

Abstract
Purpose
Mutations in the DNA damage response gene ATR (exon 10 A10 mononucleotide repeat) have been previously described in endometrial and other cancers with defective DNA mismatch repair. In vitro studies showed that endometrial cancer cell lines with A10 repeat tract truncating mutations have a failure in the ATR-dependent DNA damage response. Cell lines carrying A10 mutations fail to trigger Chk1 activation in response to ionizing radiation and topoisomerase inhibitors. We sought to determine the frequency and clinicopathologic significance of ATR mutations in patients with endometrioid endometrial cancer.
Patients and Methods
The ATR exon 10 A10 repeat was analyzed by direct sequencing in 141 tumors with microsatellite instability (MSI-positive) and 107 microsatellite stable (MSI-negative) tumors. The relationships between mutations and clinicopathologic variables, including overall and disease-free survival, were assessed using contingency table tests and Cox proportional hazard models.
Results
ATR mutations were identified in 12 cases (4.8%; three cases with insertions and nine cases with deletions). Mutations occurred exclusively in MSI-positive tumors (P = .02), with an overall mutation rate of 8.5%. Mutation was not associated with age, race, surgical stage, International Federation of Gynecology and Obstetrics grade, or adjuvant treatment. Multivariate analyses revealed a significant association with reduced overall survival (hazard ratio [HR] = 3.88; 95% CI, 1.64 to 9.18; P = .002) and disease-free survival (HR = 4.29; 95% CI, 1.48 to 12.45; P = .007).
Conclusion
Truncating ATR mutations in endometrial cancers are associated with biologic aggressiveness as evidenced by reduced disease-free and overall survival. Knowledge of ATR mutation status may hold promise for individualized treatment and targeted therapies in patients with endometrial cancer.
INTRODUCTION
Endometrial cancer is the fourth most common malignancy in women in the United States. Approximately 70% of cases present in early stage, and these account for overall long-term survivorship figures in excess of 80%. However, some patients will either present with advanced-stage disease or experience disease recurrence or progression despite an initially anticipated good prognosis. In fact, more than 7,000 women succumbed to this disease in 2008.1,2 Several clinicopathologic models have been proposed to identify patients at risk of recurrence and death from endometrial cancer. These strategies have the ultimate objective of identifying individuals who would most benefit from postoperative therapeutic interventions, such as radiation and chemotherapy alone or in combination.3,4
To date, the clinical utility of various clinical, surgical, and pathologic risk assessment models for patients with endometrial cancer remains suboptimal. Therefore, attention is being aimed at identifying molecular signatures that could predict clinical outcomes and potentially guide the development of targeted therapies. Multiple molecular alterations have been described in the histogenesis and progression of endometrioid endometrial carcinoma. These include PTEN loss and defects in DNA mismatch repair as well as mutations in KRAS2, CTNNB1, RB, and TP53.5
The phosphoinositide-3 kinase-related kinase (PIK) subfamily members ataxia-telangiectasia mutated (ATM) and mutated in ataxia-telangiectasia-Rad3 related (ATR) are well recognized for their important role in cellular responses to DNA damage via activation of cell cycle checkpoints.6 Somatic mutations in the mononucleotide repeat A10 in exon 10 of ATR have been identified in endometrioid endometrial tumors with DNA mismatch repair defects.7 The functional significance of these truncating mutations has recently been described. These mutations seem to provide a survival advantage in endometrial cancer cell lines through resistance to ionizing radiation and topoisomerase inhibitors.8 We hypothesized that heterozygous mutations in exon 10 of ATR would be associated with more aggressive phenotypes and worse clinical outcomes. The objective of this study was to determine the incidence, clinical correlates, and, specifically, the prognostic significance of ATR mutations in a large cohort of endometrioid endometrial cancer cases.
PATIENTS AND METHODS
Study Participants and Clinical Data
The Division of Gynecologic Oncology at Washington University School of Medicine has prospectively collected tumor samples from operative specimens since 1991. All enrolled participants have consented to molecular analyses and follow-up monitoring as part of ongoing Washington University's Human Research Protection Office–approved research protocols (protocols 91-507 and 93-0828).
Between November 1991 and January 2005, a total of 932 tissue samples were obtained at the time of hysterectomy. All cases were evaluated and diagnoses confirmed by experienced gynecologic pathologists at our institution. After initial pathologic review, 833 tissues collected for research were confirmed as uterine corpus malignancies. Only specimens with high neoplastic cellularity (≥ 70%) were processed. Microsatellite analyses were successfully performed in 665 tumors. One hundred ninety-five of these tumors had evidence of defects in DNA mismatch repair (DMMR) as evidenced by microsatellite instability (MSI). We excluded nonendometrioid histotypes, cases with synchronous or metachronous tumors, and 45 cases for which follow-up data was unavailable. Because previous data showed that mutations in exon 10 of ATR exclusively affect tumors with DMMR,7 we initially sequenced the region of interest in all MSI-positive cases that met inclusion criteria (n = 150). Successful sequencing was possible in 94% of these cases (n = 141).
We successfully genotyped the region of interest in ATR in a subset of unmatched microsatellite stable tumors that met inclusion criteria during the study period (n = 107).
All cases underwent primary surgery by a gynecologic oncologist at Washington University School of Medicine/Barnes–Jewish Hospital. None of these patients received preoperative radiation or chemotherapy. Surgical staging and tumor grade was assigned on the basis of International Federation of Gynecology and Obstetrics (FIGO) 1988 criteria. Clinical and pathologic information was prospectively collected and stored in a computerized database. Specific treatment was individualized and left at the discretion of the patient's attending physician. All patients were followed-up at minimum at 3-month intervals for the first 2 years and then at 6-month intervals for at least 3 years. Disease surveillance included physical examination and periodic pap smears. Diagnostic imaging was ordered and directed biopsies performed as clinically indicated. All suspected recurrences were histologically confirmed.
Tissue Processing, MSI Typing, and ATR Genotyping
Tissue specimens and blood were obtained at the time of surgery, snap-frozen, and stored at −70°C. DNA was prepared from tumors using proteinase K and phenol extraction or with the DNeasy Tissue Kit (Qiagen Inc, Valencia, CA). DNA was extracted from peripheral-blood leukocytes. When blood was not available, normal DNA was extracted from uninvolved myometrium as previously described.9–11
Microsatellite analysis was performed using five National Cancer Institute consensus microsatellite markers (BAT25, BAT26, D2S123, D5S346, and D17S250). Typing was repeated up to three times to resolve any uncertainties regarding whether abnormal polymerase chain reaction (PCR) products suggestive of MSI were present. Standard definitions for MSI positivity were applied as previously described.11–12
A 433-bp amplicon containing the A10 mononucleotide repeat in exon 10 of ATR (Ensembl No. ENSG00000175054) was amplified by PCR using the Deep VentR high-fidelity DNA polymerase (New England Biolab, Ipswich, MA) with the following primers: 5′-CACGGCATGTTTTATCTGACA-3′ (forward) and 5′-CACGGCATGTTTTATCTGACA-3′ (reverse) [Tm = 63°C]. Amplification products were purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA) and sequenced unidirectionally with the ABI Prism BigDye Terminator chemistry version 1.1 (Applied Biosystems, Foster City, CA). Mutation analysis was performed by manual inspection of individual chromatograms.
Statistical Analyses
The relationship between ATR status and covariates was performed using Fisher's exact test and t test. Overall survival (OS) was defined as the time from diagnosis to the date of death from any cause. Survivors were censored at the date of last contact. Disease-free survival (DFS) was defined as the time from surgery to recurrence or progression. The Kaplan-Meier product-limit method was used to estimate OS and DFS. Univariate and multivariate Cox proportional hazards models were fitted to check the possible effects of the covariates on OS and DFS. In the analysis of DFS, Gray's competing risk methods were also used as a sensitivity analysis to account for the potential competing effect of death.13 Factors with P values less than 0.2 in the univariate analysis were included in the corresponding multivariate model. The proportional hazards assumptions were assessed using scaled Schoenfeld residuals (Appendix Figs A1 and A2, online only).14 All analyses were two-sided, and significance was set at a P value of .05. Statistical analyses were performed using SAS (SAS Institutes, Cary, NC), as well as the cmprsk R (http://biowww.dfci.harvard.edu/∼gray) statistical packages for competing risk analysis.
RESULTS
The study group included 248 patients with endometrioid endometrial carcinoma. The median follow-up time was 68.1 months (range, 0.1 to 167.2 months). The demographics and clinicopathologic characteristics of the study group are presented in Table 1.
Table 1.
Patient Demographics and Clinicopathologic Characteristics
| Characteristic | No. of Patients (n = 248) | % |
|---|---|---|
| Age, years | ||
| Mean | 63.7 | |
| SD | 11.4 | |
| Race | ||
| White | 224 | 90.3 |
| African American | 24 | 9.7 |
| Stage | ||
| IA | 35 | 14.1 |
| IB/IC | 148 | 59.7 |
| II | 13 | 5.2 |
| III/IV | 48 | 19.4 |
| Unstaged | 4 | 1.6 |
| FIGO grade | ||
| 1 | 125 | 50.4 |
| 2 | 91 | 36.7 |
| 3 | 32 | 12.9 |
| Adjuvant treatment* | ||
| No | 174 | 71.3 |
| Yes | 70 | 28.7 |
| Events | ||
| Recurrence/progression | 38 | 15.3 |
| Death† | 87 | 35.1 |
Abbreviations: SD, standard deviation; FIGO, International Federation of Gynecology and Obstetrics.
Four cases were lost to follow-up. Adjuvant treatment included radiation (n = 51), chemotherapy (n = 12), and radiation plus chemotherapy (n = 7).
Includes five perioperative deaths (within 30 days of initial surgery).
Mutations in the A10 mononucleotide repeat in exon 10 of the ATR gene were identified in 12 cases (4.8%). Three tumors had insertion of a single A, and nine tumors had deletion of an A. All mutations occurred in MSI-positive tumors (P = .02). Figure 1 illustrates the chromatographic appearance of the wild-type sequence as well as the mutated genotypes. ATR mutations were not associated with age, race, surgical stage, FIGO grade, MLH1 methylation status, or use of adjuvant treatment.
Fig 1.

Representative examples of ATR genotypes. Mutation in A10 mononucleotide repeat in ATR exon 10. T1231: microsatellite instability (MSI)–positive tumor, A9 mononucleotide illustrates deletion (arrow). T1138: Microsatellite stable tumor, A10 mononucleotide illustrates wild type. T1062: MSI-positive tumor, A11 mononucleotide illustrates insertion (arrow). T, tumor; A, adenine; C, cytosine; T, thymine; G, guanine; N, base unrecognized by chromatography's software (ABI, Foster City, CA).
Univariate analysis (Table 2) revealed that mutations in the region of interest of ATR were indeed associated with shorter OS (hazard ratio [HR], 2.35; 95% CI, 1.08 to 5.11; P = .03). Advanced stage (HR, 3.82; 95% CI, 1.76 to 8.32; P = .0007), higher FIGO grade (HR, 2.66; 95% CI, 1.43 to 4.94; P = .002), use of adjuvant treatment (HR, 1.82; 95% CI, 1.16 to 2.84; P = .009), and, to a lesser degree, age (HR, 1.05; 95% CI, 1.03 to 1.07; P < .0001) were also associated with worse OS. MSI status and race were not associated with OS. Variables that approached significance (P < .2) were incorporated in the multivariate model (Table 3). After controlling for confounding factors, the effects of ATR mutations (HR, 3.88; 95% CI, 1.64 to 9.18; P = .002), surgical stage (HR, 1.75; 95% CI, 1.32 to 2.31; P = .0001), higher FIGO grade (HR, 1.40; 95% CI, 1.02 to 1.91; P = .04), and age (HR, 1.06; 95% CI, 1.04 to 1.08; P < .0001) on OS remained statistically significant.
Table 2.
Univariate Analyses
| Characteristic | Overall Survival |
Disease-Free Survival |
||||
|---|---|---|---|---|---|---|
| Hazard Ratio | 95% CI | P | Hazard Ratio | 95% CI | P | |
| Age | 1.05 | 1.03 to 1.07 | < .0001 | 1.02 | 0.99 to 1.05 | .16 |
| Race | ||||||
| White | 1.0 | — | 1.0 | — | ||
| African American | 1.53 | 0.76 to 3.07 | .23 | 0.83 | 0.26 to 2.69 | .75 |
| MSI | ||||||
| Negative | 1.0 | — | 1.0 | — | ||
| Positive | 1.04 | 0.67 to 1.62 | .86 | 0.75 | 0.40 to 1.42 | .38 |
| ATR mutation | ||||||
| Absent | 1.0 | — | 1.0 | — | ||
| Present | 2.35 | 1.08 to 5.11 | .03 | 3.72 | 1.45 to 9.54 | .006 |
| FIGO grade | ||||||
| 1 | 1.0 | — | 1.0 | — | ||
| 2 | 1.58 | 0.98 to 2.55 | .06 | 2.45 | 1.17 to 5.15 | .02 |
| 3 | 2.66 | 1.43 to 4.94 | .002 | 3.15 | 1.27 to 7.83 | .01 |
| Surgical stage | ||||||
| IA | 1.0 | — | 1.0 | — | ||
| IB/IC | 1.22 | 0.59 to 2.50 | .59 | 3.35 | 0.44 to 25.48 | .24 |
| II | 2.15 | 0.71 to 6.50 | .17 | 10.49 | 1.17 to 93.85 | .04 |
| III/IV | 3.82 | 1.76 to 8.32 | .0007 | 17.49 | 2.33 to 131.11 | .005 |
| Adjuvant treatment | ||||||
| No | 1.0 | — | 1.0 | — | ||
| Yes | 1.82 | 1.16 to 2.84 | .009 | 3.20 | 1.69 to 6.07 | .0004 |
Abbreviations: MSI, microsatellite instability; FIGO, International Federation of Gynecology and Obstetrics.
Table 3.
Multivariate Analyses
| Characteristic | Overall Survival |
Disease-Free Survival |
||||
|---|---|---|---|---|---|---|
| Hazard Ratio | 95% CI | P | Hazard Ratio | 95% CI | P | |
| Age | 1.06 | 1.04 to 1.08 | < .0001 | 1.03 | 1.00 to 1.06 | .08 |
| ATR mutation | 3.88 | 1.64 to 9.18 | .002 | 4.29 | 1.48 to 12.45 | .007 |
| Higher FIGO grade | 1.40 | 1.02 to 1.91 | .04 | 1.27 | 0.82 to 1.97 | .28 |
| Advanced surgical stage | 1.75 | 1.32 to 2.31 | .0001 | 2.12 | 1.44 to 3.13 | .0002 |
| Adjuvant treatment | 1.35 | 0.78 to 2.34 | .29 | 1.57 | 0.70 to 3.51 | .27 |
Abbreviation: FIGO, International Federation of Gynecology and Obstetrics.
We also evaluated the impact of ATR mutations and other clinicopathologic variables on DFS. Univariate analysis (Table 2) suggested that DFS was associated with advanced surgical stage (HR, 17.49; 95% CI, 2.33 to 131.11; P = .005), ATR mutations (HR, 3.72; 95% CI, 1.45 to 9.54; P = .006), use of adjuvant treatment (HR, 3.20; 95% CI, 1.69 to 6.07; P = .0004), and higher FIGO grade (HR, 3.15; 95% CI, 1.27 to 7.83; P = .01). MSI status, age, and race were not associated with DFS. Variables that approached significance (P < .2) were again incorporated in the multivariate model (Table 3), which indicated that both ATR mutations (HR, 4.29; 95% CI, 1.48 to 12.45; P = .007) and surgical stage (HR, 2.12; 95% CI, 1.44 to 3.13; P = .0002) were significantly associated with DFS.
Kaplan-Meier survival plots for OS and DFS according to ATR mutation status for the entire cohort are presented in Figure 2 and Appendix Figure A3 (online only). ATR mutations were significantly associated with reduced OS (log-rank test, P = .027) and DFS (log-rank test, P = .003) in this cohort of patients with endometrioid endometrial cancer.
Fig 2.

Kaplan-Meier curves for (A) overall survival and (B) disease-free survival by ATR mutation status. Vertical bars represent censored cases.
Given ATR mutations were seen only in MSI-positive tumors, we performed survival analyses including MSI-positive cases alone (Fig 3). This subgroup analyses confirmed the effects of ATR mutations on OS (HR, 3.52; 95% CI, 1.45 to 8.57; P = .005; log-rank test, P = .043) and DFS (HR, 3.01; 95% CI, 1.28 to 7.08; P = 0.01; log-rank test, P = .001).
Fig 3.
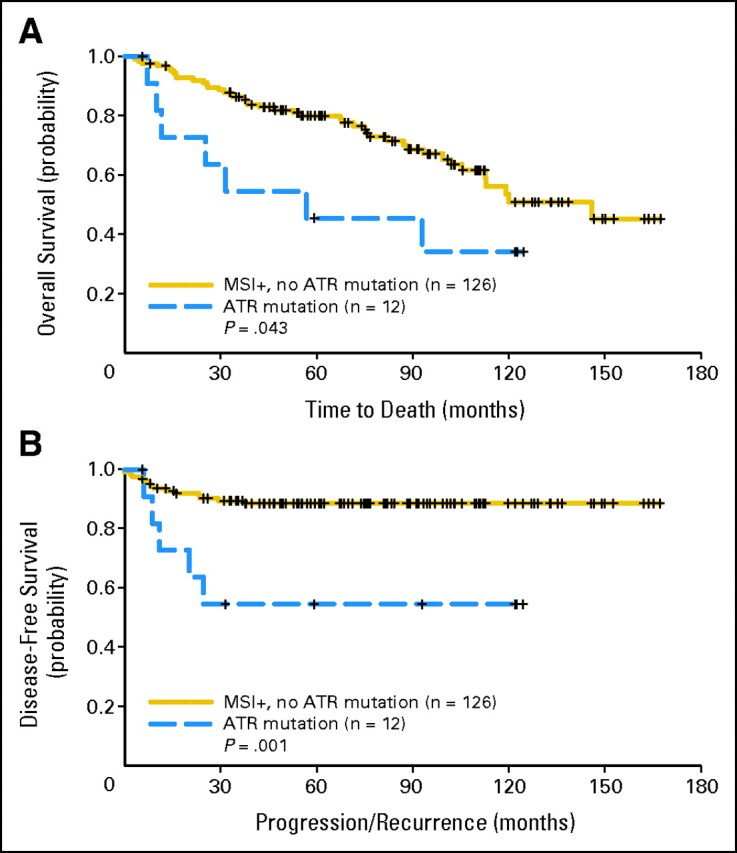
Kaplan-Meier curves for (A) overall survival and (B) disease-free survival by ATR mutation status among patients with microsatellite instability–positive (MSI+) tumors. Vertical bars represent censored cases.
DISCUSSION
The PIK superfamily members participate in diverse functions in eukaryotic cells through regulation of conserved signal transduction pathways. The PIK subfamily ATR protein has been recognized for its participation in cellular responses to DNA damage. ATR activates cell-cycle checkpoints Chk1 and Chk2 in response to DNA damage.6 ATR is structurally similar to ATM and other PIK members.15 ATM is capable of regulating p53 activity. More specifically, ATM is responsible for the DNA damage-induced phosphorylation of p53 on Ser-15. The Ser-15 residue represents an important regulatory point for p53. It is the site of interaction of p53 with its negative regulator, the oncoprotein MDM2, in vivo and in vitro. Phosphorylation at Ser-15 after DNA damage leads to reduced interaction of p53 with its negative regulator.16–18 However, Ser-15 phosphorylation and p53 accumulation have been demonstrated in ataxia-telangiectasia cell lines (deficient in ATM), suggesting that ATM is not the only protein responsible for p53 modulation in response to DNA damage.19–21 A more complicated control loop, likely involving multiple protein kinases, potentially responsive to specific types of DNA damage seems probable. ATR phosphorylates p53 at both Ser-15 and Ser-37 in vitro.17–22 ATR preferentially phosphorylates and activates the checkpoint kinase Chk1 resulting in G2 arrest in response to ionizing radiation, topoisomerase inhibitors, and cytotoxic methylation events.23–27 Modulation of ATM/ATR functions may be an attractive intervention for cancer radiotherapy and chemotherapy.
Tumors with DMMR tend to accumulate mutations in highly repetitive sequences. This mutator phenotype manifests itself as MSI. The occurrence of mutations in DNA damage response genes, such as ATR and Chk1, in tumors with DMMR defects was first described in gastric cancer.28 The A10 mononucleotide repeat in ATR (exon 10) is subject to frameshift mutations in cells with defective DMMR, resulting in premature protein truncation. Biallelic loss is incompatible with life, as seen by the lethality of ATR deletions in early developmental stages.29 However, Lewis et al8 have demonstrated that monoallelic frameshift mutation is sufficient to affect downstream ATR effectors, suggesting dominant negative effects. More specifically, in vitro ultraviolet-induced phosphorylation of Chk1 by ATR is markedly diminished in the presence of mutant ATR. The exposure of MSI-positive endometrial cancer cell lines with heterozygous truncating mutations in exon 10 of ATR to UV, ionizing radiation or topotecan, fails to trigger the expected activation of Chk1 by ATR via Ser-345 phosphorylation. These data support the functional importance of these mutations in vivo.8
Vassileva et al7 evaluated several genes involved in DNA repair in 73 cases of endometrial cancer and their adjacent areas of endometrial hyperplasia. They found that somatic ATR mutation occurred only in MSI-positive endometrial cancers and was not present in areas of hyperplasia, suggesting that ATR mutations are late events in the development of MSI-positive endometrial cancers. Their group found mutations in exon 10 of the ATR gene in two (5.3%) of 38 MSI-positive endometrial cancers (one insertion and one deletion).7
The frequency and clinical implications of ATR mutations in patients with endometrial cancer have been largely unknown. The most important strength of our study is the ability to evaluate for clinicopathologic as well as outcome associations in a large cohort with surgically staged disease. In this study, we confirmed that ATR is mutated in a subset of endometrioid endometrial tumors and that these mutations occur exclusively in tumors with DMMR defects (MSI positive). As previously suggested, the frequency of ATR mutations is approximately 5% to 10% among MSI-positive endometrioid tumors. These mutations were not associated with other prognostic demographic or pathologic variables. Importantly, however, after controlling for potential confounding variables, ATR mutation was an independent prognostic variable for both OS (HR, 3.88; 95% CI, 1.64 to 9.18; P = .002) and DFS (HR, 4.09; 95% CI, 1.41 to 11.86; P = .01). This apparent aggressive tumor behavior associated with ATR mutation could in part explain variable responses to therapy in this subgroup of patients. Unfortunately, our study is not adequately powered to evaluate specific effects of therapy among the small proportion of patients with ATR-mutated tumors.
Current understanding of ATM/ATR modulation of DNA damage responses is rapidly evolving. Recent work has suggested that ATR-mutant cancer cells with intact DNA mismatch repair systems may display enhanced sensitivity to certain chemotherapeutic drugs. Specifically, these cells may be exquisitely sensitive to certain DNA-damaging agents, such as alkylating drugs and those that inhibit DNA synthesis, but not to agents with other effects.30 Additionally, ATR- mutant cells seem to undergo stasis at the beginning of the S phase in response to ionizing radiation.31 These findings suggest that modulation of ATR in certain tumors with intact DNA mismatch repair may prove therapeutically effective. The survival effect observed in our study could be related to the compounding effect of DMMR in endometrial tumors that harbor ATR mutations. Regardless of the specific mechanism, our data suggest that ATR mutations in endometrial tumors have important prognostic implications in vivo. Further studies are needed to validate our findings and to better understand the mechanisms involved in this survival effect.
Acknowledgment
We thank Mary Ann Mallon for her technical support and the Biostatistics Core at the Siteman Cancer Center and Washington University School of Medicine for assistance with the statistical analyses.
Appendix
Fig A1.

Proportional hazards assumptions for overall survival. Specific variables are indicated on the y-axis in each panel.
Fig A2.

Proportional hazards assumptions for disease-free survival. Specific variables are indicated on the y-axis in each panel.
Fig A3.

Kaplan-Meier curves for (A) overall survival and (B) disease-free survival by ATR mutation and microsatellite instability (MSI) status. Vertical bars represent censored cases.
Footnotes
Supported by Grants No. RO1 CA71754 (P.J.G.) and Barnes-Jewish Foundation 00161-0806 (P.J.G.). The Siteman Cancer Center is supported by National Cancer Institute Cancer Center Support Grant No. P30 CA91842.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The author(s) indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: Israel Zighelboim, Paul J. Goodfellow
Financial support: David G. Mutch, Paul J. Goodfellow
Administrative support: David G. Mutch, Paul J. Goodfellow
Provision of study materials or patients: Premal H. Thaker, Matthew A. Powell, Janet S. Rader, Randall K. Gibb, David G. Mutch, Paul J. Goodfellow
Collection and assembly of data: Israel Zighelboim, Amy P. Schmidt, Matthew A. Powell, Janet S. Rader, Randall K. Gibb, David G. Mutch, Paul J. Goodfellow
Data analysis and interpretation: Israel Zighelboim, Amy P. Schmidt, Feng Gao, Premal H. Thaker, Matthew A. Powell, Janet S. Rader, Randall K. Gibb, David G. Mutch, Paul J. Goodfellow
Manuscript writing: Israel Zighelboim, Amy P. Schmidt, Feng Gao, Premal H. Thaker, Matthew A. Powell, Janet S. Rader, Randall K. Gibb, David G. Mutch, Paul J. Goodfellow
Final approval of manuscript: Israel Zighelboim, Amy P. Schmidt, Feng Gao, Premal H. Thaker, Matthew A. Powell, Janet S. Rader, Randall K. Gibb, David G. Mutch, Paul J. Goodfellow
REFERENCES
- 1.Ries LAG, Melbert D, Krapcho M, et al., editors. Based on November 2007 SEER data submission, posted to the SEER web site 2008. Bethesda, MD: National Cancer Institute; SEER Cancer Statistics Review, 1975-2005. http://seer.cancer.gov/csr/1975_2005/ [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Grigsby PW. Update on radiation therapy for endometrial cancer. Oncology. 2002;16:777–786. [PubMed] [Google Scholar]
- 4.Alvarez Secord A, Havrilesky LJ, Bae-Jump V, et al. The role of multi-modality adjuvant chemotherapy and radiation in women with advanced stage endometrial cancer. Gynecol Oncol. 2007;107:285–291. doi: 10.1016/j.ygyno.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Shiozawa T, Konishi I. Early endometrial carcinoma: Clinicopathology, hormonal aspects, molecular genetics, diagnosis, and treatment. Int J Clin Oncol. 2006;11:13–21. doi: 10.1007/s10147-005-0546-1. [DOI] [PubMed] [Google Scholar]
- 6.Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- 7.Vassileva V, Millar A, Briollais L, et al. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res. 2002;62:4095–4099. [PubMed] [Google Scholar]
- 8.Lewis KA, Mullany S, Thomas B, et al. Heterozygous ATR mutations in mismatch repair-deficient cancer cells have functional significance. Cancer Res. 2005;65:7091–7095. doi: 10.1158/0008-5472.CAN-05-1019. [DOI] [PubMed] [Google Scholar]
- 9.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lahiri DK, Nurnberger JI., Jr A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991;19:5444. doi: 10.1093/nar/19.19.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zighelboim I, Goodfellow PJ, Gao F, et al. Microsatellite instability and epigenetic inactivation of MLH1 and outcome of patients with endometrial carcinomas of the endometrioid type. J Clin Oncol. 2007;25:2042–2048. doi: 10.1200/JCO.2006.08.2107. [DOI] [PubMed] [Google Scholar]
- 12.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 13.Fine JP, Gray RJ. A proportional hazards model for the sub-distribution of a competing risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 14.Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika. 1994;81:515–526. [Google Scholar]
- 15.Cimprich KA, Shin TB, Keith CT, et al. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci U S A. 1996;93:2850–2855. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 17.Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 18.Shieh SY, Ikeda M, Taya Y, et al. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 19.Canman CE, Wolff AC, Chen CY, et al. The p53-dependent G1 cell cycle checkpoint pathway and ataxia-telangiectasia. Cancer Res. 1994;54:5054–5058. [PubMed] [Google Scholar]
- 20.Siliciano JD, Canman CE, Taya Y, et al. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu X, Lane DP. Differential induction of transcriptionally active p53 following UV or ionizing radiation: Defects in chromosome instability syndromes? Cell. 1993;75:765–778. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- 22.Tibbetts RS, Brumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, Guntuku S, Cui XS, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 24.O'Connell MJ, Walworth NC, Carr AM. The G2-phase DNA-damage checkpoint. Trends Cell Biol. 2000;10:296–303. doi: 10.1016/s0962-8924(00)01773-6. [DOI] [PubMed] [Google Scholar]
- 25.Canman CE. Replication checkpoint: Preventing mitotic catastrophe. Curr Biol. 2001;11:R121–124. doi: 10.1016/s0960-9822(01)00057-4. [DOI] [PubMed] [Google Scholar]
- 26.Cliby WA, Lewis KA, Lilly KK, et al. S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J Biol Chem. 2002;277:1599–1606. doi: 10.1074/jbc.M106287200. [DOI] [PubMed] [Google Scholar]
- 27.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menoyo A, Alazzouzi H, Espin E, et al. Somatic mutations in the DNA damage-response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res. 2001;61:7727–7730. [PubMed] [Google Scholar]
- 29.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 30.Wilsker D, Bunz F. Loss of ataxia telangiectasia mutated- and Rad3-related function potentiates the effects of chemotherapeutic drugs on cancer cell survival. Mol Cancer Ther. 2007;6:1406–1413. doi: 10.1158/1535-7163.MCT-06-0679. [DOI] [PubMed] [Google Scholar]
- 31.Hurley PJ, Wilsker D, Bunz F. Human cancer cells require ATR for cell cycle progression following exposure to ionizing radiation. Oncogene. 2007;26:2535–2542. doi: 10.1038/sj.onc.1210049. [DOI] [PubMed] [Google Scholar]