
The SNARE binding region of Syt1 localizes to the polybasic β4 strand of C2A and the Ca+2-binding loop 1 of C2B (4), whereas the calcium ions localize to a cup-like depression formed from three loops (loops 1, 2, and 3) at the apex of both C2A and C2B (9). Only loops 1 and 3 in the C2 domain contribute the conserved acidic residues that actually coordinate the cation. The apex of loops 1 and 3 of the C2 domains of Syt1 possess hydrophobic residues that insert into the phospholipid membrane (10) and may directly contribute to the fusion process (11). At the core of the C2 domain is a Greek-key folding motif that is conserved among all C2 domains (12). Although the strand connectivity is markedly different, the secondary structure and overall fold of the C2 domain is similar to other β-sandwich proteins that have structural and mechanical roles. Similar proteins include, for example, titin (13–15), fibronectin (16), and neural cell adhesion molecules (17).
The primary sequences of the C2 domains of human Syt1 are 31% identical (56% similar) to each other. The x-ray crystal structures of both the isolated C2A (12) and C2B (18) domains superimpose with a root mean-square deviation of 2.0 Å. Despite having similar structural characteristics, the tandem C2 domains of Syt1 differ in their biochemical and biological functions (19). Studies have demonstrated that the C2A and C2B domains participate at different stages in exocytosis (20), have different affinities for phospholipids (21), and differ in their selectivity for highly charged inositol compounds (22). In addition, our recent high-resolution crystal structure of human Syt1 C2AB shows that the C2B domain of Syt1 can affect the shape of the Ca+2-binding pocket of C2A, thus potentially modifying its Ca+2/phospholipid binding potential (9). To our knowledge, it is presently unclear how these disparate biological observations for binding and function can result from the tandem domain organization of Syt1.
Because the three-dimensional structure and the biochemical characterization of these two domains do not provide a clear explanation for this disparity in biological behavior, one hypothesis is that the biophysical properties of C2A and C2B are fundamentally different. In this study, we compared the mechanical properties of the C2A and C2B domains of Syt1. Stretching a construct containing a C2AB fragment resulted in two distinct unfolding force peaks. The larger force peak of ∼100 pN was identified as C2B. The second peak, which unfolded at ∼50% lower forces, was identified as C2A (∼50 pN); in addition, ∼40% of C2A domains unfold through a mechanical intermediate. Hence, our data show that C2A and C2B have significantly different mechanical properties. This feature of the molecule may be important for the C2 domains of Syt1 to respond asymmetrically to effectors such as SNAREs, phospholipids, and Ca+2.
Materials and Methods
Cloning and expression of C2A and C2AB- I27 protein chimeras for AFM experiments
The C2A domain of human Syt1(residues 140–265) and the C2AB domain of human Syt1 (residues 140–414) were amplified from our GST-Syt1 expression vector by polymerase chain reaction (PCR), using the following primers:
5′-GCGCGCGAGAAACTGGGAAAACTTCAG-3′ (forward primer for C2A and C2AB)
3′-GATCACTAGTACTTTGCAGGTCACGCCATTC-5′ (reverse primer for C2A)
3′-GATCACTAGTTTACTTCTTGACGGCCAGCA-5′ (reverse primer for C2AB)
The PCR products were gel purified and cloned into the pDrive direct cloning vector (Qiagen, Germantown, MD). Each clone was excised from pDrive with BssHII and SpeI and then subcloned into a modified version of the pRSETA vector, which includes a His-tag at the N-terminus (23). Both constructs (I272-C2A-I272 and I272-C2AB-I272) were confirmed by DNA sequencing. The expression vector was transformed into Escherichia coli BL21(DE3) cells (Novagen, Darmstadt, Germany). Transformed colonies were grown overnight at 37°C in 5 mL Luria-Bertani (LB) medium containing 100 μg/mL ampicillin. The overnight culture was reinoculated into 1 L fresh Terrific broth (TB) medium, and the cells were grown at 37°C and induced with 400 μM isopropyl-beta-D-thiogalactopyranoside (IPTG) when the OD600 reached 0.7. The cells were collected by centrifugation, quick frozen in liquid nitrogen, and stored at −80°C. A total of 5 g of cells were sonicated in 50 mL lysis buffer (1× phosphated-buffered saline (PBS), pH 7.4, 1 mg/mL lysozyme, 0.1 mg/mL DNase I) and centrifuged at 15,000 rpm for 30 min at 4°C. The proteins were purified by Ni+2 affinity chromatography, eluted with elution buffer (1× PBS, 250 mM imidazole, pH 7.4), concentrated to 6 mg/mL, and stored at 4°C.
Cloning and expression of C2AB and titin I27 domains for chemical denaturation experiments
The C2AB domain of human Syt1 (residues 140–418) was amplified from a human hippocampus QUICK-Clone cDNA library (Clontech, Mountain View, CA) by PCR using the following primers:
5′GGATCCGAGAAACTGGGAAAACTTCAGTATTCACTGGATTATG 3′
3′ TCACTATTACTTCTTGACGGCCAGCATGGC5′
The PCR reactions were gel purified and cloned into the pCR2.1 TA-cloning vector (Invitrogen). The gene was excised from pCR2.1 with BamHI and EcoRI, and then subcloned into pGEX-4T (GE Healthcare Life Sciences, Chalfont St. Giles, UK). The recombinant vector was transformed into E. coli Rosetta cells (Novagen). Heterologous gene expression was induced by adding 400 μM IPTG to a culture in 1 L TB for 4 h at 37°C. C2AB was initially purified using glutathione S-transferase (GST) affinity resin and cation-exchange chromatography. The GST tag was removed using human α-thrombin (50 U/mL total protein), and final purification was carried out using a gel filtration column (Superdex 75; GE Healthcare Life Sciences). The purified C2AB was concentrated to 24 mg/mL, divided into aliquots, and quick frozen in a liquid nitrogen bath. Samples were stored at −80°C. The titin I27 was cloned and expressed as described previously (24). The purity of the proteins was confirmed by sodium dodecyl-sulfate-polyacrylamide gel electrophoresis.
Single-molecule AFM
The mechanical properties of single proteins were studied using a homebuilt single-molecule AFM as described previously (16,24–27). The spring constant of each individual cantilever (silicon nitride gold-coated cantilevers, MLCT-AUHW; Veeco Metrology Group, Santa Barbara, CA) was calculated using the equipartition theorem (28). The cantilever spring constant varied between 30 and 50 pN/nm, and the root mean-square force noise (1 kHz bandwidth) was ∼15 pN. Unless noted, the pulling speed of the different force-extension curves was in the range of 0.5 to 0.7 nm/ms. The loading rate was calculated by multiplying the pulling speed (nanometer/second) by the cantilever spring constant (piconewton/nanometer).
Single protein mechanics
In a typical experiment, a small aliquot of the purified proteins (∼1–50 μL, 10–100 μg/mL) was allowed to adsorb to a clean glass coverslip (∼10 min) and then rinsed with PBS at pH 7.4. We also tested other substrates such as Ni-NTA-coated surfaces (29). We found that C2 protein constructs adsorbed well to glass, gold-coated glass, or Ni-NTA-coated coverslips. We obtained identical data with these different substrates. Proteins were picked up randomly by adsorption to the cantilever tip, which was pressed down onto the sample for 1 to 2 s at forces of several nanonewtons and then stretched for several hundred nanometers. The probability of picking up a protein was typically kept low (<1 in 50 attempts) by controlling the amount of protein used to prepare the coverslips (see Methods in Supporting Material).
In AFM experiments, the unfolding force depends on the pulling direction (30). The magnitude of this geometrical error can be significant for long proteins. For the polyproteins used in this study, however, the maximum error due to the pulling geometry in the measurement of the increase in contour length upon unfolding is < 1% (30).
Equilibrium denaturation of the I27 and C2AB domains
The stability of the 127 and C2AB domains was determined by using equilibrium guanidinium chloride (GdmCl) denaturation. The experiments were carried out at 28°C in PBS buffer. Protein concentration was 1–2 μM. The C2AB protein has an emission maximum ∼345 nm (data not shown), suggesting that the tryptophan residues are relatively exposed to the solvent. The C2A domain does not have a buried tryptophan residue, whereas the C2B domain does. Hence, the fluorescence emission comes from the buried tryptophan residues in C2B, and the denaturation signal results mainly from the C2B domain. Unfolding was monitored by change in fluorescence at 345 nm for C2AB and at 320 nm for I27 (excitation 290 nm) using a spectrofluorimeter (LS-50B; Perkin-Elmer, Waltham, MA). Protein samples were incubated at various GdmCl concentrations overnight to ensure that equilibrium was achieved. The emission spectra were stable after 12 h, demonstrating that the fraction of folded and unfolded molecules had reached equilibrium. In addition, the emission spectra for C2AB at a GdmCl concentration >3 M did not change, indicating that the majority of the molecules were unfolded above this concentration.
Steered molecular dynamics (SMD) simulations
We simulated the unfolding of C2AB using steered molecular dynamics (SMD) as implemented in NAMD (31,32). Coulombic forces were restricted using the switching function from 10 Å to a cutoff at 12 Å. The CHARMM22 force field was used throughout the simulations. C2AB (Protein Data Bank code 2R83) was solvated in a water sphere with a boundary of 15 Å. The system was charge neutralized by adding Na+ and Cl− ions. The total ionic strength of the system corresponded to a final concentration of 0.1 M. This simulation contained a total of 13762 atoms. The system was then minimized with 1000 steps of conjugate gradient minimization from an initial temperature of 310 K. This step was followed by a 400 ps MD simulation to equilibrate the entire system (protein, water, and ions). For the SMD portion of the simulation, a spring constant κ of 10 kBT Å−2 was used. Simulated force was applied by fixing one termini of the protein and moving the SMD atom with constant velocity along a predetermined vector. The trajectories were recorded every 2 fs and analyzed with VMD. The C2AB fragment of Syt1 was stretched at a constant velocity of 0.001 Å ps−1 and was followed for ∼260 Å. We ran three simulations of the extension of C2AB in both pulling directions (N→C and C→N) with similar results.
Results
Equilibrium denaturation of C2AB and titin I27 domains
As a first step to analyze the stability of the C2AB domains, we used chemical denaturation with GdmCl and steady-state fluorescence techniques to determine the thermodynamic stability of the domains. As a reference, we used the titin I27 domain, which has been extensively studied using both chemical and mechanical denaturation techniques (24,33–35). Furthermore, both the I27 domain and the C2 domain are similarly sized β-sheet domains that are constructed around a central Greek-key folding motif (12,36). In the crystal structure of C2AB (Fig. 1 A), two of its three tryptophan residues are exposed to the solvent (yellow). C2A possesses a single solvent-exposed tryptophan (W259), whereas C2B has both a solvent-exposed tryptophan (W404) and a partially buried tryptophan (W358). Therefore, the main contribution to the fluorescence intensity and emission arises from the buried tryptophan in C2B (orange). The I27 domain of titin has a single, buried tryptophan residue (Fig. 1 B). As shown by the denaturation curve in Fig. 1 C, the C2AB protein readily denatures when exposed to GdmCl (black squares). The fluorescent signal rapidly changes between ∼1 and 2 M GdmCl, with a [D]50% ∼1.6 M. In contrast, the denaturation curve for I27 shows [D]50% ∼2.7 M GdmCl (Fig. 1 C (37)). The simplest explanation for this ∼twofold difference in [D]50% is that the C2B domains are thermodynamically less stable than the titin I27 domain.
Mechanical stability of domain C2AB
To measure the single-molecule mechanical properties of the C2AB domains of Syt1, we constructed a protein chimera consisting of a C2AB module flanked on the N- and C-termini by two I27 domains (I27)2-C2AB-(I27)2. Fig. 2 A shows that stretching of the (I27)2-C2AB-(I27)2 polyprotein results in a force-extension curve with a characteristic sawtooth pattern with several force peaks. We found that most recordings showed two levels of unfolding forces (Fig. 2 A): low force peaks (∼10–150 pN) and high force peaks (150–250 pN). To establish a molecular fingerprint for each domain in the protein chimera, we analyzed the spacing between peaks in the sawtooth patterns. We used the worm-like chain (WLC) model for polymer elasticity, which predicts the entropic restoring force F generated upon the extension x of a polymer (38,39). The thin red and black lines in Fig. 2 A correspond to fits of the WLC equation to the curve that precedes each force peak. The I27 domains have been shown to unfold at forces of ∼200 pN and produce an increase in contour length (ΔLc)of ∼28 nm upon unfolding (13,24). Hence, in this recording, the last four force peaks must correspond to the titin I27 domain and the first two force peaks to C2 domains. As shown in Fig. 2 B, we observed a wide range of ΔLc values (from ∼25 to 60 nm) for C2 domains, with a mean ΔLc of 43 ± 8 nm (n = 98). An unfolding force histogram (Fig. 2 C) showed that C2 domains unfold at forces of 62 ± 30 pN (n = 279; red), a value that is threefold lower than the titin I27 domain (188 ± 29 pN, n = 264; black). Furthermore, a loading rate-dependence plot comparing the I27 domain with the C2 domains confirms that this threefold lower unfolding force is maintained over a wide range of loading rates (Fig. 2 D). These data demonstrate that C2 domains have a lower mechanical stability than titin I27 domains.
Figure 2.
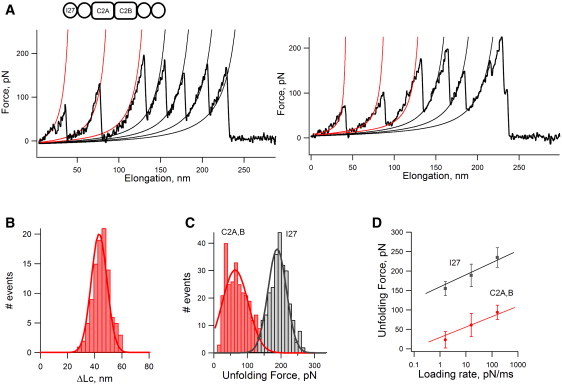
Mechanical properties of C2AB. (A) Typical force-extension curve obtained after stretching a I272-C2AB-I272 protein. The I27 domains unfold at forces of ∼190 pN and produces an increase in contour length (ΔLc) of ∼28 nm. These values were identified by measuring the spacing between force peaks using the WLC equation (thin black lines). In this recording, four I27 domains are unfolded, and the first force peaks must correspond to the unfolding of C2 domains. (B) For C2 domains, we measured a ΔLc of ∼43 ± 8 nm (n = 98). (C) An unfolding force histogram shows that C2 domains must unfold at forces of 62 ± 30 pN (n = 279; red), a value that is threefold lower than that for I27 (188 ± 29 pN, n = 264; black). (D) A plot of unfolding force versus the loading rate shows that the threefold difference between C2 and I27 domains is maintained over a wide range of loading rates.
Mechanical stability of C2A domains
Although we can confidently discriminate between C2 domains and the I27 titin domain with these data, the identity of the individual C2 domains in these recordings cannot be established at this point. To unambiguously identify the force peaks from each C2 domain, we constructed a polyprotein chimera that contains one repeat of C2A and two flanking titin I27 domains. Fig. 3 A shows two examples of force-extension curves obtained after stretching I272-C2A-I272 proteins. We demonstrated that most C2A domains unfolded in a two-state manner, with a ΔLc of ∼40 nm (39.6 ± 4.5 nm, n = 51). This finding corresponds well with the predicted ΔLc, which is ∼40 nm. This calculation was carried out by assuming that the length of a single stretch of amino acids (aa) is ∼0.36 nm (40), which corresponds to the separation of the Cα atoms of two adjacent aa in the extended conformation. For C2A, therefore, we calculated 124 aa × 0.36 nm/aa = 44.6 nm. We subtracted the diameter of the folded domain (4 nm) from this value, giving a theoretical contour length of 40.6 nm, which is very close to the experimental one. This value is also similar to that measured for C2A polyproteins (41). The recording shown in the left panel (Fig. 3 A, left) is a representative example. In these recordings, the C2A unfolded at forces of ∼50 pN (51 ± 14 pN; n = 78; Fig. 3 C). However, we also observed that ∼40% of C2A domains unfolded in a more complex pattern, as shown in the right panel of Fig. 3 A. In these cases, the domain unfolded in two steps: The first step elongated the protein by ∼7 nm (7.4 ± 3.5 nm, n = 31; blue bars) and the second step elongated the protein by ∼36 nm. We interpret this pattern as the C2A domains unfolding through a mechanical unfolding intermediate: native to intermediate state (N → I) followed by an intermediate to unfolded state (I → U). The origin of this unfolding intermediate remains unclear, but it may correspond to the first two β-strands of C2A unfolding before the core (see below).
Figure 3.
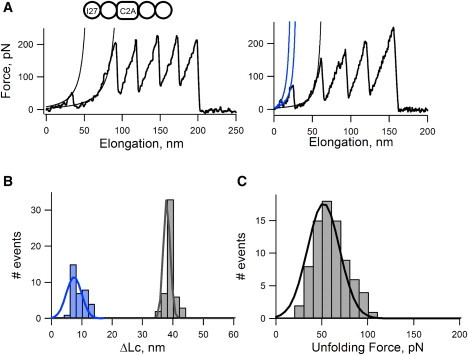
Mechanical properties of C2A. (A) Two examples of force-extension curves obtained after stretching I272-C2A-I272 proteins. The thin lines correspond to fits to the WLC equation. In the example shown on the right, the C2A force peak precedes a low force peak. (B) Histogram of increases in contour lengths measured for the C2A domain. Most domains unfold in an all-or-none fashion in which the ΔLc is ∼40nm (39.6 ± 4.5 nm, n = 51). However, we also observed ∼38% (31 of 82 recordings) of domains unfolded through an intermediate, which contributes to an increase in contour length of ∼7 nm (7.4 ± 3.5 nm, n = 31; blue bars). (C) An unfolding force histogram shows that C2A domains have unfolding forces of 51 ± 14 pN (n = 78).
Speed dependence of C2A versus C2B
Now that we have established the mechanical fingerprint for C2A, we can reanalyze the C2AB data shown in Fig. 2. The C2A domain unfolds at forces of ∼50 pN and, at times, through a mechanical unfolding intermediate. This observation describes the mechanical “fingerprint” of the C2A domain. For example, stretching the (I27)2-C2AB-(I27)2 protein results in a sawtooth pattern with six force peaks (Figs. 2 A and 4 A). We identify the first force peak as the C2A domain, the second peak as the C2B domain, and the last four peaks as I27 domains. Using this mechanical fingerprinting approach, we find that the C2B domain unfolds at higher forces than the C2A domain. Fig. 4 B shows a histogram for unfolding forces for the peaks identified as C2A (gray bars) and C2B (red bars); the average unfolding forces are ∼50 pN (49 ± 18 pN, n = 91) and ∼100 pN (106 ± 23 pN, n = 110), respectively. There is some overlap between the force histograms of C2A and C2B. Because of the stochastic nature of the unfolding patterns, the C2 domains may sometimes be misassigned. This error can be minimized by analyzing sawtooth patterns where the force peaks corresponding to C2 domains are distinct, as shown in Fig. 4 A. In this example, the first two unfolding peaks in the sawtooth pattern are clearly different: The first peak (marked with a dotted black arrow) shows an unfolding intermediate, and the main unfolding event is observed at a lower force than the second force peak (marked with a dotted red arrow). Here, we assign the first peak as the C2A domain and the second peak as the C2B domain.
Figure 4.
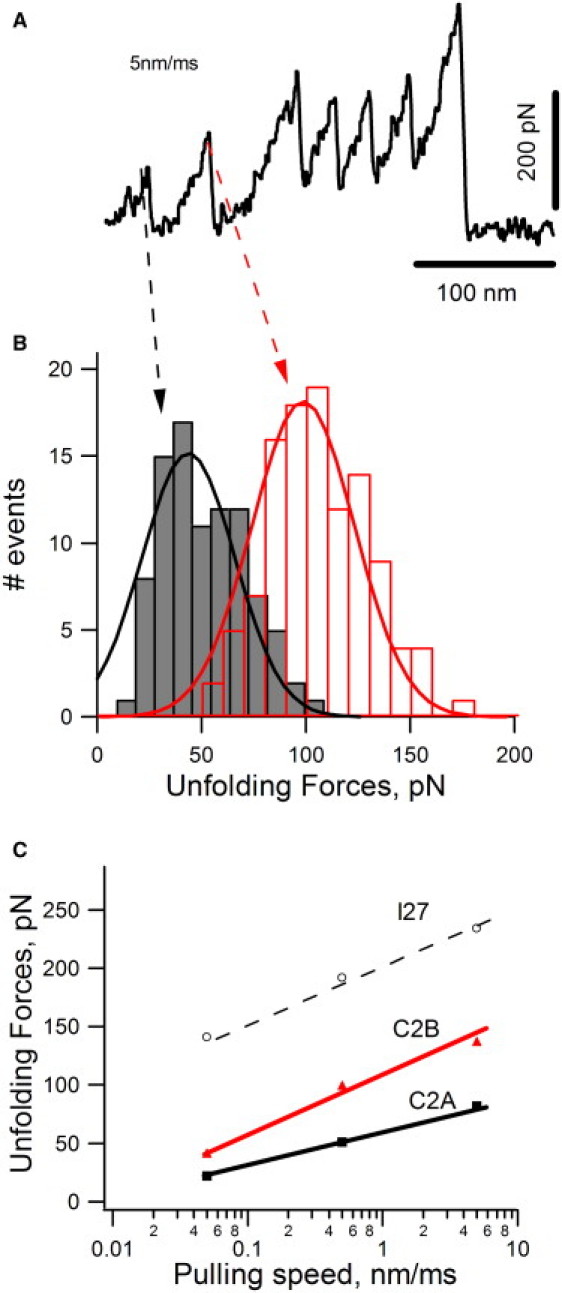
Mechanical properties of C2A versus C2B domains. (A) Force-extension curve obtained after stretching a I272-C2AB-I272 protein at 5 nm/ms. We identified the first peak as the mechanical unfolding of the C2A domain and the second as the unfolding of the C2B domain. (B) Unfolding force histogram for C2A (black) and C2B (red) domains. The average unfolding forces are ∼50 pN (49 ± 18 pN, n = 91) and ∼100 pN (106 ± 23 pN, n = 110), respectively. (C) Plot of unfolding forces versus loading rate for C2A (black squares), C2B (red triangles), and I27 (open circles). A 100-fold increase in loading rate increases the unfolding forces by 60 pN for C2A, 96 pN for C2B, and 93 pN for I27.
We now can study the mechanical properties of each domain separately under different conditions. One important variable is how the unfolding forces vary with the loading rate. Fig. 4 A shows an experiment carried out at 250 pN/ms, which is 10× faster than the normal loading rate. The sawtooth pattern is similar to that obtained at 25 pN/ms except that the unfolding forces are ∼1.5× higher. A plot of the loading rate dependence (Fig. 4 C) shows that a 100-fold increase in loading rate increases the unfolding forces by 60 pN for C2A and 96 pN for C2B. These results further demonstrate that the C2A and C2B domains of Syt1 have distinct mechanical properties.
Molecular basis for differences between C2A and C2B
To formulate a domain-level explanation for the differences between C2A and C2B domains, we simulated the extension of C2AB using SMD (32,42). For this simulation, we fixed the Cα position of residue 140 in C2A and extended the chain by applying an external pulling force on residue 414 in C2B. These forces were applied by harmonically restraining the C-terminal Cα atom of C2B and moving that point at a constant velocity along a defined vector. In Fig. 5, we pulled the human Syt1-C2AB structure at a constant velocity of 0.001 Å ps−1 for 200 Å. The initial events noted in this simulation are β1 and β2 decoupling from the one side of the β-sheet in C2A. Because β2 is linked to loop 1, the shape of the Ca+2-binding pocket was severely distorted. Loop 1 is one of the three Ca+2/phospholipid binding loops in Syt1, because it has two of the five Ca+2 binding residues of C2A (Asp-172 and Asp-178) and one of the hydrophobic residues known to interact with phospholipids (Met-174). The C2B domain was not distorted to the same degree over the same time period. We obtained similar results regardless of pulling direction and speed. In this series of simulations, we demonstrated that C2A denatures first, followed by C2B, which is consistent with our force spectroscopy results.
Figure 5.
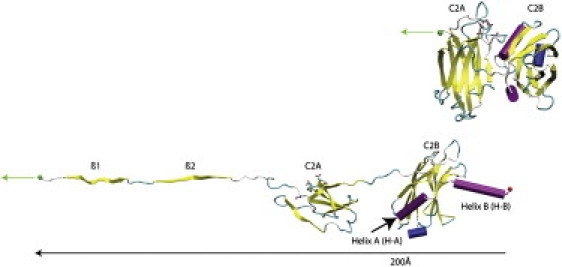
SMD simulation of C2A and C2B domains. Two snapshots of the extension C2AB (residues 140–414) pulled at a constant velocity of 0.001Å ps−1 over 20 nm. The fixed atom was in residue 414 in C2B. The top structure represents the initial conformation of C2AB, and the bottom structure represents the conformation after 200 Å in the simulation. The red sphere corresponds to the fixed atom, and the green sphere and arrow correspond to the moving atom. The Ca+2-binding residues of C2A are highlighted as sticks. The Figure was rendered using VMD and Tachyon.
Discussion
The C2A and C2B domains of Syt1 are similar with respect to their overall sequence similarity and their three-dimensional structures. In addition, they bind Ca+2 and phospholipids with similar affinities, yet they play different roles in exocytosis (19). This observation implies that there are marked differences between the two domains that are crucial to function, yet not obvious with respect to their primary or tertiary structure. To probe these differences, we analyzed the C2 domains of Syt1 using single-molecule AFM. Our analysis shows that 1), the C2 domains are thermodynamically less stable compared to other β-sheet domains; and 2), the C2A and C2B domains have different mechanical stabilities. The C2B domain is relatively strong and unfolds at ∼100 pN in an all-or-none manner. The C2A domain, in contrast, is significantly weaker and tends to unfold in two steps through a mechanical unfolding intermediate.
The stability of human Syt1-C2 domains
To examine the stability of the C2AB portion of Syt1 relative to control domains, we analyzed the C2AB domains of Syt1 and the I27 domain of titin using steady-state chemical denaturation. Our analysis of the C2AB domains of Syt1 shows that the C2 domains of Syt1 are thermodynamically less stable than the I27 domain of titin. Because the C2A domain lacks an appropriate environmentally sensitive fluorophore, we relied on the partially buried Trp in C2B. This experiment showed that the C2B domain, as a part of C2AB, is significantly less stable than titin I27 domains. This finding is an interesting result because the I27 domain and both C2 domains are of similar size and overall fold.
Our single-molecule AFM results show that C2 domains are also less stable than I27 titin domains. The relatively low resistance to mechanical forces of C2 domains lies in their topologies. The C2 domain topology lacks the force-bearing terminal β-strand architecture of the I27 domain (42). The hydrogen bonds holding C2 domains together are parallel to the axis of extension, and so they are in a “zipper-like” configuration (42). This finding implies that the bonds must break sequentially, causing the strands to separate at a lower force.
C2B has a higher mechanical stability than C2A
Because the two C2 domains of Syt1 possess similar topology, one would expect both to possess similar mechanical properties; however, we found that C2A unfolds at ∼50 pN, whereas C2B unfolds at ∼100 pN. One possible explanation for these differences is the presence of the helix A within the C2B fold (Fig. 5). Based on the SMD simulations, we discovered that the helix A stabilizes β8 by contributing additional H-bonds and hydrophobic interactions with core packing residues. β8 forms backbone H-bonds with β1 to form one edge of one β-sheet of the C2B domain; stabilizing this interaction, therefore, would add strength to the entire domain. A total of 16 different isoforms of synaptotagmin have been identified within the human genome, and all have residues that correspond to the helix A in their respective C2B domains (43). Therefore, it is likely that the difference in mechanical responsiveness between the C2A and C2B domains is a common property of all synaptotagmin isoforms.
Mechanical unfolding intermediate in C2A
We observed that ∼40% of C2A domains unfolded in a more complex pattern, as shown in the right panel of Fig. 3 A. In these cases, the domain unfolds in two steps: The first step elongates the protein by ∼7 nm (7.4 ± 3.5 nm, n = 31; blue bars), and the second step elongates the protein by ∼36 nm. We interpret this pattern as the C2A domains unfolding through a mechanical unfolding intermediate: native to intermediate state (N → I) followed by an intermediate to unfolded state (I → U).
Mechanical unfolding intermediates have been observed in other β-sandwich folds such as FNIII (44) and filamin (45). By analyzing both the wild-type and mutant forms of 10FNIII, Li et al. (44) demonstrated that the unfolding intermediate observed in 10FNIII was due to the A and G β-strands detaching from the domain followed by the unfolding of the remainder of the protein. They concluded that the intermediate in FNIII could protect the domain from complete unfolding in response to an applied force (44). In contrast, our data suggest that the unfolding intermediate is not very stable, because the initial peak has a low force value (<30 pN). C2A may readily respond to stretching forces by partially unfolding the first ∼20 aa, which may correspond to β1 and β2 in Fig. 5. Interestingly, because β2 is linked to the Ca+2-binding loop 1, this unfolding event might be important to modulate Ca+2/phospholipid binding in C2A but not in C2B.
Potential effects of mechanical forces on Syt1 C2 domain Ca+2-binding loops
How can the different stabilities between C2A and C2B fit into the exocytotic pathway? Although synaptotagmin has been identified as a Ca+2 trigger for exocytosis (46–48), there is also evidence that it participates in a more direct way by mediating the final step in fusion pore formation (49–51). The force difference that we measured by single-molecule AFM could mimic the strain that Syt1 experiences during regulated exocytosis. In the simplest scenario, we assume that one end (the N-terminal) is anchored to the synaptic vesicle membrane and that the other end is bound to the plasma membrane lipids and the SNARE complex through the C2 domains. When the fusion machinery pulls a vesicle toward the presynaptic terminus, the Syt1 protein experiences a strain due to its linkage between the vesicle and its interactions with the target phospholipid bilayer and/or the SNARE complex. This linkage could restrain motion of the C2 domains of Syt1, thereby inducing mechanical strain. The induced strain could have a significant effect on Syt1 function. For example, a force applied to C2A could easily perturb the structure of loop 1 in C2A after it binds to the target membrane, thus delocalizing any calcium ions within the Ca+2-binding pocket. SMD simulations of 10FNIII suggest that a force applied to one terminus of the domain results in a deformation of the integrin-binding loop. A shortening of this loop could potentially reduce the accessibility to its binding partners, thereby modulating its affinity (52). In the case of Syt1, the divalent cation-binding pocket of C2A provides the negatively charged quenching field to cancel the charge from the Ca+2. This quenching field would be dependent on the tertiary structure of the domain (53). Modification of this quenching field would spontaneously cause a buildup of a strong, localized electric field within the membrane at the site of exocytosis (54,55). Based on our SMD results, which are shown in Fig. 5, we hypothesize that a relatively small stretching force may be sufficient to deform the Ca+2-binding loop and SNARE bindings regions of C2A. These built-in mechanical sensitive switches may be important in modulating the affinity for Syt1 binding partners.
Acknowledgments
The authors thank Tzintzuni Garcia for help with the artwork in Fig. 1. We also thank Dr. Jane Clarke for providing the multimeric I27-pRSET expression vector.
This work was funded by a Career Award in the Biomedical Sciences from the Burroughs-Wellcome Fund (to R.B.S.), in part by the National Institutes of Health grants R21MH070589 and R01DK073394, training fellowship T32GM008280 from the Houston Area Molecular Biophysics Program (to K.F.), the John Sealy Memorial Endowment Fund for Biomedical Research, and by the Polycystic Kidney Foundation (grant 116a2r, to A.F.O.).
Contributor Information
R. Bryan Sutton, Email: rbsutton@utmb.edu.
Andres F. Oberhauser, Email: afoberha@utmb.edu.
Supporting Material
References
- 1.Rothman J.E., Sollner T.H. Throttles and dampers: controlling the engine of membrane fusion. Science. 1997;276:1212–1213. doi: 10.1126/science.276.5316.1212. [DOI] [PubMed] [Google Scholar]
- 2.Jahn R., Scheller R.H. SNAREs–engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 3.Liu W., Montana V., Bai J., Chapman E.R., Mohideen U. Single molecule mechanical probing of the SNARE protein interactions. Biophys. J. 2006;91:744–758. doi: 10.1529/biophysj.105.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lynch K.L., Gerona R.R., Larsen E.C., Marcia R.F., Mitchell J.C. Synaptotagmin C2A loop 2 mediates Ca2+-dependent SNARE interactions essential for Ca2+-triggered vesicle exocytosis. Mol. Biol. Cell. 2007;18:4957–4968. doi: 10.1091/mbc.E07-04-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutton R.B., Ernst J.A., Brunger A.T. Crystal structure of the cytosolic C2A–C2B domains of synaptotagmin III. Implications for Ca(+2)-independent snare complex interaction. J. Cell Biol. 1999;147:589–598. doi: 10.1083/jcb.147.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugita S., Shin O.H., Han W., Lao Y., Sudhof T.C. Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J. 2002;21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yersin A., Hirling H., Steiner P., Magnin S., Regazzi R. Interactions between synaptic vesicle fusion proteins explored by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 2003;100:8736–8741. doi: 10.1073/pnas.1533137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li F., Pincet F., Perez E., Eng W.S., Melia T.J. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat. Struct. Mol. Biol. 2007;14:890–896. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- 9.Fuson K.L., Montes M., Robert J.J., Sutton R.B. Structure of human synaptotagmin 1 C2AB in the absence of Ca2+ reveals a novel domain association. Biochemistry. 2007;46:13041–13048. doi: 10.1021/bi701651k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapman E.R., Davis A.F. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J. Biol. Chem. 1998;273:13995–14001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- 11.Frazier A.A., Roller C.R., Havelka J.J., Hinderliter A., Cafiso D.S. Membrane-bound orientation and position of the synaptotagmin I C2A domain by site-directed spin labeling. Biochemistry. 2003;42:96–105. doi: 10.1021/bi0268145. [DOI] [PubMed] [Google Scholar]
- 12.Sutton R.B., Davletov B.A., Berghuis A.M., Sudhof T.C., Sprang S.R. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell. 1995;80:929–938. doi: 10.1016/0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- 13.Rief M., Gautel M., Oesterhelt F., Fernandez J.M., Gaub H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 14.Li H., Linke W.A., Oberhauser A.F., Carrion-Vazquez M., Kerkvliet J.G. Reverse engineering of the giant muscle protein titin. Nature. 2002;418:998–1002. doi: 10.1038/nature00938. [DOI] [PubMed] [Google Scholar]
- 15.Fowler S.B., Best R.B., Toca Herrera J.L., Rutherford T.J., Steward A. Mechanical unfolding of a titin Ig domain: structure of unfolding intermediate revealed by combining AFM, molecular dynamics simulations, NMR and protein engineering. J. Mol. Biol. 2002;322:841–849. doi: 10.1016/s0022-2836(02)00805-7. [DOI] [PubMed] [Google Scholar]
- 16.Oberhauser A.F., Badilla-Fernandez C., Carrion-Vazquez M., Fernandez J.M. The mechanical hierarchies of fibronectin observed with single-molecule AFM. J. Mol. Biol. 2002;319:433–447. doi: 10.1016/S0022-2836(02)00306-6. [DOI] [PubMed] [Google Scholar]
- 17.Carl P., Kwok C.H., Manderson G., Speicher D.W., Discher D.E. Forced unfolding modulated by disulfide bonds in the Ig domains of a cell adhesion molecule. Proc. Natl. Acad. Sci. USA. 2001;98:1565–1570. doi: 10.1073/pnas.031409698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Y., Sequeira S.M., Malinina L., Tereshko V., Sollner T.H. Crystallographic identification of Ca2+ and Sr2+ coordination sites in synaptotagmin I C2B domain. Protein Sci. 2004;13:2665–2672. doi: 10.1110/ps.04832604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai J., Chapman E.R. The C2 domains of synaptotagmin–partners in exocytosis. Trends Biochem. Sci. 2004;29:143–151. doi: 10.1016/j.tibs.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Nishiki T., Augustine G.J. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J. Neurosci. 2004;24:8542–8550. doi: 10.1523/JNEUROSCI.2545-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arac D., Chen X., Khant H.A., Ubach J., Ludtke S.J. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- 22.Mikoshiba K., Fukuda M., Ibata K., Kabayama H., Mizutani A. Role of synaptotagmin, a Ca2+ and inositol polyphosphate binding protein, in neurotransmitter release and neurite outgrowth. Chem. Phys. Lipids. 1999;98:59–67. doi: 10.1016/s0009-3084(99)00018-3. [DOI] [PubMed] [Google Scholar]
- 23.Steward A., Toca-Herrera J.L., Clarke J. Versatile cloning system for construction of multimeric proteins for use in atomic force microscopy. Protein Sci. 2002;11:2179–2183. doi: 10.1110/ps.0212702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrion-Vazquez M., Oberhauser A.F., Fowler S.B., Marszalek P.E., Broedel S.E. Mechanical and chemical unfolding of a single protein: a comparison. Proc. Natl. Acad. Sci. USA. 1999;96:3694–3699. doi: 10.1073/pnas.96.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bullard B., Linke W.A., Leonard K. Varieties of elastic protein in invertebrate muscles. J. Muscle Res. Cell Motil. 2002;23:435–447. doi: 10.1023/a:1023454305437. [DOI] [PubMed] [Google Scholar]
- 26.Miller E., Garcia T., Hultgren S., Oberhauser A.F. The mechanical properties of E. coli type 1 pili measured by atomic force microscopy techniques. Biophys. J. 2006;91:3848–3856. doi: 10.1529/biophysj.106.088989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oberhauser A.F., Marszalek P.E., Erickson H.P., Fernandez J.M. The molecular elasticity of the extracellular matrix protein tenascin. Nature. 1998;393:181–185. doi: 10.1038/30270. [DOI] [PubMed] [Google Scholar]
- 28.Florin E.L., Rief M., Lehmann T., Ludvig M., Dornmair C. Sensing specific molecular interactions with the atomic force microscope. Biosens. Bioelectron. 1995;10:895–901. [Google Scholar]
- 29.Klein D.C., Stroh C.M., Jensenius H., van Es M., Kamruzzahan A.S. Covalent immobilization of single proteins on mica for molecular recognition force microscopy. ChemPhysChem. 2003;4:1367–1371. doi: 10.1002/cphc.200300844. [DOI] [PubMed] [Google Scholar]
- 30.Carrion-Vazquez M., Marszalek P.E., Oberhauser A.F., Fernandez J.M. Atomic force microscopy captures length phenotypes in single proteins. Proc. Natl. Acad. Sci. USA. 1999;96:11288–11292. doi: 10.1073/pnas.96.20.11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu H., Isralewitz B., Krammer A., Vogel V., Schulten K. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys. J. 1998;75:662–671. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright C.F., Steward A., Clarke J. Thermodynamic characterisation of two transition states along parallel protein folding pathways. J. Mol. Biol. 2004;338:445–451. doi: 10.1016/j.jmb.2004.02.062. [DOI] [PubMed] [Google Scholar]
- 34.Wright C.F., Lindorff-Larsen K., Randles L.G., Clarke J. Parallel protein-unfolding pathways revealed and mapped. Nat. Struct. Biol. 2003;10:658–662. doi: 10.1038/nsb947. [DOI] [PubMed] [Google Scholar]
- 35.Li H., Oberhauser A.F., Fowler S.B., Clarke J., Fernandez J.M. Atomic force microscopy reveals the mechanical design of a modular protein. Proc. Natl. Acad. Sci. USA. 2000;97:6527–6531. doi: 10.1073/pnas.120048697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamill S.J., Steward A., Clarke J. The folding of an immunoglobulin-like Greek key protein is defined by a common-core nucleus and regions constrained by topology. J. Mol. Biol. 2000;297:165–178. doi: 10.1006/jmbi.2000.3517. [DOI] [PubMed] [Google Scholar]
- 37.Fowler S.B., Clarke J. Mapping the folding pathway of an immunoglobulin domain: structural detail from Phi value analysis and movement of the transition state. Structure. 2001;9:355–366. doi: 10.1016/s0969-2126(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 38.Bustamante C., Marko J.F., Siggia E.D., Smith S. Entropic elasticity of lambda-phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 39.Marko J.F., Siggia E.D. Stretching DNA. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 40.Dietz H., Rief M. Protein structure by mechanical triangulation. Proc. Natl. Acad. Sci. USA. 2006;103:1244–1247. doi: 10.1073/pnas.0509217103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carrion-Vazquez M., Oberhauser A.F., Fisher T.E., Marszalek P.E., Li H. Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog. Biophys. Mol. Biol. 2000;74:63–91. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 42.Lu H., Schulten K. Steered molecular dynamics simulations of force-induced protein domain unfolding. Proteins. 1999;35:453–463. [PubMed] [Google Scholar]
- 43.Craxton M. Synaptotagmin gene content of the sequenced genomes. BMC Genomics. 2004;5:43. doi: 10.1186/1471-2164-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L., Huang H.H., Badilla C.L., Fernandez J.M. Mechanical unfolding intermediates observed by single-molecule force spectroscopy in a fibronectin type III module. J. Mol. Biol. 2005;345:817–826. doi: 10.1016/j.jmb.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 45.Schwaiger I., Kardinal A., Schleicher M., Noegel A.A., Rief M. A mechanical unfolding intermediate in an actin-crosslinking protein. Nat. Struct. Mol. Biol. 2004;11:81–85. doi: 10.1038/nsmb705. [DOI] [PubMed] [Google Scholar]
- 46.Koh T.W., Bellen H.J. Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci. 2003;26:413–422. doi: 10.1016/S0166-2236(03)00195-4. [DOI] [PubMed] [Google Scholar]
- 47.Yoshihara M., Littleton J.T. Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron. 2002;36:897–908. doi: 10.1016/s0896-6273(02)01065-6. [DOI] [PubMed] [Google Scholar]
- 48.Chapman E.R. Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat. Rev. Mol. Cell Biol. 2002;3:498–508. doi: 10.1038/nrm855. [DOI] [PubMed] [Google Scholar]
- 49.Martens S., Kozlov M.M., McMahon H.T. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- 50.Kertz J.A., Almeida P.F., Frazier A.A., Berg A.K., Hinderliter A. The cooperative response of synaptotagmin I C2A. A hypothesis for a Ca2+-driven molecular hammer. Biophys. J. 2007;92:1409–1418. doi: 10.1529/biophysj.106.087197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang C.T., Bai J., Chang P.Y., Chapman E.R., Jackson M.B. Synaptotagmin-Ca2+ triggers two sequential steps in regulated exocytosis in rat PC12 cells: fusion pore opening and fusion pore dilation. J. Physiol. 2006;570:295–307. doi: 10.1113/jphysiol.2005.097378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krammer A., Lu H., Isralewitz B., Schulten K., Vogel V. Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch. Proc. Natl. Acad. Sci. USA. 1999;96:1351–1356. doi: 10.1073/pnas.96.4.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shao X., Li C., Fernandez I., Zhang X., Sudhof T.C. Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch. Neuron. 1997;18:133–142. doi: 10.1016/s0896-6273(01)80052-0. [DOI] [PubMed] [Google Scholar]
- 54.Rosenheck K. Evaluation of the electrostatic field strength at the site of exocytosis in adrenal chromaffin cells. Biophys. J. 1998;75:1237–1243. doi: 10.1016/S0006-3495(98)74043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luitel P., Schroeter D.F., Powell J.W. Self-electroporation as a model for fusion pore formation. J. Biomol. Struct. Dyn. 2007;24:495–503. doi: 10.1080/07391102.2007.10507138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.