

Abstract
Purpose
To perform a large-scale gene profiling of the liver in a mouse model of fatty liver induced by high carbohydrate (sucrose) diet (HCD) to gain a deeper insight into potential mechanisms of diet-induced hepatic steatosis.
Methods
C57BL/6 male mice were fed either a purified, control diet or a HCD for 16 weeks. HCD feeding led to marked liver steatosis without inflammation or necrosis. The expression of 42,500 genes/sequences was assessed.
Results
A number of genes (471) underwent significant expression changes in HCD- as compared to standard diet-fed mice (n = 5/group; P < 0.01). Of these genes, 211 were down- and 260 up-regulated. The latter group includes 20 genes encoding enzymes involved in carbohydrate conversion to fat. The genes that underwent expression changes perform a large variety of molecular functions, and the vast majority of these have never been tested before in non-alcoholic fatty liver of nutritional origin. They reveal novel aspects of the disease and allow identification of candidate genes that may underlie the initiation of hepatic steatosis and progression to non-alcoholic steatohepatitis.
Conclusions
HCD-fed laboratory animals provide a model of early non-alcoholic fatty liver disease resembling the disease in humans. The genome wide gene profiling of the liver reveals the complexity of the disease, unravels novel aspects of HCD-induced hepatic steatosis, and helps elucidate its nature and mechanisms.
Electronic supplementary material
The online version of this article (doi:10.1007/s12072-007-9025-2) contains supplementary material, which is available to authorized users.
Keywords: Fatty liver, Gene profiling, High carbohydrate diet, Mouse
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a spectrum of pathology ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), and in some instances progressing to cirrhosis and even hepatocellular carcinoma [1]. NAFLD is by far the most frequent cause of abnormal liver enzymes in the United States. Therefore, there is great interest in the potential pathogenesis, prevention, and/or treatment of this disease. Multiple factors have been considered and identified as causes of hepatic steatosis, and they can be classified in two major groups: exogenous and endogenous. Among exogenous factors, hepatotoxic drugs, hepatitis C infection, malnutrition, and, perhaps the most frequently encountered factor, composition and amount of food, have been confirmed as causes of nonalcoholic steatosis. Because of its high prevalence, the disease has become a focus of intensive research and, although some 1,000 studies dealing with the disease have been published in the last 25 years, the mechanisms underlying its occurrence and progression to NASH are not fully elucidated.
Experimental models based on voluntary food intake have been developed to induce hepatic steatosis in laboratory animals [2–8]. The nutritional models using complete diets resemble the human condition in that they contain amounts of lipids or carbohydrates that exceed the energy needs of the body. As a consequence, the body processes and deposits the excessive nutrients as fat regardless of their original chemical nature. No matter the source, the body deposits the fat preferentially in subcutaneous and visceral areas and, to a lesser extent, in the liver. The biochemical pathways involved in fat processing and deposition differ as a function of the origin of fat. The deposition of the excessive dietary fat involves several biochemical events, including digestion, reconstitution in the intestinal epithelium, assembling, transport, and deposition [9]. However, excessive amounts of ingested carbohydrates, mainly hexoses, can be stored only as glycogen and, in significant amounts, only in skeletal muscle and the liver. The glycogen content of these organs at the saturating levels is around 5–6% of their mass. Therefore, after such levels are attained, the excessive carbohydrates cannot be stored further as glycogen; instead the body converts them into fat. The metabolic pathways accomplishing carbohydrate conversion into fat are well known and they were well characterized in earlier studies [10, 11].
During the last 15 years it has become increasingly apparent that macronutrients such as long-chain fatty acids and glucose act as signaling molecules leading to changes in gene expression. Therefore, gene profiling of organs as affected by macronutrients may provide important information on the mechanisms underlying disturbances such as liver steatosis, overweight, obesity, insulin resistance, and others. To our knowledge, no comprehensive, genome-wide gene profiling of hepatic steatosis induced by a high-carbohydrate diet (HCD), without the complications of steatohepatitis, has been reported in either animals or humans. Therefore, this study was undertaken to (i) gain a deeper insight into the biochemical and cell physiological mechanisms associated with HCD-induced liver steatosis and (ii) identify potential “hidden” genes/pathways that may contribute to the progression of liver steatosis to NASH.
The model of HCD-induced liver steatosis used in this study consists of long-term (16 weeks) feeding of an HCD to mice, and resembles the disease in humans. Human clinical investigations have demonstrated that a diet low in fat and rich in carbohydrates (closely resembling the HCD used in our mouse study), even when administered for short periods of time, for example, 5 or 25 days, can lead to occurrence of uncomplicated fatty liver [12–15]. Thus, this mouse model is a highly relevant means of investigating mechanisms of hepatic steatosis.
Comprehensive gene profiling of the liver was performed using the microarray DNA technology, which allows simultaneous assessment of the expression of 42,500 genes/sequences. A number of genes that underwent significant changes were classified according to their function and selected genes were analyzed from the viewpoint of their potential participation in various cellular processes related to nonalcoholic hepatic steatosis.
Materials and methods
Animals and their treatment
The animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, USA, 1996) as approved by the Institutional Animal Care and Use Committee of the University of Louisville (Louisville, KY). Male C57BL/6 mice (Harlan, Indianapolis, IN), weighing 23.5 ± 0.8 g were maintained under standard conditions for 7 days before initiation of study diets. Thereafter, the mice were divided in two groups of 10 individuals each and started on two different diets: high-carbohydrate diet (HCD) and a purified, control diet named herein standard diet (SD; both from Harlan Teklad, Madison, WI). The composition of the HCD was identical to that used by Feldstein et al. [8], that is (in g kg−1), 650 sucrose, 200 casein, 50 corn oil, 40 mineral mixture (AIN-93G-MX), 10 vitamin mixture (AIN-93-VX), 2.5 choline bitartrate, 3.0 dl-methionine, and 10 cellulose.
Animal killing and tissue sampling
After 16 weeks of feeding HCD or SD, the mice were fasted from 2,200 to 8,000 h, anesthetized with urethane (100 mg kg−1 body weight, intraperitoneally) and the abdominal cavity opened. Blood (0.5–0.7 mL) was drawn from the inferior vena cava with citrate-containing syringes, immediately centrifuged, and the plasma was collected. The liver was perfused through the portal vein with 3–5 mL of ice-chilled phosphate-buffered saline (pH 7.4), with the inferior vena cava severed to remove the blood. The left one-third of the left lobe was immersed in formalin while the rest was placed immediately in liquid nitrogen.
Liver histology
Liver sections were stained with hematoxylin-eosin and analyzed for the presence of fat, polymorphonuclear infiltration, and necrotic areas.
Biochemical assays
The following assays were performed using commercial kits; in plasma: glucose, triacylglycerols (TAG), free fatty acids, alanine-2-oxoglutarate aminotransferase (ALT) (Infinity, Thermo Electron Corp., Melbourne, Australia), adiponectin and TNF-α (R&D Systems, Minneapolis, MN), and insulin (Crystal Chem. Inc., Downers Grove, IL); in the liver: TBARS according to Quintanilha et al. [16] and TAG as above. Total RNA was extracted from the liver using a kit (Ambion, Austin, TX, Cat. No. 1924) and purified with Qiagen minicolumns (Qiagen, Valencia, CA; Cat. No. 74104). RNA quality was assessed using Agilent 2100 bioanalyzer and reagents supplied by the manufacturer (Agilent Technologies, Inc., Palo Alto, CA). About 10 μg of total RNA were processed for mRNA expression using the Affymetrix GeneChip MGU 430 2.0 Array (Affymetrix, Santa Clara, CA) and Affymetrix technology. This chip allows testing 42,500 transcripts for their expression.
Quantitative real-time PCR
The amount of mRNA for 10 randomly selected genes was measured by quantitative real-time (RT) PCR for five mice in each group. The Taqman Gold RT-PCR kit (Applied Biosystems, Inc., Foster City, CA) was used for all of the reactions and the manufacturer’s protocol for GAPDH control was followed. For each mouse, 2μg of total RNA and random hexamer primers were used in the initial reverse transcription reaction. Each gene was detected by a revalidated Taqman Gene Expression Assay probe set that was labeled with 6FAM and the amplification step was done in triplicate for each gene in two variants: (i) template and reverse transcriptase present, (ii) no template present, and (iii) no reverse transcriptase present. The PCR amplification was analyzed by an iCycler iQ Real-Time Detection System (BioRad Laboratories, Inc., Hercules, CA) and the resulting expression ratios were calculated by the 2−ΔΔCt method as described in the Technical Bulletin of Gene Expression (Applied Biosystems, 2002).
Assay of protein abundance
The total liver protein extraction, gel electrophoresis, immunoblotting and band visualization were performed using reagents and technology provided by Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The following antibodies were used: GCK (H-88)—sc7908 (Santa Cruz), EGFR (Cell Signaling Technology, Inc., Cat. No. 2232, Danvers, MA), and cytochrome P450 reductase (Abcam, Cat. No. ab13513, Cambridge, MA).
Gene data processing and statistics
Gene data were analyzed with the Affymetrix Microarray Suite 5.0 algorithm to generate signal value and detection label. Only genes that generated 5 present calls in each group (n = 5, in each group, in a one chip—one animal design) were taken into consideration for further statistics and classification. Also, a false discovery rate was set at 10% and calculated for all probe sets. The genes whose expression was changed 1.5-fold or greater in either direction (up or down), in the HCD-fed mice as compared to the SD-fed mice, were given priority in ascribing a potential significance. Finally, the comparison between SD and HCD-fed mice was made on the basis of a P-value of 0.01. A detailed presentation of the gene statistics procedure used in this study was given in an earlier publication from our laboratory [17].
Results
Body weight
At the end of the feeding period, the body weight of the HCD-fed mice was 29% greater than that of the SD-fed mice (P < 0.05). SD-fed mice gained 28% body weight over the initial time point while the HCD-fed mice gained 65% (P < 0.05; Table 1).
Table 1.
Body weight and biochemical parameters of the plasma and liver in SD- and HCD-fed mice at the end of the feeding period
| Parameter/marker | Mean ± SEM* | P | |
|---|---|---|---|
| Standard diet | HCD | ||
| Body weight (g) | 30.1 ± 0.3** | 38.8 ± 1.2** | <0.05 |
| Glucose (mM) | 9.3 ± 0.25 | 14.0 ± 0.75 | <0.001 |
| ALT (mU mL−1) | 28.3 ± 3.0 | 31.2 ± 2.9 | NS |
| Free fatty acids (plasma, mEq dL−1) | 0.50 ± 0.02 | 0.33 ± 0.021 | <0.001 |
| Cholesterol (plasma, mg dL−1) | 36.4 ± 0.86 | 67.4 ± 2.6 | <0.001 |
| Insulin (pg mL−1) | 582 ± 54 | 2,037 ± 360 | <0.001 |
| Triacylglycerols (liver, mg g−1) | 60.2 ± 6.7 | 159.4 ± 14.6 | <0.001 |
| Triacylglycerols (plasma, mg dL−1) | 101.0 ± 9.8 | 92.9 ± 8.3 | NS |
| TBARS (nmol g−1 wet weight) | 90.4 ± 14.5 | 170.9 ± 16.8 | <0.010 |
| TNF-α (pg mL−1) | 40.3 ± 13.4 | 37.6 ± 18.7 | NS |
| Adiponectin (ng mL−1) | 23.7 ± 0.48 | 23.2 ± 2.0 | NS |
* Means ± SEM were calculated for 5 animals in each group
** The initial body weight was 23.5 ± 0.8 g (n = 10)
Blood biochemistry
The data in Table 1 show that, at the time of killing, the HCD-fed animals had increased levels of glucose, cholesterol, and insulin in plasma. Free fatty acids in plasma were significantly lower than in control, SD-fed mice. No significant changes were observed in plasma adiponectin, TNF-α, TAG, and ALT levels.
Liver histology
The histological appearance of the livers was normal in SD-fed mice while in HCD-fed mice the liver displayed fat infiltration with no inflammation or necrosis. The fat infiltration score was estimated to be 60% (3+, according to the method of Järveläinen et al. [18] (Fig. 1a and b).
Fig. 1.

Liver sections of standard diet- (a) and high carbohydrate diet- (b) fed mice. Note the normal appearance of the liver in A and fat accumulation in B. Note that fat accumulation in the hepatocyte has the shape of macrovacuole (arrows). Magnification: 200×
Liver biochemistry
TAG content in the liver of HCD-fed mice was significantly higher (P < 0.001) than in the livers of SD-fed mice. TBARS were also increased (P < 0.01; Table 1).
All these changes demonstrate that the HCD used in these experiments induced typical hepatic steatosis and, for the feeding period employed, the liver did not display markers of hepatitis (inflammation, necrosis, and others). Some of these data resemble, in part, the results reported by Feldstein et al. using HCD feeding [8]. Whether feeding this diet for longer periods of time may lead to NASH remains to be established.
Liver genomics
A numerical account of the genomics data obtained in our experiments is given in the self-explanatory diagram of Fig. 2. Selected genes that underwent a change in expression of 1.5-fold or more in either direction are presented in Tables 2 and 3. The genes in Table 2 were classified according to Bulera et al. [19], with slight modifications [20]. Also a group of genes was selected and tabulated, comprising several glutathione S-transferases, because of their potential participation in the progression of liver steatosis to NASH (Table 4). Two Tables I and II are presented in Microsoft Excel as supplementary material. Table I presents all gene changed by HCD feeding while Table I contains an expanded listing of genes involved in carbohydrate and fat metabolism processed using the DAVID Functional Annotation Chart for gene ontology (GO) and Kegg.
Fig. 2.

Flow chart of numeric distribution of genes and sequences detected in the liver. We propose that the number of genes in the dark ovals should be taken into consideration for future analysis of the genomics in the experimental model used in this study
Table 2.
Selected genes that underwent 2-fold or higher change in their expression in either direction (up or down) in the liver of HCD-fed mice as compared to the liver of SD-fed mice
| Functional group and gene name | Gene bank accession code | Fold change | Expression* | |
|---|---|---|---|---|
| Up | Down | |||
| Apoptosis | ||||
| Phosphatidylserine receptor | AK017622.1 | 2.5 | 489 ± 40 | |
| Cell motility | ||||
| Eps8 (Epidermal growth factor receptor pathway substrate 8 or EGF receptor kinase substrate 8) | NM_007945 | 2.0 | 67 ± 12 | |
| Cell proliferation | ||||
| Cold inducible RNA binding protein | NM_007705.1 | 2.5 | 328 ± 19 | |
| Channels/Transporters | ||||
| Amiloride-sensitive cation channel 5, intestinal | NM_021370.1 | 2.4 | 182 ± 6 | |
| Lipocalin 13 (precursor) (retinoid carrier protein) | BC027556.1 | 2.4 | 256 ± 24 | |
| Complex lipid metabolism | ||||
| Galactocerebrosidase (galactosylceramidase; precursor) (EC 3.2.1.46). | NM_008079.1 | 3.1 | 71 ± 6 | |
| Cytochromes P450 | ||||
| Cytochrome P450, family 17, subfamily a, polypeptide 1 (catalyzes 17-α hydroxylase and 17,20-lyase activities) (EC 1.14.99.9) | NM_007809.1 | 2.6 | 326 ± 23 | |
| P450 (cytochrome) oxidoreductase (EC 1.6.2.4) | NM_008898.1 | 26 | 4,896 ± 306 | |
| Cytokines/Cytokine receptors | ||||
| Emr4 (EGF-like module containing, mucin-like, hormone receptor like sequence 4) | AY032690.1 | 2.9 | 85±20 | |
| Chemokine (C–C) motif ligand 9 (CCL-9; small inducible cytokine A9; macrophage inflammatory protein 1-gamma | AF128196.1 | 2.2 | 2,034 ± 130 | |
| Chemokine (C–C) motif ligand 2 | AF065933.1 | 10.2 | 218 ± 26 | |
| Macrophage inflammatory protein-related protein-2 (MRP-2) | NM_011338 | 2.2 | 4,897 ± 231 | |
| Glutathione metabolism | ||||
| Glutathione S-transferase, μ 3 (EC 2.5.1.18) | JO3953.1 | 3.7 | 5,970 ± 490 | |
| Glutathione S-transferase, alpha 2 (Yc2) (EC 2.5.1.18) | NM_008182 | 8.3 | 9,268 ± 966 | |
| Nucleic acid metabolism | ||||
| Deoxyribonuclease II alpha (EC 3.1.22.1). A role in DNA degradation in apoptosis. | NM_010062.1 | 3.4 | 8,015 ± 529 | |
| Nucleotide metabolism | ||||
| Cytidine deaminase (EC 3.5.4.5) | AK008793.1 | 2.2 | 475 ± 41 | |
| Protein metabolism | ||||
| Ubiquitin specific protease 18 | NM_011909.1 | 2.5 | 332 ± 27 | |
| Serine (or cysteine) proteinase inhibitor, clade B, member 1a | AB030426 | 3.4 | 520 ± 122 | |
| Secretory products | ||||
| Intestinal trefoil factor (TFF3 precursor) | NM_011575.1 | 4.5 | 256 ± 36 | |
| Signaling/signal transduction | ||||
| Membrane anchored glycoprotein RECK (inhibitor of tumour invasion, regulator of MMP-9) | NM_016678.1 | 4.5 | 187 ± 23 | |
| Heat shock protein 1 | NM_013560.1 | 2.1 | 5,538 ± 995 | |
| Guanine nucleotide binding proptein, alpha 14 | NM_008137.1 | 2.3 | 187 ± 27 | |
| Macrophage expressed gene 1 (shares a distant ancestry to perforin) | L20315.1 | 2.5 | 1,453 ± 79 | |
| Calcium/calmodulin-dependent kinase II gamma (Camk2g) | BC025597.1 | 2.1 | 133 ± 6 | |
| LIM homebox protein 2 (Lhx2) | NM_010710.1 | 2.1 | 63 ± 5 | |
| Methyl-CpG binding domain protein 1 | AK007371.1 | 4.5 | 385 ± 37 | |
| Retinoic acid early transcript 1, alpha (Rae-1 alpha) (early mammalian embryogenesis) | NM_009016. 1 | 3.5 | 2,613 ± 460 | |
| STAT-induced STAT inhibitor-2 mRNA | BB244736 | 2.3 | 1545 ± 87 | |
| DNAJ (Hsp40) homolog, subfamily B, member 1(Heat shock 40 kDa protein 1) | NM_018808.1 | 3.7 | 8175 ± 314 | |
| SH3-binding kinase 1 (Sbk) | BC025837.1 | 2.3 | 137 ± 9 | |
| Inhibin E (INHBE) | NM_007945.1 | 2.7 | 67 ± 11 | |
| Xenobiotic metabolism | ||||
| Sulfotransferase 1C1 (cytosolic) | NM_026935.1 | 2.1 | 214 ± 19 | |
* Expressed in intensity reading units (Mean ± SEM, for five animals in each group), for the SD-fed mice. To find the absolute value for the high carbohydrate-diet fed mice, the value given in this column will be multiplied by the value in the column Up or divided by the value in the column Down. The difference between the two groups with regard to gene expression is significant at P < 0.01. A complete Table I) comprising all 451 genes that underwent changes of 1.5-fold or more is available as supplemental material to this article
Table 3.
Selected genes that likely have direct relevance to the biochemical mechanisms underlying HCD-induced fatty liver in the mouse
| Functional group and gene name | Gene bank accession code | Fold change | Expression* |
|---|---|---|---|
| Up | |||
| Carbohydrate metabolism | |||
| Solute carrier family 2 (facilitated glucose transporter, member 5) | NM_019741.1 | 2.7 | 165 ± 8 |
| Glucokinase (Hexokinase D, EC 2.7.1.1) | BC011139.1 | 2.4 | 1,614 ± 202 |
| Phosphoglucomutase 3 (EC 5.4.2.6) | AK013402.1 | 1.6 | 492 ± 53 |
| UDP-glucose pyrophosphorylase 2 (EC 2.7.7.9) | AF424698.1 | 1.6 | 10,426 ± 455 |
| Ketohexokinase (EC 2.7.1.3) | BC013464.1 | 1.7 | 11,278 ± 326 |
| Malic enzyme, supernatant (cytosolic) (EC 1.1.1.40) | NM_008615.1 | 1.8 | 4,292 ± 440 |
| Glucose phosphate isomerase (EC 5.3.1.9) | NM_008155.1 | 1.8 | 3,742 ± 147 |
| Pyruvate dehydrogenase kinase, isoenzyme 4 (EC 2.7.1.99) | NM_013743.1 | 1.8 | 472 ± 24 |
| Pyruvate dehydrogenase kinase, isoenzyme 1 (EC 2.7.1.99) | BC027196.1 | 1.7 | 1,261 ± 61 |
| Glycerolphosphate dehydrogenase 2, mitochondrial (EC 1.1.1.8) | NM_010274.1 | 1.6 | 2,412 ± 240 |
| Dihydrolipoamide S-acetyltransferase (E2 component of pyruvate dehydrogenase complex) (EC 2.3.1.12) | BC026680.1 | 1.6 | 1,815 ± 126 |
| Glucose 6-phosphate dehydrogenase X-linked (EC 1.1.1.49) | NM_008062.1 | 1.8 | 265 ± 38 |
| Fatty acid and complex lipid metabolism | |||
| ATP citrate lyase (EC 4.1.3.8) | BI456232 | 1.9 | 9,127 ± 1072 |
| Fatty acid desaturase 2 (EC 1.14.99.6) | BB430611 | 1.9 | 5,419 ± 390 |
| Glycerol-3-phosphate acyltransferase, mitochondrial (EC 2.3.1.15) | NM_008149.1 | 1.9 | 8,022 ± 395 |
| Monoglyceride lipase (EC 3.1.1.23) | NM_011844.2 | 1.6 | 2,362 ± 128 |
| Peroxisome proliferator-activated receptor alpha | BC016892.1 | 1.7 | 6,062 ± 714 |
| Stearoyl-CoA desaturase (EC 1.14.99.5) | NM_009127.1 | 1.9 | 9,793 ± 1,908 |
| Fatty acid desaturase 2 (Delta-6 desaturase) (EC 1.14.99.5) | NM_019699.1 | 2.5 | 6,486 ± 270 |
| Sterol regulatory element binding factor 1 (SREBP-1) | AI326423 | 1.6 | 5,679 ± 257 |
| Lipocalin 13 (precursor) (retinoid carrier protein) | BC027556.1 | 2.4 | 256 ± 24 |
| Fatty acid binding protein 5, epidermal | BC002008.1 | 2.5 | 3,714 ± 448 |
| Adiponutrin (a triacylglycerol lipase and acylglycerol O-acyltransferase) (EC 3.1.1.3, and EC 2.3.1.-) | NM_054088.1 | 5.6 | 144 ± 8 |
| ELOVL family member 6 (elongation of very long chain fatty acids; a lipogenic enzyme regulated by SREBPs) | NM_130450.1 | 2.4 | 2,787 ± 185 |
* Expressed in intensity reading units (Mean ± SEM, for five animals in each group), for the SD-fed mice. To find the absolute value for the high carbohydrate-diet fed mice, the value given in this column should be multiplied by the value in the column Up. The difference between the two groups with regard to gene expression is significant at P < 0.01. An expanded list of genes involved in carbohydrate and fat metabolism is given in Table I as supplementary material
Table 4.
Down-regulation of glutathione S-transferases in the liver of mice fed a high carbohydrate diet
| Gene name | Access code | Change (-fold) in HCD group |
|---|---|---|
| Glutathione S-transferase μ 1 | J03952.1 | Down 1.8 |
| Glutathione S-transferase θ 3 | BC003903.1 | Down 2.0 |
| Glutathione S-transferase μ 3 | J03953.1 | Down 3.7 |
| Glutathione S-transferase, α 2 (Yc2) | NM_008182.1 | Down 8.0 |
| Glutathione S-transferase α 4 | NM_010357.1 | Down 3.4 |
The QRT-PCR data (Table 5) confirmed changes in gene expression (for 10 genes in each experimental group) obtained with microarray technology. Only minor quantitative differences were observed between the two methods. Such differences are routinely observed in many studies.
Table 5.
Comparison of gene expression changes for control and HCD-fed mice as determined by quantitative RT-PCR and cDNA microarray
| Gene code | Applied Biosystems assay identification | Gene name | Change in expression | |
|---|---|---|---|---|
| Microarray | Q-RT-PCR | |||
| AK003441.1 | Mm00614943_m1 | Ankyrin repeat and KH Domain containing 1 | 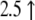 |
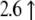 |
| AW489168 | Mm00519268_m1 | Bcl-2 binding component 3 | 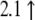 |
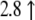 |
| AF065933.1 | Mm00441242_m1 | Chemokine (C-C motif) ligand 2 | 10.2↓ | 2.7↓ |
| NM_009998.1 | Mm00456591_m1 | Cytochrome P450, family 2, subfamily b, poly-peptide 10 | 20.0↓ | ND* |
| NM_010062.1 | Mm00438463_m1 | Deoxyribonuclease II alpha | 3.4↓ | 2.1↓ |
| NM_018808.1 | Mm00444519._m1 | DnaJ (HSP40) Homolog, subfamily b, member 1 | 3.7↓ | 5.8↓ |
| NM_007945 | Mm00514752_m1 | Eps8 (Epidermal growth factor receptor pathway substrate 8) | 2.0↓ | 1.8↓ |
| NM_008182 | Mm00833353_m1 | Glutathione S-transferase α 2 | 8.3↓ | 12.5↓ |
| U72881.1 | Mm00803317_m1 | Regulator of G protein signaling 16 | 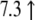 |
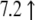 |
| NM_008898.1 | Mm00435876_m1 | P450 Cytochrome oxidoreductase | 26.0↓ | ND* |
* ND, not detected. The relative expression for Q-RT-PCR was normalized to the 18S rRNA copy level. Arrows indicate the direction of change
Protein abundance
The gel images in Fig. 3 show that three proteins randomly selected to be tested for their abundance—glucokinase (BC011139.1), cytochrome-P450 oxidoreductase (NM_008898.1), and Emr4 (AY032690.1; EGF-like module containing mucin-like, hormone receptor-like sequence 4)—changed in the same direction as their transcriptome. A fourth protein, MCP-1 (AF128196.1), tested using both the Western blot method and ELISA, could not be detected. This cytokine, however, was not assayed in plasma. Interpretation of genomic data is mainly based on changes in transcriptome expression rather than in protein abundance. It is generally assumed that changes in protein abundance parallels changes in gene expression. However, if a gene or a set of genes are studied closely, the assessment of their protein products must be performed before functional significance is ascribed to expression changes.
Fig. 3.

Gel images illustrating the Western blot assay of protein abundance. The following genes were tested for their protein abundance: GCK, glucokinase; EGFR, epidermal growth factor-like module containing mucine-like, hormone receptor-like receptor sequence 4 (or Emr4); Cyp450 reducatse, and β-actin. Other abbreviations: SD, standard diet; HCD, high carbohydrate diet. The following values apply for the HCD-to-SD ratio: GCK, 2.00 ± 0.18; Cyp450 reductase, 0.16 ± 0.08; EGFR, 1.23 ± 0.28
Discussion
In this study, 471 genes (Fig. 2) were identified whose expression was changed in the livers of HCD-fed animals. These genes include the ones that encode the enzymes, approximately 20, involved in glucose and fructose metabolism and their conversion to fat [10, 12, 15] (Table 3 and Table I of supplementary material, and Fig. 4). The large number of genes that underwent changes in expression, taken together with the direction of change, and with the functional diversity they belong to, demonstrate that fat accumulation in the hepatocyte in HCD-induced liver steatosis is associated with alterations of a much wider spectrum of biochemical and molecular processes than expected on the basis of the data available thus far. Such a conclusion could have emerged only from the large-scale gene profiling data and supports the usefulness of this tool in unraveling multifaceted mechanisms of disease.
Fig. 4.

Schematic representation of the metabolic pathways involved in carbohydrate conversion to fat in the liver. Represented are glycolysis, part of the citric acid cycle, citrate cleavage enzyme, fatty acid synthase, fatty acyl-CoA desaturase, pentosephosphate pathway and triacylglycerol synthesis. The following nonstandard abbreviations are used: Fru, fructose; Gluc, glucose; Fru-1P, fructose 1-phosphate; Gluc-6-P, glucose 6-phosphate; TrP, triosephosphates; OxAc, oxaloacetate; Citr, citrate; Mal, malonyl-; CAC, citric acid cycle; Facyl-, fatty acyl; –(C=C)–, monounsaturated, long-chain fatty acid; TAG, triacylglycerol; PPP, pentosephosphate pathway. Red triangles denote enzymes or other proteins whose gene expression was upregulated at least 1.5-fold, and they are as follows: 1, glucose transporter 5; 2, ketohexokinase; 3, glucokinase; 4, components of pyruvate dehydrogenase complex (pyruvate dehydrogenase kinase isoenzymes); 5, citrate cleavage enzyme; 6, glucose 6-phosphate dehydrogenase; 7, stearoyl (fatty acyl)-CoA desaturase; 8, acylglycerol O-acyltransferase; 9, malic enzyme (cytosolic and NADP-dependent). The pentosephosphate pathway (PPP) is represented here at the lower right side of the figure by the reaction catalyzed by glucose 6-phosphate dehydrogenase, which was upregulated in HCD-fed animals. Genes encoding enzymes involved in fatty acid β-oxidation, a pathway that may contribute to triacylglycerol accumulation in the liver, were not found to be changed. Also, some of the genes listed in Table 4 are not represented in the figure in order to keep a certain degree of simplicity. Enzyme classification (EC) for the enzymes in the map is given in Table 3
Importantly, a large number of genes involved in carbohydrate conversion to fat were upregulated by the HCD. A group of genes that are of particular interest for understanding potential mechanisms of HCD-induced hepatic steatosis are presented in Table 3. The data in this table demonstrate that, as expected, HCD upregulates several genes directly involved in carbohydrate conversion to fat [11, 21, 22]. These genes have been displayed within the context of the metabolic pathways to which they belong (Fig. 4) in an attempt to facilitate understanding of their role(s) in HCD-induced liver steatosis. In addition, one gene, sterol regulatory element-binding protein (SREBP)-1c, which controls the transcription of genes involved in fatty acid synthesis [23], and whose transcription is regulated by insulin [24], was upregulated. Another gene, peroxisome proliferator-activated receptor (PPAR)-α, also a transcription factor, but encoding enzymes involved in fatty acid oxidation [25], was likewise upregulated.
Of interest, the vast majority of genes identified in this study (Tables 2 and 5, and Table I of supplementary material) cannot be directly linked to the biochemical or molecular processes leading to fatty liver. Changes in expression of these genes likely reflect alterations in cellular processes caused by, rather than leading to, hepatic steatosis. Owing to space limitations, these genes will not be discussed in any detail.
NASH is thought to evolve through a 2-hit process in which the first hit is steatosis. The second hit or hits include multiple factors such as oxidative stress, proinflammatory cytokines, mitochondrial dysfunction, insulin resistance, and even industrial exposures [1]. An important issue is whether the data reported herein provide insights into genes that may predispose the liver to a “second hit,” thus leading to NASH. Since no changes in transcriptome expression of proinflammatory or profibrotic cytokines (e.g., TNF-α, IL-1β, IL-18, TGF-β, and others), classically thought to mediate liver injury including fibrosis, were identified in the HCD-fed mouse liver, it seems that these classic proinflammatory cytokines, at least those secreted in the liver, may not be critically involved in the early aspects of this disease. The lack of TNF-α participation in dietary-induced NASH was recently suggested by Deng et al. [7], who demonstrated that knocking out the TNF-α receptor 1 does not prevent NASH induced by force-feeding a fat-enriched diet. Similar studies by Dela Pena et al. [26] showed that TNFR1 knockout mice still develop hepatic steatosis when fed a methionine-restricted, choline-deficient diet. Moreover, studies in children with NAFLD demonstrate normal serum TNF but decreased adiponectin as early events [27]. Our study does show that the transcriptome of two macrophage inflammatory proteins, MCP-1 and MCP-2 (Table 2), were upregulated, which may predict potential facilitation of extrahepatic cell infiltration into the liver and the initiation of inflammation. Several genes, other than proinflammatory cytokines, may be considered as plausible candidates for a potential role in the progression to NASH. One of these is the macrophage-expressed gene 1 (L20315.1), a relative of perforin (granzyme B), which was upregulated 2.5-fold (Table 2). Another gene is methyl-CpG-binding domain protein 1 (AK007371.1, known as MBD1), a member of a family of five mammalian methyl-CpG-recognizing proteins, which plays a key role in maintaining a transcriptionally inactive state of methylated promoters [28, 29]. This gene was downregulated 4.5-fold. Its downregulation may facilitate expression of genes that otherwise would be in a state of restricted transcription.
Another group of genes of interest for potential progression of the steatotic liver to NASH is represented by glutathione S-transferases (Table 5), which were downregulated in the steatotic liver. Downregulation of these enzymes may lead to a decreased capacity of the liver to detoxify xenobiotics, thereby increasing the susceptibility of the liver to undergo pathologic changes including necrosis. Such changes may be triggered by chemical agents from the environment, and there are well-documented examples of industrial NASH, such as that caused by petrochemical exposure [30, 31].
Lastly, the increased circulating levels of both glucose and insulin recorded in this study suggest the existence of a certain degree of insulin resistance. These findings raise the question of whether the steatotic liver induced by HCD feeding is insulin resistant. The concept of an obligatory association of the steatotic liver with insulin resistance has been challenged by experimental and clinical data. Thus, it has been demonstrated that NASH can occur in the absence of overt insulin resistance [32–36]. On the basis of the data presented in this and other studies, we surmise that, in the model of hepatic steatosis used in this study, and at the moment of animal killing, the liver is not insulin resistant. Thus, (i) the insulin response element-binding protein (IREBP-1) [37, 38], a target of insulin signal transduction downstream of the PI-3K/protein kinase B (Akt) pathway, regulates the expression of many enzymes involved in carbohydrate conversion to fat; this factor can only be active in the presence of an intact insulin signaling cascade, (ii) the enzymes involved in carbohydrate conversion to fat, including glucokinase (EC 2.7.1.1), ketohexokinase (EC 2.7.1.3), glucose-6-phosphate dehydrogenase (EC 1.1.1.49), and others, were upregulated in the liver of the mouse model used in our study (Table 3); such an upregulation could not be accomplished otherwise than through an adequate response of the liver cells to insulin, (iii) nonenzymatic factors involved in lipid synthesis, such as SREBP-1, are also under the control of insulin [24, 39]; the expression of this factor that, in turn, controls several major enzymes involved in fatty acid synthesis [40], was upregulated 1.6-fold (Table 3). Taken together, the gene expression data of this and of cited studies are not compatible with an insulin-resistant liver during the phase of NAFLD in which our animals were killed. Further research is required to study the evolution of the fatty liver, including potential progression to steatohepatitis, in the model used in this study.
In conclusion, our study (i) demonstrates the usefulness of the mouse model of HCD-induced hepatic steatosis for the study of the fatty liver of nutritional origin, (ii) emphasizes the importance of using large-scale gene profiling of the liver in identifying potential causes and understanding the mechanisms underlying the disease, and (iii) offers a database for further investigation of the mechanisms underlying the hepatic steatosis of dietary origin and its potential progression to NASH.
Electronic supplementary material
Below are the link to the electronic supplementary material. Two tables (Microsoft Excel), one comprising all the genes that underwent changes in their expression with statistical processing and another comprising gene ontology for the genes involved in carbohydrate and lipid metabolism.
Acknowledgements
The work reported in this study was supported by National Institutes of Health grants (to I.V.D., Z.S., S.S.B., T.B.K., A.V.S., and C.J.M.) and a Department of Veterans Affairs grant (to C.J.M.).
Abbreviations
- ALT
Alanine-2-oxoglutarate amino transferase (EC 2.6.1.2)
- HCD
High-carbohydrate diet
- NAFLD
Nonalcoholic fatty liver disease
- NASH
Nonalcoholic steatohepatitis
- TBARS
Thiobarbituric acid reactive substances
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s12072-007-9025-2) contains supplementary material, which is available to authorized users.
Ion V. Deaciuc and Zhenyuan Song are contributed equally to this study.
Contributor Information
Ion V. Deaciuc, Phone: +1-502-8523873, FAX: +1-502-5620486, Email: ion.deaciuc@louisville.edu
Zhenyuan Song, Email: z0song0z@louisville.edu.
Xuejun Peng, Email: xpeng@tgrd.com.
Shirish S. Barve, Email: shirish.barve@louisville.edu
Ming Song, Email: m0song03@louisville.edu.
Qiang He, Email: q0he0002@louisville.edu.
Thomas B. Knudsen, Email: thomas.knudsen@louisville.edu
Amar V. Singh, Email: avsing01@louisville.edu
Craig J. McClain, Email: craig.mcclain@louisville.edu
References
- 1.McClain CJ, Mokshagundam PL, Barve SS, Song Z, Hill DB, Chen T, Deaciuc I. Mechanisms of non-alcoholic steatohepatitis. Alcohol 2004;34:67–79. [DOI] [PubMed]
- 2.Den Boer M, Voshol PG, Kuipers F, Havekes LM, Romijn JA. Hepatic steatosis: a mediator of the metabolic syndrome. Lessons from animal models. Artherioscl Thromb Vasc Biol 2004;24:644–9. [DOI] [PubMed]
- 3.Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis 2001;21:89–104. [DOI] [PubMed]
- 4.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, et al. Model of non-alcoholic steatohepatitis. Am J Clin Nutr 2004;79:502–9. [DOI] [PubMed]
- 5.Nanji AA. Animal models of nonalcoholic fatty liver disease and steatohepatitis. Clin Liver Dis 2004;8:559–74. [DOI] [PubMed]
- 6.Portincasa P, Grattagliano I, Palmieri VO, Palasciano G. Nonalcoholic steatohepatitis: recent advances from experimental models to clinical management. Clin Biochem 2005;38:203–17. [DOI] [PubMed]
- 7.Deng QG, She H, Cheng JH, French SW, Koop DR, Xion S, et al. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology 2005;42:905–14. [DOI] [PubMed]
- 8.Feldstein AE, Canbay A, Guicciardi ME, Higuchi H, Bronk SF, Gores GJ. Diet associated steatosis sensitizes to Fas mediated liver injury in mice. J Hepatol 2003;39:978–83. [DOI] [PubMed]
- 9.Friedman HA, Nylund B. Intestinal fat digestion, absorption and transport. Am J Clin Nutr 1980;33:1108–39. [DOI] [PubMed]
- 10.Foufelle F, Girard J, Ferre P. Regulation of lipogenic enzyme expression by glucose in liver and adipose tissue: a review of the potential cellular and molecular mechanisms. Adv Enzyme Regul 1996;36:199–226. [DOI] [PubMed]
- 11.Flatt JP. Conversion of carbohydrate to fat in the adipose tissue: an energy-yielding and, therefore, self-limiting process. J Lip Res 1970;11:131–43. [PubMed]
- 12.Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab 2005;2:5. doi:10.1186/1743-7075-2-5. [DOI] [PMC free article] [PubMed]
- 13.Diraison F, Yankah V, Letexier D, Dusserre E, Jones P, Beylot M. Differences in the regulation of adipose tissue and liver lipogenesis by carbohydrates in humans. J Lipid Res 2003;44:846–53. [DOI] [PubMed]
- 14.Hudgins L, Hellerstein M, Seidman C, Seidman C, Neese R, Diakun J, et al. Human fatty acid synthesis is stimulated by eucaloric low fat, high carbohydrate diet. J Clin Invest 1996;97:2081–91. [DOI] [PMC free article] [PubMed]
- 15.Schwartz JM, Linfoot P, Dare D, Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low carbohydrate and low-fat, high carbohydrate isoenergetic diets. Am J Clin Nutr 2003;77:43–50. [DOI] [PubMed]
- 16.Quintanilha AT, Packer L, Davies JM, Racanelli TM, Davies KJ. Membrane effects of vitamin E deficiency: bioenergetic and surface charge density studies of skeletal muscle and liver mitochondria. Ann NY Acad Sci 1982;393:32–47. [DOI] [PubMed]
- 17.Deaciuc IV, Peng X, D’Souza NB, Shedlofsky SI, Burikhanov R. Microarray gene analysis of the liver in a rat model of chronic, voluntary alcohol uptake. Alcohol 2004;32:113–27. [DOI] [PubMed]
- 18.Järveläinen HA, Fang C, Ingelman-Sundberg M, Lindros KO. Effect of chronic administration of endotoxin and ethanol on rat liver pathology and proinflammatory and antiinflammatory cytokines. Hepatology 1999;29:1602–14. [DOI] [PubMed]
- 19.Bulera SJ, Eddy SM, Ferguson E, Jatkoe TA, Reindel JF, Bleavins MR, et al. RNA expression in the early characterization of hepatotoxicants in Wistar rats by high-density DNA microarrays. Hepatology 2002;33:1239–58. [DOI] [PubMed]
- 20.Deaciuc IV, Doherty DE, Burikhanov R, Lee EY, Stromberg AJ, Peng X, et al. Large-scale gene profiling of the liver in a mouse model of chronic, intragastric ethanol infusion. J Hepatol 2004;40:219–27. [DOI] [PubMed]
- 21.Spencer AF, Lowenstein JM. The supply of precursors for the synthesis of fatty acids. J Biol Chem 1962;237:3640–8. [PubMed]
- 22.Wise EM, Ball EG. Malic enzyme and lipogenesis. Proc Natl Acad Sci USA 1964;52:1255–63. [DOI] [PMC free article] [PubMed]
- 23.Müller-Wieland D, Kotzka J. SREBP-1: gene regulatory key to syndrome X? Ann NY Acad Sci 2002;967:19–27. [DOI] [PubMed]
- 24.Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 2004;86:839–48. [DOI] [PubMed]
- 25.Tan NS, Michalik L, Desvregne B, Wahli W. Multiple expression control mechanisms of peroxisome proliferator-activated receptors and their target genes. J Steroid Biochem Mol Biol 2005;93:99–105. [DOI] [PubMed]
- 26.Dela Pena A, Leclerq I, Field J, George J, Jones B, Farrell G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepetitis. Gastroenterology 2005;129:1663–74. [DOI] [PubMed]
- 27.Louthan MV, Barve S, McClain CJ, Joshi-Barve S. Decreased serum adiponectin: an early event in pediatric non-alcoholic fatty liver disease. J Pediatr 2005;147:835–8. [DOI] [PubMed]
- 28.Ballestar E, Wolffe AP. Methyl-CpG-binding proteins. Targeting specific gene repression. Eur J Biochem 2001;268:1–6. [DOI] [PubMed]
- 29.El-Osta A, Wolffe AP. DNA methylation and histone deacetylation in the control of gene expression: basic biochemistry to human development and disease. Gene Expr 2000;9:63–75. [DOI] [PMC free article] [PubMed]
- 30.Mehlman MA. Dangerous and cancer-causing properties of products and chemicals in the oil refining and petrochemical industry. VIII. Health effects of motor fuels: carcinogenicity of gasoline—scientific update. Environ Res 1992;59(1):238–49. [DOI] [PubMed]
- 31.Cave M, Deaciuc IV, Mendez C, Song Z, Joshi-Barve S, Barve S, et al. Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem 2007;18:184–95. [DOI] [PubMed]
- 32.Adams LA, Angulo P, Lindor KD. Nonalcoholic fatty liver disease. Can Med Assoc J 2005;172:899–905. [DOI] [PMC free article] [PubMed]
- 33.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147–52. [DOI] [PMC free article] [PubMed]
- 34.Browning JD, Szczepaniak LS, Dobbins R, Nurember P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004;40:1387–95. [DOI] [PubMed]
- 35.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med 2004;10:1–7. [DOI] [PubMed]
- 36.Hamaguchi M, Kojima T, Takeda N, Taniguchi H, Fujii K, Omatsu T, et al. The metabolic syndrome as a predictor of non-alcoholic fatty liver disease. Ann Intern Med 2005;143:722–8. [DOI] [PubMed]
- 37.Villafuerte BC, Phillips LS, Rane MJ, Zhao W. Insulin-response element-binding protein 1. A novel Akt substrate involved in transcriptional action of insulin. J Biol Chem 2004;279:36650–9. [DOI] [PubMed]
- 38.Villafuerte BC, Kaytor EN. An insulin-response element-binding protein that ameliorates hyperglycemia in diabetes. J Biol Chem 2005;280:20010–20. [DOI] [PubMed]
- 39.Foufelle F, Ferre P. Regulation of carbohydrate metabolism by insulin: role of transcription factor SREBP-1c in the hepatic transcriptional effects of the hormone. J Soc Biol 2001;195:243–8. [PubMed]
- 40.Griffin MJ, Sul HS. Insulin regulation of fatty acid synthase gene transcription: roles of USF and SREBP-1c. Life 2004;56:595–600. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below are the link to the electronic supplementary material. Two tables (Microsoft Excel), one comprising all the genes that underwent changes in their expression with statistical processing and another comprising gene ontology for the genes involved in carbohydrate and lipid metabolism.