

Abstract
Objective
A naturally-occurring mutation in cardiac calsequestrin (CASQ2) at amino acid 307 was discovered in a highly inbred family and hypothesized to cause Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT). The goal of this study was to establish a causal link between CASQ2D307H and the CPVT phenotype using an in vivo model.
Methods and Results
Cardiac-specific expression of the CASQ2D307H transgene was achieved using the α-MHC promoter. Multiple transgenic (TG) mouse lines expressing CASQ2D307H from 2- to 6-fold possess structurally normal hearts without any sign of hypertrophy. The hearts displayed normal ventricular function. Myocytes isolated from TG mice had diminished ICa-induced Ca2+ transient amplitude and duration as well as increased Ca2+ spark frequency. These myocytes, when exposed to isoproterenol and caffeine, displayed disturbances in their rhythmic Ca2+ oscillations and membrane potential and exhibited delayed afterdepolarizations. ECG monitoring revealed that TG mice challenged with isoproterenol and caffeine developed complex ventricular arrhythmias, including non-sustained polymorphic ventricular tachycardia.
Conclusions
The findings of the present study demonstrate that expression of mutant CASQ2D307H in the mouse heart results in abnormal myocyte Ca2+ handling and predisposes to complex ventricular arrhythmias similar to the CPVT phenotype observed in human patients.
1. Introduction
CASQ2 is the most abundant Ca2+ buffering protein present in the lumen of the cardiac sarcoplasmic reticulum (SR). It allows calcium to be stored at total concentrations of up to 20 mM, while the free concentration remains ~1 mM 1–4. It binds calcium with high capacity (40–50 Ca2+/CASQ2) and moderate affinity (kd ~1 mM) 1,3–5. Increasingly, it has become apparent that CASQ2 might also play an important role in regulating the SR Ca2+ release 6,7. Studies have suggested that CASQ2 can interact functionally with the cardiac ryanodine receptor (RyR2) in vitro, either by binding with the RyR2 directly or through its interactions with junctin and triadin 8–11. It is hypothesized that this interaction allows CASQ2 to be localized to the terminal cisternae of the junctional SR (jSR) 8,10,12,13. Also, it was proposed that through interaction with triadin, CASQ2 might be able to communicate with the RyR2 6,14. However, the nature of this interaction and its exact role in Ca2+ release remain to be understood.
The importance of CASQ2 is highlighted by the fact that mutations in the CASQ2 gene have been recently linked to ventricular arrhythmias and sudden cardiac death 15–17. In particular, a naturally-occurring mutation in cardiac CASQ2 at amino acid 307 (CASQ2D307H) was discovered in seven Bedouin families 18,19, as a cause for Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) 19. CPVT is a disease that is characterized by adrenergically-mediated ventricular tachycardia that can rapidly progress into ventricular fibrillation and sudden cardiac death 18. The characteristic symptoms are recurrent syncope, seizures, or sudden death triggered by exercise or emotional stress, both of which raise the levels of catecholamine 18.
Sequencing of DNA obtained from CPVT patients showed that CASQ2 exon 9 had a G→C substitution at nucleotide 1038 19. This substitution resulted in a change from an aspartate residue to a histidine at position 307 in CASQ2 19. This aspartate residue is invariant both in vertebrate and invertebrate CASQ 19. Acute expression of this mutant protein in isolated adult rat cardiomyocytes by adenoviral gene transfer resulted in decreased SR Ca2+ storage, Ca2+ channel-gated Ca2+ release (Ca2+ transients) and local Ca2+ release events (Ca2+ sparks) 20. In addition, the rhythmicity of the Ca2+ transients was disrupted in these myocytes upon catecholamine stimulation.
In the present study, we wished to determine the effect of CASQ2D307H on heart function by chronically expressing this mutant protein in vivo. This was accomplished by generating TG mouse lines that express CASQ2D307H in the heart. Transgenic expression of mutant protein does not lead to cardiac hypertrophy. Cardiomyocytes isolated from the TG mice displayed in vitro arrhythmias that are consistent with the CPVT phenotype. Most importantly, complex ventricular arrhythmias, including non-sustained ventricular tachycardia, are triggered in the hearts of TG mice when challenged with isoproterenol and caffeine. Our findings in the TG mouse suggest that CASQ2D307H mutation can contribute to the CPVT phenotype in humans.
2. Materials and Methods
2.1. Generation of TG mice expressing the CASQ2D307H mutant protein
The D307H mutation was introduced into the CASQ2 open reading frame, using PCR method. Once the D307→H mutation was confirmed by DNA sequencing, the coding region of the mutant protein was sub-cloned into a vector containing the α-MHC promoter as described previously 21–23. TG mice (FVB/N) expressing mutant CASQ2D307H were generated by the TG core facility. This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication # 85-23, revised 1996).
2.2. Western blotting analyses
Western blotting was performed following standard protocols 24.
2.3. Isolation of mouse myocytes and electrophysiological recordings
Myocytes were isolated using established protocols 25. Transmembrane ionic currents were measured using whole-cell patch-clamp recordings, as described previously 26, with Axopatch 200B amplifier (Axon Instruments, USA) and pClamp-9 software. The external solution contained 140 mM NaCl, 5.4 mM KCl, 1.0 mM CaCl2, 0.5 mM MgCl2, 10 mM Hepes, and 5.6 mM glucose (pH 7.3). Patch pipettes (tip resistance of 1–3 MΩ) were filled with a solution that contained 90 mM K-aspartate, 50 mM KCl, 3 mM Na2ATP, 3.5 mM MgCl2, 10 mM Hepes, and 0.05 mM Fluo-3 K-salt (pH 7.3). The myocytes were stimulated by application of 400-ms-long voltage pulses to specified membrane potentials from a holding potential of −50 mV at 1-min intervals. For the measurements of calcium currents potassium ions was replaced by cesium.
2.4. Confocal Ca2+ measurements
Intracellular Ca2+ imaging was performed by using an Olympus Fluoview 1000 laser scanning confocal microscope equipped with an Olympus 60 Ч 1.4NA oil objective. Fluo-3 was excited by the 488-nm line of an argon-ion laser, and the fluorescence was acquired at wavelengths >510 nm in the line-scan mode of the confocal system at the rate of 2 ms per scan. Ca2+ spark parameters were quantified with a detection/analysis computer algorithm 26.
2.5. Calcium sparks analysis
To measure Ca2+ sparks in intact myocytes, the myocytes were permeabilized by exposure to saponin (0.01% for 45–60 s). The permeabilization solution contained 100 mM K aspartate, 20 mM KCl, 3 mM MgATP 0.5 mM EGTA, 0.114 mM CaCl2 (free [Ca2+], ~100 nM), 0.81 mM MgCl2 (free [Mg2+], ~1 mM), 10 mM phosphocreatine, 10 mM HEPES, 5 U/ml creatine phosphokinase, and 8% dextran (40,000), pH 7.2. The control experimental solution contained 100 mM K aspartate, 20 mM KCl, 3 mM MgATP, 0.5 mM EGTA, 0.114 mM CaCl2 (free [Ca2+], ~100 nM), 0.81 mM MgCl2 (free [Mg2+], ~1 mM), 10 mM phosphocreatine, 10 mM Hepes, 0.03 mM Fluo-3 potassium salt (TefLabs, Austin, TX), and 5 U/ml creatine phosphokinase, pH 7.2.
The free [Ca2+] and [Mg2+] at given total Ca2+, Mg2+, ATP, and EGTA concentrations were calculated using WinMAXC 3.2 (Stanford University, Stanford, CA).
2.6. Analysis of cardiac function by echocardiography
Echocardiograms to assess systolic function were performed by using M-mode and two-dimensional measurements. The measurements represented the average of six selected cardiac cycles from at least two separate scans performed in random-blind fashion with papillary muscles used as a point of reference for consistency in the level of scan. End diastole was defined as the maximal left ventricular diastolic dimension and end systole was defined as the peak of posterior wall motion. Fractional shortening (FS), a surrogate of systolic function, was calculated from left ventricle dimensions as follows: FS = ((EDD − ESD)/EDD) ×100%.
2.7. ECG monitoring and induction of arrhythmia
ECG recordings were obtained from mice as previously described 27–29. Mice over-expressing CSQD307H (n=8) and their wild-type littermates (n=8) were anesthetized with isoflurane (1–1.5%) at minimum effective concentrations, and placed on a heating pad (Braintree Scientific, Inc.) to maintain normothermia. ECGs were recorded using a physiologic data acquisition system (MP 100, Biopac Systems) with a sampling rate of 2 kHz for 84±17 min. After baseline recording (10 min), 5 pairs of mice (5 WT and 5 TG: 2 pairs each from 2- and 4-fold lines, and 1 pair from 6-fold line) received 4 doses of isoproterenol (2 mg/kg IP) in 10–20 min intervals, the first 2 of which were combined with caffeine (120 mg/kg IP; see refs. 30,31). To ensure that the observed arrhythmias were adrenergically mediated, another group of mice (3 pairs from 2-fold line) received incremental doses of isoproterenol alone every 10 min (cumulative dose of 8.75±0.02 mg/kg IP). Both TG and age-matched WT littermates were tested the same day using the same drug protocol.
2.8. Statistical analysis
Cross tabulations with chi-square and yale correction factor or z-test were used as appropriate for categorical variables using a statistical software package (Sigmastat 2.03, Jandel Scientific).
3. Results
3.1. Generation and characterization of TG mice expressing CASQ2D307H
Cardiac-specific expression of the CASQ2D307H transgene was achieved using the α-MHC promoter 21–23. Out of five positive TG lines containing the transgene, three TG lines (F8, F17, and F21) were propagated for further characterization. Three- to six-month-old mice were used for the studies, except as indicated.
The level of CASQ2D307H protein expression in the three TG lines was determined by western blotting using an antibody specific for cardiac CASQ2 (Fig. 1A). Quantification of CASQ2 bands showed that the CASQ2D307H protein is expressed 2-, 4- and 6-fold in TG lines F21, F8 and F17, respectively (Fig. 1A and data not shown). These data confirmed that we obtained multiple TG lines with graded expression of the CASQ2D307H protein, thus allowing us to compare the effect of expressing different amounts of mutant protein. To determine whether expression of CASQ2D307H protein induced alterations in other SR proteins, we determined the levels of RyR2, L–type calcium channel (DHPR 2α), Na+/Ca2+ exchanger (NCX1), triadin, SERCA 2a and phospholamban (PLB) in the 4- and 6-fold lines (Fig. 1B and data not shown). Quantitation and correction for loading variations indicated that there were no significant changes in the expression levels of these proteins. Therefore 2- to 6-fold expression of CASQ2D307H does not alter the expression levels of related SR Ca2+ transport proteins. For most of the studies described below we compared the 2-fold and 6-fold overexpressors.
Fig. 1. (A–B) Expression of SR proteins in TG (2- and 6-fold CASQ2D307H) and WT hearts.


(A) 5–10 μg of whole heart homogenates (2- and 6-fold CASQ2D307H) were separated on 10% SDS-PAGE gels and probed with antibodies against CASQ2, SERCA 2a, triadin, α-actin and α-actinin. (B) 4–8 μg (10–30 μg for RyR2) of whole heart homogenates (6-fold CASQ2D307H) were separated on SDS-PAGE gels and probed with antibodies against RyR2 (4–20% gradient gel), SERCA 2a (10% gel), phospholamban (PLB; 15% gel), L-type calcium channel (DHPR 2α; 10% gel) and Na+/Ca2+ exchanger (NCX1, 10% gel). The 120-kDa full length NCX1 protein is shown 44. (C) CASQ2 immunofluorescence in TG and WT myocytes. Mice ventricular cardiomyocytes from WT littermates and TG (6-fold CASQ2D307H) were immunostained with mouse anti α-actinin (1:500) or rabbit anti-calsequestrin (1:200), followed by Alexa 488-conjugated goat anti-mouse (1:300) and Cy™3-conjugated Donkey anti-rabbit (1:300 dilution) antibodies.
3.2. TG cardiac myocytes show subtle structural changes
To determine whether the mutant CASQ2D307H was being properly localized to the junctional SR, confocal and electron microscopy (EM) were performed. Imaging of the stained cells revealed the expected patterns of fluorescence for CASQ2 and α-actinin (Fig. 1C). The CASQ2 staining co-localized with the α-actinin staining, consistent with the fact that the Z-line (α-actinin) and junctional SR (CASQ2) are in close proximity to one another. The only difference between the TG and WT staining pattern was that the intensity of CASQ2 staining appeared brighter in the TG myocytes, which is consistent with the over-expression of CASQ2D307H in the 6-fold line.
The ultrastructure of myocytes expressing CASQ2D307H was very similar to that of WT littermate cells but subtle changes were observed. We observed variations in the size and internal configuration of the jSR cisternae associated with the transverse tubules. In WT myocardium, the jSR is typically in the form of flat cisternae with a dense content, due to calsequestrin, that is condensed into periodic densities (Fig. 2A). Most of the cisternae in the mutant hearts have the same general structure as WT ones (Figs. 2B and C). Some however, show a decrease in the dense content, a general loosening of the content disposition and some widening of the cisternae (Fig. 2E and F). Some TG myocytes show evidence for a disturbance of T tubules. Normally, T tubules run transversely at the Z lines and have occasional longitudinal branches. In thin sections, each intermyofibrillar space has either none or a single T tubule profile. Some myocytes from CASQ2D307H TG hearts show multiple T tubule profiles, indicating a convoluted tubule (data not shown).
Fig. 2. jSR alterations in CASQ2D307H (6-fold) myocytes.
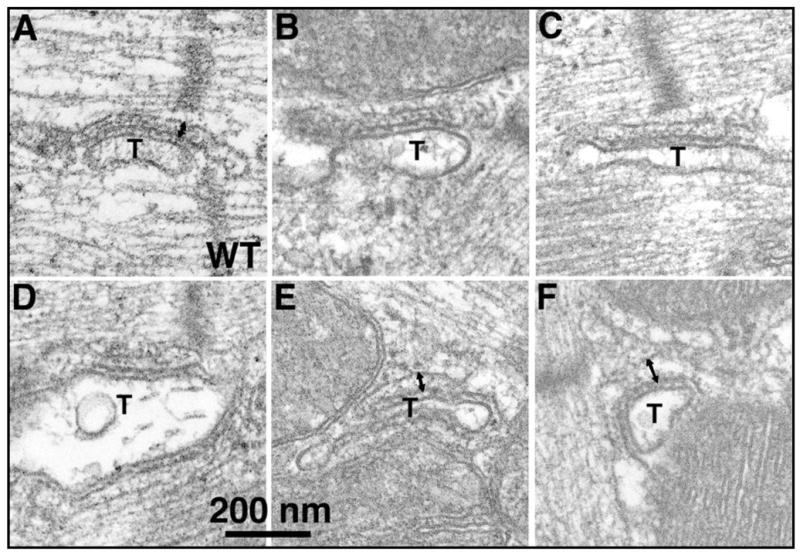
(A) In WT myocardium, the jSR cisternae are narrow, they contain a condensed form of CASQ2 and are closely apposed to wide T tubule profiles (T). (B,C) Most of the jSR cisternae in the mutant myocytes are quite similar to those in WT. (D–F) Alterations of jSR in mutant myocytes include a looser appearance of the CASQ2 in the lumen and an increase in width of the cisternae. The double arrows in A, E and F indicate the widening of terminal cisternae in TG compared to WT myocytes.
3.3. TG hearts expressing CASQ2D307H show normal overall structure and contractility
We also wanted to make sure that expression of mutant CASQ2D307H does not cause cardiac hypertrophy/pathology that may complicate our analysis. Several lines of evidence suggest that the TG hearts are essentially normal. First, there was no change in the heart weight-to-body weight ratios of TG (4.4±0.7 mg/g) compared to WT (4.3±0.3 mg/g) mice. Second, hearts from TG and WT littermates were subjected to standard histological analysis by staining with hematoxylin/eosin. These hearts appear normal and do not exhibit hypertrophy or fibrosis. Myocyte structure is similar in appearance in all hearts (Fig. 3A). Third, we examined the expression level of genes that are known markers of hypertrophy: atrial natriuretic factor (ANF), β-myosin heavy chain (β-MHC) and skeletal α-actin by RT-PCR. These genes are typically re-activated under conditions of cardiac hypertrophy, but we did not see activation of these genes in TG hearts (data not shown).
Fig. 3. TG hearts show normal morphology and function is unaltered.

(A) Hearts from TG mice (~1 year old) were subjected to standard histological analysis by staining with hematoxylin/eosin. (B) Examples of M-mode echocardiography in 4–6 month old WT and TG mice (6-fold CASQ2D307H) showing normal cardiac chamber and contractility with no evidence of structural abnormalities. Summary of data are shown in Table 1.
M-mode echocardiography in WT (Fig. 3B, top panel) and TG (lower panel) mice showed normal cardiac chamber and contractility with no evidence of structural abnormalities. There were no differences in the left ventricular end systolic dimension (LV-ESD) and end diastolic dimension (LV-EDD) and fractional shortening (FS) in TG mice compared to their littermate controls (see Table 1).
Table 1.
Echocardiographic analysis of WT and TG hearts.
| WT (n = 8) |
TG (n = 9) |
|
|---|---|---|
| LV-ESD (cm) | 0.15±0.02 | 0.14±0.01 |
| LV-EDD (cm) | 0.32±0.02 | 0.33±0.01 |
| ES-PW (cm) | 0.14±0.01 | 0.13±0.01 |
| ED-PW (cm) | 0.07±0.01 | 0.07±0.01 |
| FS% | 54±3.3 | 58.1±3.6 |
Data shown represent mean ± s.e.m.; left ventricular end systolic dimension (LV-ESD), end diastolic dimension (LV-EDD), posterior wall thickness at end systole and end diastole (ES-PW and ED-PW, respectively) and fractional shortening (FS), a surrogate of systolic function.
3.4. SR Ca2+ content and release
Calsequestrin is a Ca2+ storage protein and expression of mutant CASQ2D307H could affect calcium store and release properties. Therefore, we studied the calcium handling properties of myocytes isolated from TG and WT hearts. Caffeine applications (10 mM) were used to assess changes in the total SR Ca2+ content in each group of isolated myocytes. The relative amounts of Ca2+ released from the SR after caffeine administration were assessed from changes in both Fluo-3 fluorescence and Na+-Ca2+ exchange current (INCX) in myocytes dialyzed with the Ca2+ indicator Fluo-3 (Fig. 4A). Neither the caffeine-induced transients [2.39±0.24 (WT) vs. 2.06±0.22 (TG)], nor the INCX current [0.55±0.12 (WT) vs. 0.52±0.15 (TG)] changed significantly (Fig.4B–C).
Fig. 4. Intracellular Ca2+ load, ICa and Ca2+ transients in WT and TG myocytes (6-fold CASQ2D307H).

(A) Patch clamp recordings were used to measure Ca2+ transients (top traces) and NCX currents (bottom traces); pooled data for caffeine-induced Ca2+ transients (B) and NCX currents (C); (D) representative recordings of ICa (bottom traces) and calcium transients (top traces) evoked by depolarization steps from −50 mV MP to 0 mV; voltage dependencies of Ca2+ transients (E) and ICa (F) in ventricular myocytes, isolated from WT (squares and solid lines) and TG (6-fold CASQ2D307H) hearts (circles and dashed lines).
3.5. Ca2+ sparks, Ca2+ transients and ICa
The effects of expressing mutant CASQ2D307H on ICa and intracellular [Ca2+] transients in patch-clamped myocytes are illustrated in Figure 4D–F. There were no apparent changes in the parameters of ICa in myocytes expressing CASQ2D307H and cells from WT littermates (Fig. 4F, Table 2). The peak amplitude of ICa was nearly identical for all experimental groups of cells. In addition, the time course of ICa decay was similar (Table 2). Thus, expression of CASQ2D307H did not change the characteristics of the Ca2+ trigger for Ca2+ release from the SR. However, Ca2+ transients from TG mice were smaller and had reduced τdecay compared to WT myocytes (Fig. 4D–E and Table 2). Measurement of spontaneous Ca2+ sparks in permeabilized myocytes from TG mice and their WT littermates showed a trend toward decreased Ca2+ spark amplitude and a significant increase in Ca2+ spark frequency (Supplemental Figure and Table 3).
Table 2.
Parameters of ICa and Ca transients.
| ICa | Ca transients | N of cells | |||||
|---|---|---|---|---|---|---|---|
| Peak Amplitude (pA/pF) |
Tfast (ms) |
Tslow (ms) |
F/F0 | Rise Time (ms) |
Tdecay (ms) |
||
| WT | −4.88±0.55 | 15.7±4.7 | 76±25 | 1.96±0.16 | 28±4 | 320±16 | 12 (7 animals) |
| TG | −4.19±0.55 | 16.1±4.2 | 75±23 | 1.61±0.14* | 24±3 | 268±15* | 9 (6 animals) |
Significantly different at P<0.05, compared with WT
Table 3.
Characteristics of spontaneous Ca2+ sparks recorded in permeabilized myocytes isolated from WT mouse hearts and TG mice expressing CASQ2D307H.
| Amplitude ΔF/F0 | Rise time, ms | HA Width, μm | Frequency, 100 μm−1 s−1 | N of Sparks | N of Cells | N of Mice | |
|---|---|---|---|---|---|---|---|
| WT | 0.78±0.01 | 8.0±0.1 | 2.20±0.02 | 7.7±0.5 | 1152 | 80 | 6 |
| TG | 0.75±0.01 | 8.7±0.1* | 2.24±0.02 | 10.2±0.8* | 1077 | 62 | 3 |
Significantly different at P<0.05, One Way ANOVA.
3.6. Ca2+ cycling in rhythmically paced myocytes
The effects of isoproterenol treatment (1 μmol/L) on periodic Ca2+ transients in WT and TG myocytes are illustrated in Figure 5. To induce arrhythmia in myocytes, we used a modified protocol 30 where the bath solution contains 100 μM of caffeine. The myocytes were stimulated at 1 Hz, and membrane potential (MP) changes were recorded in the current-clamp mode. The exposure of WT myocytes to isoproterenol caused an increase in the amplitude of Ca2+ transients without any apparent disturbances in periodic Ca2+ cycling (Fig. 5A–B; results are representative of six myocytes). However, in TG myocytes expressing CASQ2D307H, the amplitude of Ca2+ current-induced Ca2+ transients was reduced (Fig. 5A). The AP duration between control and TG myocytes was not significantly different (APD90 is 57±24 for control animals and 45±19 for TG). Isoproterenol administration in the presence of caffeine caused profound disturbances in Ca2+ cycling manifested by extra systolic, spontaneous Ca2+ transients. As seen in the line-scan images (Fig. 5B), spontaneous release usually originated locally and then propagated through the cell as a regenerative Ca2+ wave. Importantly, the membrane potential traces showed delayed afterdepolarizations (DADs) and the occasional generation of action potentials at the time of spontaneous Ca2+ transients. Similar results were obtained in four other myocytes. These results indicate that expression of CASQ2D307H destabilizes the Ca2+ release mechanism, leading to spontaneous, premature discharges of SR Ca2+ stores in myocytes undergoing periodic pacing.
Fig. 5. TG myocytes (6-fold CASQ2D307H) show spontaneous Ca2+ transients.

Recording of membrane potential (top traces), along with line-scan confocal images (middle traces) and temporal profiles (bottom traces) in myocytes before (A) and after superfusion (B) with 1 μM of ISO and 100 μM of caffeine.
3.7. Complex ventricular arrhythmias are triggered in TG hearts by isoproterenol and caffeine
Continuous ECG monitoring prior to drug challenge did not reveal any significant spontaneous ventricular arrhythmias in WT mice. In contrast, CASQ2D307H mice had more spontaneous supraventricular ectopic beats at baseline than WT mice (cumulative numbers of observations were 378 vs. 8 in TG and WT mice, respectively, P<0.001).
After challenge with isoproterenol alone or in combination with caffeine, mutant CASQ2D307H mice developed significantly more supraventricular and junctional ectopic beats compared to WT (cumulative numbers of observations for junctional ectopic beats was 2862 vs. 39 in TG and WT mice, respectively, P<0.001). CASQ2D307H mice did not have an increased number of simple ventricular arrhythmias compared to WT (Fig. 6A). However, after drug challenge, the number of total complex ventricular arrhythmias was increased 4-fold (P<0.001) in CASQ2D307H mice compared to WT mice (Fig. 6A). Furthermore, there was a significant increase (P<0.05) in the number of CASQ2D307H mice that developed complex ventricular arrhythmias (Fig. 6B), including non-sustained ventricular tachycardia, compared to WT mice (Fig. 6C+D).
Fig. 6. Mice expressing mutant CASQ2D307 displayed more complex forms of ventricular arrhythmia, including non-sustained ventricular tachycardia, compared to aged-matched WT mice.

(A) Cumulative number of simple and complex ventricular arrhythmias in TG (n=8) from 2-, 4- and 6-fold over-expression and age-matched WT (n=8) mice. Under light anesthesia, mice were challenged with IP isoproterenol injection alone or in combination with caffeine injection. Simple ventricular arrhythmias included single isolated ventricular ectopic beats. Complex ventricular arrhythmias included: couplet, bigeminy, trigeminy and non-sustained ventricular tachycardia. ** P< 0.001 TG vs. WT mice. (B) Higher proportion of mice expressing CASQD307H (n=8), which displayed complex forms of ventricular arrhythmias compared to WT (n=8), following treatment with isoproterenol alone or in combination with caffeine injection. * P< 0.05 TG vs. WT mice. (C) Representative ECG tracings (lead 1 top and lead 3 bottom) in a TG mouse (6-fold CASQ2D307H) displaying a run of non-sustained polymorphic ventricular tachycardias (NSVT). Please note the presence of 2 couplets before and after the run of NSVT, which can be considered as an attempted, but aborted VT. (D) Magnification of section A of the ECG tracings (lead 1 top and lead 3 bottom) displaying 2 non-sustained polymorphic ventricular tachycardias. Please note a junctional escape beat (e.g., compensatory pause) following each NSVT. S: sinus beat; P: compensatory pause; J: Junctional premature beat. VT: ventricular tachycardia.
4. Discussion
CPVT is a potentially devastating condition that can lead to sudden cardiac death even in very young children after physical or emotional stress. CPVT can be caused by mutations in either the ryanodine receptor (RyR2) or the CASQ2 gene 15,17,19,32–35. There are several different RyR2 mutations that are linked to CPVT and they are all dominant, while there are fewer known CASQ2 mutations and these are all recessive 15,17.
In the present study, we chose to introduce increasing amounts of mutant CASQ2D307H to compete with the endogenous WT CASQ2 protein to determine its effect on Ca2+ handling and heart function. Since the CASQ2D307H mutation is recessive, we predicted that low amounts of CASQ2D307H would not significantly affect heart function, while higher amounts of CASQ2D307H would out-compete the endogenous WT CASQ2 and lead to ventricular arrhythmias. Our findings show that TG mice did not develop a significant amount of spontaneous ventricular arrhythmias under resting conditions. However, when challenged with catecholamines, the TG hearts developed a much higher level of complex ventricular arrhythmias compared to WT hearts (Fig. 6A). These complex VEBs included couplets, bigeminy, trigeminy, as well as non-sustained ventricular tachycardia, both monomorphic and polymorphic, consistent with the CPVT phenotype observed in patients in that they are triggered by catecholamines. We observed arrhythmias in all 3 TG lines and did not observe a graded response between lines.
The predisposition to complex ventricular arrhythmias in our TG mice could be due to the D307H mutation itself and/or increased expression of CASQ2 in the heart. Recent studies showed that over-expression of WT mouse CASQ2 (20-fold) and canine CASQ2 (10-fold) in the mouse hearts can cause mild to severe cardiac hypertrophy and result in an increase in SR Ca2+ storage capacity 36,37. Despite an increase in Ca2+ storage capacity, Ca2+ release was severely compromised in these mice. These data suggested that excessive amounts of CASQ2 can also lead to abnormally high levels of Ca2+ storage but reduced Ca2+ release. In contrast, we did not observe cardiac hypertrophy or pathology by increasing CASQ2 levels from 2- to 6-fold in TG hearts. This was supported by several lines of evidence, including histological data showing normal morphology and structure. In addition, expression of CASQ2D307H did not cause an increase in SR Ca2+ storage. These facts represent a striking difference between the CASQ2WT and CASQ2D307H expression models suggesting that the mutant protein behaves very differently from the WT protein.
Biochemical studies on purified CASQ2D307H mutant protein showed that the D307H mutation affects the Ca2+-dependent conformation of the protein and the ability of CASQ2 to interact with junctin and triadin 38. An additional possibility is that the mutation may also affect the ability of CASQ2 to polymerize into long polymers that appear to be crucial to its buffering capacity 39,40. Our findings suggest that the expression of CASQ2D307H destabilizes the Ca2+ release mechanism, possibly by interfering with the CASQ2-triadin and CASQ2-junctin interactions that regulate RyR2 function 6, leading to spontaneous, premature discharges of SR Ca2+ stores in myocytes undergoing periodic pacing. Indeed, the membrane potential traces of TG myocytes showed delayed afterdepolarizations (DADs) and the occasional generation of action potentials at the time of spontaneous Ca2+ transients (Fig. 5). Increased spontaneous Ca2+ release leads to the development of DADs through activation of depolarizing membrane currents via the Na-Ca exchange. These oscillations in membrane potential are thought to be the underlying cause of ventricular arrhythmias 20,41–43 and would also appear to be the mechanism underlying the complex ventricular arrhythmias observed in our TG mice.
In conclusion, the findings of the present study demonstrate that over-expression of CASQ2D307H in the mouse heart results in abnormal myocyte Ca2+ handling and complex ventricular arrhythmias that could contribute to the CPVT phenotype observed in human patients. Our study provides important evidence to establish a link between a mutation in CASQ2 and complex ventricular arrhythmias that are consistent with the CPVT phenotype.
Limitations
A potential limitation associated with our methodology was the lack of evaluation of arrhythmias in conscious mice. However, one of the main advantages of recording ECG in anesthetized mice is the ability to record from multiple leads to improve the diagnostic specificity, which can be technically very difficult with telemetric devices in conscious and exercising mice.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health grant HL-64140 (to MP), HL-74045 and HL-63043 (to SG) and American Heart Association.
Abbreviations
- SR
Sarcoplasmic reticulum
- CASQ2
Calsequestrin
- CPVT
Catecholaminergic polymorphic ventricular tachycardia
- WT
Wild-type
- TG
Transgenic
- RyR2
Cardiac ryanodine receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Cala SE, Jones LR. Rapid purification of calsequestrin from cardiac and skeletal muscle sarcoplasmic reticulum vesicles by Ca2+-dependent elution from phenyl-sepharose. J Biol Chem. 1983;258:11932–6. [PubMed] [Google Scholar]
- 2.Campbell KP, MacLennan DH, Jorgensen AO, Mintzer MC. Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53,000 dalton glycoprotein. J Biol Chem. 1983;258:1197–04. [PubMed] [Google Scholar]
- 3.Scott BT, Simmerman HK, Collins JH, Nadal-Ginard B, Jones LR. Complete amino acid sequence of canine cardiac calsequestrin deduced by cDNA cloning. J Biol Chem. 1988;263:8958–64. [PubMed] [Google Scholar]
- 4.Yano K, Zarain-Herzberg A. Sarcoplasmic reticulum calsequestrins: structural and functional properties. Mol Cell Biochem. 1994;135:61–70. doi: 10.1007/BF00925961. [DOI] [PubMed] [Google Scholar]
- 5.Ostwald TJ, MacLennan DH, Dorrington KJ. Effects of cation binding on the conformation of calsequestrin and the high affinity calcium-binding protein of sarcoplasmic reticulum. J Biol Chem. 1974;249:5867–71. [PubMed] [Google Scholar]
- 6.Györke I, Hester N, Jones LR, Györke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–8. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szegedi C, Sarkozi S, Herzog A, Jona I, Varsanyi M. Calsequestrin: more than ‘only’ a luminal Ca2+ buffer inside the sarcoplasmic reticulum. Biochem J. 1999;337:19–22. [PMC free article] [PubMed] [Google Scholar]
- 8.Beard NA, Laver DR, Dulhunty AF. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog Biophys Mol Biol. 2004;85:33–69. doi: 10.1016/j.pbiomolbio.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Franzini-Armstrong C, Kenney LJ, Varriano-Marston E. The structure of calsequestrin in triads of vertebrate skeletal muscle: a deep-etch study. J Cell Biol. 1987;105:49–56. doi: 10.1083/jcb.105.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jorgensen AO, Shen AC, Arnold W, McPherson PS, Campbell KP. The Ca2+-release channel/ryanodine receptor is localized in junctional and corbular sarcoplasmic reticulum in cardiac muscle. J Cell Biol. 1993;120:969–80. doi: 10.1083/jcb.120.4.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi YM, Alseikhan BA, Jones LR. Localization and characterization of the calsequestrin-binding domain of triadin 1. Evidence for a charged beta-strand in mediating the protein-protein interaction. J Biol Chem. 2000;275:17639–46. doi: 10.1074/jbc.M002091200. [DOI] [PubMed] [Google Scholar]
- 12.Franzini-Armstrong C, Protasi F. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol Rev. 1997;77:699–29. doi: 10.1152/physrev.1997.77.3.699. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389–97. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- 14.Terentyev D, Cala SE, Houle TD, Viatchenko-Karpinski S, Györke I, Terentyeva R, et al. Triadin overexpression stimulates excitation-contraction coupling and increases predisposition to cellular arrhythmia in cardiac myocytes. Circ Res. 2005;96:651–8. doi: 10.1161/01.RES.0000160609.98948.25. [DOI] [PubMed] [Google Scholar]
- 15.di Barletta MR, Viatchenko-Karpinski S, Nori A, Memmi M, Terentyev D, Turcato F, et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006;114:1012–9. doi: 10.1161/CIRCULATIONAHA.106.623793. [DOI] [PubMed] [Google Scholar]
- 16.Eldar M, Pras E, Lahat H. A missense mutation in the CASQ2 gene is associated with autosomal-recessive catecholamine-induced polymorphic ventricular tachycardia. Trends Cardiovasc Med. 2003;13:148–51. doi: 10.1016/s1050-1738(03)00025-2. [DOI] [PubMed] [Google Scholar]
- 17.Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Györke I, et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res. 2006;98:1151–8. doi: 10.1161/01.RES.0000220647.93982.08. [DOI] [PubMed] [Google Scholar]
- 18.Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Khoury A, et al. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13–21. Circulation. 2001;103:2822–7. doi: 10.1161/01.cir.103.23.2822. [DOI] [PubMed] [Google Scholar]
- 19.Lahat H, Pras E, Olender T, Avidan N, Ben Asher E, Man O, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–84. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viatchenko-Karpinski S, Terentyev D, Györke I, Terentyeva R, Volpe P, Priori SG, et al. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res. 2004;94:471–7. doi: 10.1161/01.RES.0000115944.10681.EB. [DOI] [PubMed] [Google Scholar]
- 21.Gulick J, Subramaniam A, Neumann J, Robbins J. Isolation and characterization of the mouse cardiac myosin heavy chain genes. J Biol Chem. 1991;266:9180–5. [PubMed] [Google Scholar]
- 22.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem. 1991;266:24613–20. [PubMed] [Google Scholar]
- 23.Subramaniam A, Gulick J, Neumann J, Knotts S, Robbins J. Transgenic analysis of the thyroid-responsive elements in the alpha-cardiac myosin heavy chain gene promoter. J Biol Chem. 1993;268:4331–6. [PubMed] [Google Scholar]
- 24.Babu GJ, Zheng Z, Natarajan P, Wheeler D, Janssen PM, Periasamy M. Overexpression of sarcolipin decreases myocyte contractility and calcium transient. Cardiovasc Res. 2005;65:177–86. doi: 10.1016/j.cardiores.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 25.Zhao W, Frank KF, Chu G, Gerst MJ, Schmidt AG, Ji Y, et al. Combined phospholamban ablation and SERCA1a overexpression result in a new hyperdynamic cardiac state. Cardiovasc Res. 2003;57:71–81. doi: 10.1016/s0008-6363(02)00609-0. [DOI] [PubMed] [Google Scholar]
- 26.Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Györke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–20. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y, Zhang Z, Timofeyev V, Sharma D, Xu D, Tuteja D, et al. The effects of intracellular Ca2+ on cardiac K+ channel expression and activity: novel insights from genetically altered mice. J Physiol. 2005;562:745–58. doi: 10.1113/jphysiol.2004.076216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, et al. Functional roles of Cav1.3 (α1D) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res. 2002;90:981–7. doi: 10.1161/01.res.0000018003.14304.e2. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z, He Y, Tuteja D, Xu D, Timofeyev V, Zhang Q, et al. Functional roles of Cav1.3(α1D) calcium channels in atria: insights gained from gene-targeted null mutant mice. Circulation. 2005;112:1936–44. doi: 10.1161/CIRCULATIONAHA.105.540070. [DOI] [PubMed] [Google Scholar]
- 30.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, et al. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 31.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292–8. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 32.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–90. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 33.Laitinen PJ, Swan H, Kontula K. Molecular genetics of exercise-induced polymorphic ventricular tachycardia: identification of three novel cardiac ryanodine receptor mutations and two common calsequestrin 2 amino-acid polymorphisms. Eur J Hum Genet. 2003;11:888–91. doi: 10.1038/sj.ejhg.5201061. [DOI] [PubMed] [Google Scholar]
- 34.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–00. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 35.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 36.Jones LR, Suzuki YJ, Wang W, Kobayashi YM, Ramesh V, Franzini-Armstrong C, et al. Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J Clin Invest. 1998;101:1385–93. doi: 10.1172/JCI1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato Y, Ferguson DG, Sako H, Dorn GW, Kadambi VJ, Yatani A, et al. Cardiac-specific overexpression of mouse cardiac calsequestrin is associated with depressed cardiovascular function and hypertrophy in transgenic mice. J Biol Chem. 1998;273:28470–7. doi: 10.1074/jbc.273.43.28470. [DOI] [PubMed] [Google Scholar]
- 38.Houle TD, Ram ML, Cala SE. Calsequestrin mutant D307H exhibits depressed binding to its protein targets and a depressed response to calcium. Cardiovasc Res. 2004;64:227–33. doi: 10.1016/j.cardiores.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Park H, Wu S, Dunker AK, Kang C. Polymerization of calsequestrin. Implications for Ca2+ regulation. J Biol Chem. 2003;278:16176–82. doi: 10.1074/jbc.M300120200. [DOI] [PubMed] [Google Scholar]
- 40.Wang S, Trumble WR, Liao H, Wesson CR, Dunker AK, Kang CH. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat Struct Biol. 1998;5:476–83. doi: 10.1038/nsb0698-476. [DOI] [PubMed] [Google Scholar]
- 41.Bers DM. Dordrecht. 2. The Netherlands: Kluwer Academic Publishers; 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. [Google Scholar]
- 42.Cranfield PF, Aronson RS. Cardiac Arrhythmias: The Role of Triggered Activity and Other Mechanisms. Mount Kisco, NY: Futura Publishing Company, Inc; 1988. [Google Scholar]
- 43.Priori SG, Corr PB. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol. 1990;258:H1796–1805. doi: 10.1152/ajpheart.1990.258.6.H1796. [DOI] [PubMed] [Google Scholar]
- 44.Philipson KD, Longoni S, Ward R. Purification of the cardiac Na+-Ca2+ exchange protein. Biochim Biophys Acta. 1988;945:298–06. doi: 10.1016/0005-2736(88)90492-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.