

Abstract
Phototropins are plant blue-light photoreceptors containing two light-, oxygen-, or voltage-sensitive (LOV) domains and a C-terminal kinase domain. The two LOV domains bind noncovalently flavin mononucleotide as a chromophore. We investigated the photocycle of fast-recovery mutant LOV2-I403V from Arabidopsis phototropin 2 by step-scan Fourier transform infrared spectroscopy. The reaction of the triplet excited state of flavin with cysteine takes place with a time constant of 3 μs to yield the covalent adduct. Our data provide evidence that the flavin is unprotonated in the productive triplet state, disfavoring an ionic mechanism of bond formation. An intermediate adduct species was evident that displayed changes in secondary structure in the helix or loop region, and relaxed with a time constant of 120 μs. In milliseconds, the final adduct state is formed by further alterations of secondary structure, including β-sheets. A comparison with wild-type adduct spectra shows that the mutation does not interfere with the functionality of the domain. All signals originate from within the LOV domain, because the construct does not comprise the adjacent Jα helix required for signal transduction. The contribution of early and late adduct intermediates to signal transfer to the Jα helix outside of the domain is discussed.
Introduction
Phototropins are blue-light photoreceptors in plants that regulate phototropism, stomatal opening, chloroplast relocation, rapid inhibition of hypocotyl elongation, and promotion of cotyledon expansion (1). In the green alga Chlamydomonas reinhardtii, phototropin is responsible for completion of the sexual life cycle (2). Phototropins contain two light-, oxygen-, or voltage-sensitive domains (LOV1 and LOV2) that noncovalently bind flavin mononucleotide (FMN) as a chromophore (3). The LOV domains are connected to a C-terminal serine/threonine kinase domain via a linker region with a conserved Jα helix (4).
Absorption of blue light by the LOV domain induces a photocycle in which the triplet state of the FMN (LOV715 or LOV660) is formed within a few nanoseconds (5,6). LOV715 reacts within 10 μs with a conserved cysteine to form the photoadduct LOV390. This adduct is characterized by a covalent bond between the C4a atom of FMN and the cysteine's sulfur (Fig. 1) (7,8). Ultraviolet-visible (UV/Vis) spectroscopy has provided some information on the reaction pathway from the triplet to the adduct state. The fine structure of the LOV715 spectrum was interpreted to originate mainly from a protonated triplet excited state by nanosecond proton transfer from the cysteine (5). In contrast, the initial formation of a sulfur-carbon bond without any proton transfer was proposed, based on a shift in the spectrum of the adduct at 77 K from 390 to 405 nm (9). Indications that an initial electron transfer takes place come from the trapping of a flavin anionic radical at temperatures below 150 K in an unreactive fraction of the LOV domain (10). Therefore, the protonation state of the triplet excited state and the mechanism of adduct formation remain unclear.
Figure 1.
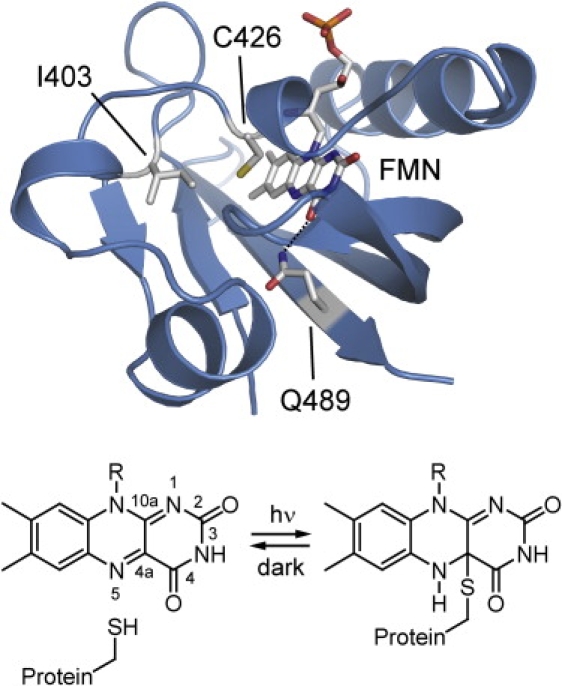
(Top) Structural model of Arabidopsis phot2-LOV2 domain in the dark state. The structure was generated by homology modeling with Swiss Model (http://swissmodel.expasy.org), using the crystal structure of Adiantum neo-LOV2 (Protein Data Bank entry 1G28). Amino-acid side chains of C426, I403, and Q489 are highlighted as sticks. C426 is the reaction partner of the chromophore flavin mononucleotide (FMN). I403 was exchanged with valine in this study. The side chain of Q489 was shown to flip upon light activation of LOV2 (details in text). (Bottom) Blue light induces formation of an adduct between FMN and cysteine. The adduct decays within seconds or minutes back to the dark form.
LOV390 decays completely back to the dark state within several seconds to minutes, depending on the biological origin. In response to LOV2 domain activation, the Jα helix in the linker to the kinase domain dissociates from the LOV domain surface (4). Finally, the kinase domain autophosphorylates phototropin (11). A permanent displacement of the Jα helix by mutation leads to a constitutively active kinase (12).
The processes involved in signal transfer from the chromophore to the Jα helix are only partly elucidated. A light-induced flip of a glutamine side chain hydrogen-bonded to the flavin C4=O was proposed (Fig. 1), based on an interpretation of crystal structure data (13). This proposal was supported by shifts in the C4=O vibrational frequency upon exchange of the glutamine to leucine, as detected by Fourier transform infrared (FTIR) spectroscopy (14). Crystal-structure analysis of LOV1 and LOV2 (13,15) and NMR spectroscopy (4) demonstrated that structural changes in LOV390, compared with the dark state, are restricted to the immediate vicinity of the chromophore. The diffusion coefficient of the LOV domain does not change after microsecond adduct formation, as demonstrated by transient grating (TG) spectroscopy (16). Pronounced structural changes were only detected when extended LOV domain constructs were studied. This is the case for circular dichroism (CD) spectroscopy (8,17), small-angle x-ray scattering (18), time-resolved optical rotatory dispersion spectroscopy (19), and TG spectroscopy (20). Observed changes were attributed to the Jα helix or the complete linker region outside of the LOV domain. The only indications of secondary-structure changes within the LOV domain came from low-temperature FTIR difference spectroscopy on Adiantum neo-LOV2 (21). Iwata et al. (21) interpreted temperature-dependent changes in the spectra to a loosening of turns, followed by β-sheet tightening and changes in α-helical content. In their study, the Jα helix was included in the construct, which complicates the assignment.
FTIR spectroscopy is an established technique for studying the secondary structure of proteins via the amide I vibration, which is primarily a coupled C=O stretching mode of the backbone. Different elements can be distinguished by their frequency, with β-sheets absorbing at 1620–1640 cm−1 with a weak contribution at around 1680 cm−1, helices and loops absorbing at 1640–1660 cm−1, and turn elements absorbing at 1660–1690 cm−1 (22,23).
We applied time-resolved FTIR difference spectroscopy to a LOV domain without Jα extension. To accelerate recovery time to the dark state, we introduced a I403V mutation into the LOV2 domain of Arabidopsis phototropin 2. The fast-cycling system enabled an application of the step-scan technique with microsecond time resolution. Light-induced changes after excitation by a nanosecond laser pulse were followed, to investigate adduct formation from the triplet excited state and subsequent structural changes. The spectrum of the productive triplet state at 2 μs shows an unprotonated flavin directly before its reaction with the cysteine. An intermediate adduct state LOV390-I was detected in the early microsecond time domain, which can be distinguished from the final adduct LOV390-II by the secondary structure of its apoprotein. This finding is discussed in the framework of other time-resolved approaches that did not characterize this intermediate within the LOV domain.
Materials and Methods
Sample preparation
The LOV2 domain of Arabidopsis thaliana phototropin 2, with extensions comprising amino acids D363 to Q500, was expressed and purified as previously described (18), with the following modifications. Escherichia coli cells were grown at 310 K in lysogeny broth medium containing 100 mg L−1 ampicillin until A600 had reached 0.5, and were then incubated with 0.1 mM isopropyl-β-d-thiogalactopyranoside for 20 h at 293 K in the dark. After cell lysis and resuspension, the supernatant was collected and mixed with glutathione-sepharose 4B (Amersham Biosciences, Piscataway, NJ). The resin was washed with 8 mM Na2HPO4, 1.5 mM KH2PO4, 140 mM NaCl, and 2.7 mM KCl (pH 7.4). The domain was cleaved off from its glutathione S-transferase tag with thrombin protease at 293 K for 16 h. This procedure leaves five extra amino-acid residues, Gly-Ser-Pro-Glu-Phe, at the N-terminus of the domain. The cleaved LOV2 domain was further purified by gel chromatography with Sephacryl S-100 HR (Amersham Pharmacia Biotech, Tokyo, Japan) and a buffer solution containing 100 mM NaCl, 25 mM Tris-HCl, and 1 mM Na2-EDTA (pH 7.8). The purity of the protein was >99%, as judged by Coomassie brilliant blue staining after sodium dodecyl sulfate poly-acrylamide gel electrophoresis.
The amino-acid substitution from Ile-403 to Val was introduced by using a Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA), following the manufacturer's instructions, and was verified by DNA sequencing. The following mutagenic primers were used for the polymerase chain reaction: 5′-GATAATCCCGTTATCTTTGCA-3′ and 5′-TGCAAAGATAACGGGATTATC-3′. Expression and purification were performed as described above.
The proteins were transferred into 10 mM phosphate buffer (pH 8) by repeated ultrafiltration, using a Vivaspin 500 filter device with a 10-kDa cutoff (Sartorius, Göttingen, Germany), and were concentrated to 4 mM. A droplet of 1 μL was placed on a BaF2 window and sealed by a second window. The sample was measured in solution; no drying process was applied.
FTIR spectroscopy
We performed FTIR spectroscopy using an IFS 66v/S spectrometer (Bruker, Ettlingen, Germany) equipped with a step-scan option at a spectral resolution of 4.5 cm−1. A broadband interference filter (Spectrogon, Täby, Sweden) after the cuvette restricted the spectral range to 1974–1200 cm−1, which allowed for an undersampling of data points and efficiently blocked stray light. For photo excitation, a light pulse of 445-nm wavelength, 3-mJ/cm2 energy density, and 10-ns duration was generated by a tunable optical parametric oscillator system (Opta, Bensheim, Germany), which was pumped by the third harmonic of a Nd:YAG laser (Quanta-Ray Lab-150, Spectra Physics, Darmstadt, Germany). Homogeneous illumination of the sample was achieved using a diffuser plate. The repetition rate of the laser at 10 Hz was adjusted by an optical shutter to 0.5 Hz for sample excitation. The temperature of the sample was maintained at 40°C by a circulating water bath. For the step scan, 1000 equidistant time slices of 5 μs were sampled up to 5 ms. Eighty-eight coadditions of 449 mirror positions were averaged. The sample was exchanged every 898 excitation pulses, to minimize photodegradation. The integrity of the sample was monitored by comparing rapid scan spectra before and after each step-scan experiment. The intensity of difference bands decreased to 50–75% of the initial value. In the step-scan approach, data points recorded late in the experiment, i.e., far from the center burst, mainly contribute to spectral resolution and noise level. Data points recorded at the beginning of an experiment are close to the center burst of the interferogram, and therefore carry most of the significant information. The first of the time-dependent spectra was scaled in intensity by a factor of 1.29 to account for the detector rise time and for a delay of 1 μs between laser excitation and data acquisition, as determined using bacteriorhodopsin as standard. A rapid scan was performed before and after step scan on the same sample. Thirty-eight of these measurements of 32 scans were averaged, with a scan duration in forward backward mode of 38 ms and a scanner velocity of 280 kHz.
In a separate experiment, a light-emitting diode (Luxeon Star, Philips Lumileds, San Jose, CA), with an emission maximum at 455 nm and a power of 20 mW/cm2, was used for comparison of the difference spectra of the wild-type and mutant. The slower-cycling wild-type was illuminated for 1 s, and the faster-cycling I403V mutant was illuminated for 0.5 s. Immediately after illumination, rapid scans were recorded for a duration of 3.2 s and 0.8 s, respectively. We averaged 2048 light-minus-dark spectra for both samples.
Data analysis
Time-resolved data were analyzed by applying a global fit procedure. Starting with the simplest kinetic model, a sequence of irreversible first-order transitions without branching was taken (24). Decay times were obtained from a nonlinear least squares fit with the sum of three exponentials, using the SolvOpt toolbox within MATLAB (The MathWorks, Natick, MA). The fitted data matrix was reconstructed by pseudo-inversion of the matrix of the fitted exponential functions (25). In the next step, the concentration profile of each species was determined, using the integrated solutions of the kinetic differential equations. Species-associated difference spectra were obtained by pseudo-inversion of the concentration profile matrix. An increase in the number of exponentials did not further improve the quality of the fit. In our experiment, indications for a branching reaction, such as secondary products or a broadening of product bands, were not evident.
Results
The photocycling time of LOV domains is in the range of several seconds to minutes or even hours (26,27). To apply the FTIR step-scan technique with its necessary extensive averaging (28), the LOV domain must complete the photocycle within a few seconds. The LOV2 domain of Arabidopsis phototropin 2 (phot2) exhibits the fastest decay reported so far of LOV390 to the dark state, with t1/2 = 5 s (26). An increase in the back reaction rate was achieved by raising the temperature to 40°C. To accelerate the photocycle further, isoleucine 403 was changed to a valine (Fig. 1). The same conservative mutation in phot1-LOV2 from Avena sativa (I16V) led to an increase in the adduct decay rate by one order of magnitude, without altering the UV/Vis spectrum (29). An additional effect of this mutation is a twofold decrease of the adduct formation rate. Both effects were assigned to a loss of steric support because of the absence of one CH2 group in the vicinity of the chromophore.
The influence of the mutation was investigated by applying FTIR experiments to the I403V mutant (Fig. 2, upper curve) and wild-type phot2-LOV2 from Arabidopsis (Fig. 2, lower curve). The light-minus-dark difference spectra of the wild-type and mutant are identical, except for a downshift of the positive band at 1732 cm−1 by 1 cm−1, and a small increase in band intensity at 1677 and 1663 cm−1. Moreover, the positive band at 1663 cm−1 and the negative band at 1642 cm−1 of the wild-type spectrum are shifted by <3 cm−1 by the mutation. The variations in frequency are much smaller than those recorded within the LOV domain family (21,30–32). We conclude from the high similarity of the spectra of mutant and wild-type that the domain remains fully functional, and that the photocycle is only altered with respect to its kinetics by the replacement of isoleucine 403 by valine. The accelerating effect of the mutation, therefore, enables time-resolved studies, and helps avoid the less defined effects of accelerating additives such as imidazole (33).
Figure 2.

Comparison of light-induced FTIR difference spectra of Arabidopsis phot2-LOV2-I403V mutant (upper trace) and the wild-type (lower trace). Signals of mutant and wild-type were averaged for 0.8 s and 3.2 s after illumination, respectively. Positive bands arise from changes in chromophore and apoprotein structure in the adduct LOV390, and negative bands originate from the dark state. The spectra are essentially congruent, which points to a negligible influence on the structure of the LOV domain by the amino-acid exchange.
A three-dimensional data set of time-resolved FTIR difference spectra was recorded with the step-scan technique on the phot2-LOV2-I403V mutant. The first spectrum covered a time range of 4 μs after the laser flash. The resulting difference spectrum (Fig. 3 A, upper curve) shows positive signals attributable to the photo-induced formation of an intermediate state, and negative signals that originated from the dark state of the LOV domain. Prominent bands of the intermediate were detected at 1657 cm−1, 1622 cm−1, 1485 cm−1, and 1435 cm−1. These spectral features resemble marker bands of the flavin triplet excited state in the LOV2 domain of Adiantum phy3 (34). Moreover, very similar features at 1652 cm−1, 1484 cm−1, and 1436 cm−1 were assigned to the triplet state of riboflavin tetraacetate by time-resolved dispersive infrared spectroscopy (35). Because of the improvement in spectral resolution from 16 cm−1 to 4.5 cm−1 in our approach, a low-frequency shoulder of the band at 1652 cm−1 was resolved as a second band at 1622 cm−1. These two bands can be assigned to carbonyl vibrations of the flavin (35), which show a distinct downshift from 1712 cm−1 and 1676 cm−1 in LOV2 (see (38)) because of the strong change in electron-density distribution in the transition from ground state to triplet excited state. The agreement between the triplet spectra of riboflavin tetraacetate and LOV2 is very high, considering the influence of the apoprotein. We conclude from this comparison that flavin in LOV715 is unprotonated at the N5-position. A protonation of 3FMN would lead to three additional vibrational modes of freedom and a charged molecule. These changes would strongly alter the band pattern, including that of carbonyl vibrations at 1652 cm−1 and 1622 cm−1. We can rule out that the reference spectrum shows a protonated riboflavin, because the triplet spectra were recorded in deuterated acetonitrile (35). This solvent exhibits very a low proton-donating/deuteron-donating ability, considering the pKa of acetonitrile in water of 29 (36).
Figure 3.

(A) Upper trace depicts light-induced FTIR difference spectrum of LOV2-I403V at 2 μs after laser excitation. Lower trace shows species-associated difference spectrum of the first intermediate, as derived from global fitting with τ = 3.3 μs. Positive bands are assigned to triplet excited state of FMN. (B) Decay kinetics of triplet excited state at characteristic wavenumbers (solid lines, global fit; dots, experimental data).
The global fit to the data set provided a species-associated difference spectrum with a decay time constant of 3.3 μs (Fig. 3 A, lower curve). The microsecond time constant is in agreement with previous results on triplet state decay in LOV1 of C. reinhardtii phot (37) and LOV2 of A. sativa phot1 (6). A shorter decay time of 1 μs was obtained by TG spectroscopy on wild-type Arabidopsis phot2-LOV2 (16). The deceleration in our experiment may be an effect of the introduced mutation (29). The traces of the global fit follow the measured kinetic traces at selected wavenumbers of the marker bands (Fig. 3 B). The bands at around 1650 cm−1 only partially decay, whereas the bands at 1485 cm−1 and 1435 cm−1 decay to a constant level close to zero. According to the concentration profiles of the fitted species, the first difference spectrum (Fig. 3 A, upper curve) contains ∼50% contribution from the triplet excited state and ∼50% of the subsequent species.
The triplet excited state reacts with cysteine to yield the adduct state LOV390 (6). Accordingly, the kinetic trace at 1731 cm−1 shows the appearance of a band assigned to the C4=O vibration of the adduct (30,32,38) within a few microseconds (Fig. 4 A). At the position of the strongest adduct band at 1537 cm−1, the kinetics reflect a rise until 12 μs, followed by a partial decay of the band (Fig. 4 A). These kinetics indicate the formation of an intermediate state that we denote as LOV390-I. The global fit yielded a decay constant of 120 μs for LOV390-I. Spectra in a time range of 7–90 μs were averaged, where the contribution of this intermediate dominated. The spectrum of LOV390-I was compared with the final adduct spectrum of LOV390-II at 52 ms after excitation, which was obtained by the rapid-scan technique (Fig. 4 B, curve b). The difference spectrum at 52 ms shows typical light-induced difference bands of the adduct state LOV2390 (31). The comparison shows that all positive marker bands originating from the flavin chromophore in LOV390 are identical, such as the bands at 1731 cm−1, 1429 cm−1, and 1379 cm−1 (Fig. 4 B, curve a) (30,32). However, the comparison reveals significant differences in the spectra in the region of 1615–1695 cm−1 (amide I) and at around 1550 cm−1 (amide II). Changes in these regions indicate changes in the secondary structure of the protein. To illustrate these differences, a double-difference spectrum of the 52-ms spectrum minus the LOV390-I spectrum was calculated (Fig. 4 B, b minus a). The double difference shows bands in the amide II region at around 1550 cm−1, and distinct signals in the amide I region at 1686 cm−1, 1677 cm−1, 1646 cm−1, and 1629 cm−1. For comparison, the noise level was determined as a difference absorption covering 85 μs without excitation (Fig. 4 B, curve c). The vibrational differences provide evidence that the two spectra from the microsecond and millisecond time domains do not arise from the same intermediate. The differences in amide regions point to a different secondary structure of the apoprotein in the early LOV390-I intermediate, compared with the late LOV390-II.
Figure 4.

(A) Kinetics at characteristic wavenumbers after laser excitation show formation and decay of intermediate LOV390-I (solid lines, global fit; dots, experimental data). The band at 1731 cm−1 is not influenced by decay of LOV390-I because it represents a pure chromophore mode not overlapped by vibrations from the apoprotein. (B) Light-induced FTIR difference spectrum of the LOV390-I intermediate obtained by averaging across the time range between 7–90 μs (a). The spectrum at 52 ms after laser excitation is shown for comparison, representing LOV390-II (b). Numerical subtraction of the LOV390-II spectrum minus the LOV390-I spectrum yields the double-difference spectrum, depicted as lower trace (b minus a). The noise level is represented by a difference spectrum obtained by averaging across 85 μs before excitation (c). Vibrational differences in the amide I and amide II regions are evident, indicating changes in secondary structure.
Rapid-scan experiments were performed under conditions identical to those in the step-scan experiments, to investigate the thermal back reaction of the LOV390-II state to the dark-state LOV445 (Fig. 5). The data were well-reproduced by a global fit with a single time constant of 0.58 s. All difference bands of the chromophore and apoprotein decayed simultaneously. The protein fully recovered to the dark state within 2 s. Under the same conditions, the wild-type LOV2 domain showed a dark recovery constant of τ = 3.3 s (data not shown).
Figure 5.

Series of difference spectra in the millisecond time range of LOV2-I403V after laser excitation, showing the back reaction of LOV390–II to the dark state. The mono-exponential fit to the decay yields a time constant of 0.58 s. The protein fully recovers within 2 s.
Discussion
We report here on the first time-resolved infrared spectra of the triplet excited state of flavin in a protein. For flavin-containing blue-light receptors, this is the second microsecond time-resolved IR study after our investigation of the sensor of blue light using flavin adenine dinucleotide (BLUF) domain (39). Previous studies of the LOV domain by NMR (4) and x-ray (13,15) analysis did not reveal significant changes in secondary structure, if excluding the adjacent Jα helix element. These studies were restricted by a static view of the dark and light states. We applied step-scan FTIR spectroscopy to gain a dynamic view of adduct formation from the triplet state and of transient structural changes within the LOV domain.
Protonation state of flavin in LOV715 and mechanism of adduct formation
We present the vibrational spectrum of the productive triplet state LOV715 (Fig. 3). Similar spectra were obtained for the triplet state of Adiantum neo-LOV2 (34). However, these spectra were recorded at 77 K either on the C966A mutant, which does not form the LOV390 state, or after prolonged illumination of the wild-type. In the latter case, the difference spectrum represented a remaining unreactive fraction of the sample. These spectra differ from the spectrum of LOV715 in a shift of the band at 1493 cm−1 or 1497 cm−1 to 1485 cm−1. The molecular basis of this shift is unclear. Further experiments and interpretation by a theoretical approach must be conducted to evaluate whether this shift represents a characteristic difference between productive and unproductive triplet states.
The protonation state of flavin in LOV715 has been under debate because of its central role in the mechanism of adduct formation. An ionic mechanism with a 3FMNH+ was proposed (40) and supported by an interpretation of UV/Vis spectroscopic data (5). On the other hand, FTIR spectroscopy indicated that the proton-donor cysteine is not deprotonated upon formation of the unproductive triplet excited state (34). In our experiment, prominent bands of LOV715 (Fig. 3 A) provided evidence for an unprotonated, productive 3FMN, disfavoring an ionic mechanism.
The spectra of LOV715 and LOV390-I were readily assigned to the triplet and adduct states of the flavin, respectively. Intermediate species in the reaction were not detected. It was proposed that the protonated sulfur forms a bond to flavin C4a before proton transfer (9). The resulting zwitterionic structure would lead to significant alterations in flavin vibrations that were not evident (Figs. 3 A and 4 B). Alternatively, the involvement of radical species was proposed (41). Formation of a flavin anionic radical species would result in a single band at 1636 cm−1 (35), instead of the two bands detected at 1657 cm−1 and 1622 cm−1. An admixture of a neutral flavin radical to the triplet-state spectrum cannot be excluded, but its spectrum differs from that of a triplet state by the presence of a strong band at around 1532 cm−1 and the lack of bands at 1485 cm−1 and 1435 cm−1 (35). It should be noted that the time resolution of our experiment was limited.
The difference spectrum of LOV715 shows a negative band at 1695 cm−1 that originates from the apoprotein (Fig. 3 A). The only flavin vibrations of the dark state in this spectral region are the C4=O vibration at 1712 cm−1 and the C2=O vibration at 1676 cm−1 (38). The kinetic trace at 1695 cm−1 shows a decay in absorbance to a constant level with adduct formation (data not shown), which indicates that the band may already be present in the triplet state. This presence implies a reorganization of the protein environment as a result of the change in electron-density distribution in the isoalloxazine ring. The exceptionally high frequency of the band points to a change in turn structure (22,23) or to a glutamine side chain (42), such as the one hydrogen bonded to flavin C4=O. A light-induced rotation of this glutamine side chain and a loss of its hydrogen bond to the flavin C4=O was postulated (13). The FTIR data reveal that the vibrational band of flavin C4=O shifts to 1731 cm−1 at the time of cysteinyl adduct formation (Fig. 4 A). Based on the subsequent constancy of the 1731 cm−1 band intensity over time, we conclude that the flip of the glutamine and the loss of its hydrogen bond are completed with the formation of LOV390-I.
Response of the apoprotein to adduct formation in the microsecond and millisecond time domain
The FTIR difference spectrum of LOV390-I immediately after adduct formation shows bands in the amide I region at 1662(+)/1642(−) cm−1 (Fig. 4 B). These bands are assigned to a pronounced loosening of hydrogen bonds in helical or loop-structure elements, in agreement with low-temperature experiments at 77 K (21). They reflect the immediate response of the apoprotein to adduct formation. Other bands in this spectral region at 1686(+)/1676(−) cm−1 are assigned to the C2=O vibration of the chromophore (38). Difference spectra recorded at 77 K also showed prominent features at 1692(+), 1687(+), and 1673(−) cm−1, which decayed above 150 K, and were assigned to turn structures (21). These structural changes were not evident in our time-dependent experiments, which is explained either by the assignment to the linker region outside the LOV domain, or to the discrepancy between low-temperature and time-dependent measurements. Such a discrepancy was reported for the photoactive yellow protein because of the strong restriction in movement of the chromophore and protein at low temperatures (43).
The band at 1537 cm−1 (Fig. 4 B) represents a marker for secondary-structure changes as it shifts to 1521 cm−1 upon global 13C-labeling of the apoprotein (38). This amide II band also receives strong contributions of the C10a=N1 vibration of flavin (30,32). The kinetics of the band at 1537 cm−1 exhibits maximum intensity at 12 μs with the formation of LOV390-I. It decays with a time constant of 120 μs, until a stationary level is formed between 500 μs and 5 ms (Fig. 4 A). A loss of intensity in an absorption difference spectrum indicates a relaxation toward the dark state. In this case, it is attributed to a relaxation of strain in secondary structure, as induced by the formation of LOV390-I. The stationary level after 500 μs is attributable to the contribution of flavin vibrations that are left over in the difference spectrum. Unfortunately, the secondary structure of the relaxed state could not be fully characterized because of instabilities in the signal in the water-absorbing region around 1650 cm−1 later than 100 μs. These instabilities are due to the high water content of the sample and the low signal intensity. The absorbance at 1537 cm−1 rises again after 5 ms, as deduced from the spectrum of LOV390-II at 52 ms. Changes in secondary structure take place that lead to an even higher absorbance than that of LOV390-I (see below).
The relaxation time of LOV390-I of 120 μs is in the same time range found using other time-resolved techniques. However, those measurements were performed on LOV domains with Jα-helical extensions and at different temperatures. A loss in α-helicity was demonstrated by optical rotatory dispersion on A. sativa phot1-LOV2 with τ = 90 ± 36 μs (19). From TG and transient lens experiments, a change in the diffusion constant in an entropically guided process with τ = 140 μs or 300 μs was reported (16,20,44). The change in diffusion coefficient was assigned to the Jα helix outside the LOV domain, suggesting a linkage to the decay of LOV390-I observed by us in the LOV core.
The spectrum of the final adduct state LOV390-II according to steady-state and millisecond rapid-scan experiments (Figs. 4 B and 5) differs in the spectral regions of the amide I and II vibrations from that of LOV390-I. This difference is typical for changes in secondary structure. The double-difference spectrum shows prominent bands at 1686(+), 1677(−), 1646(−), and 1629(+) cm−1. The latter two bands were observed in low-temperature measurements on the Adiantum neo-LOV2-linker above 250 K, and were interpreted as a tightening of the β-sheet hydrogen-bond network (21). This interpretation was supported by the global 13C-labeling of apoprotein or FMN (38). Our study allows an assignment of these changes to the LOV domain core. They occur later than 5 ms, as inferred from the absence of any changes in the amide II region at 1537 cm−1 (Fig. 4 A). The bands at 1677(−) and 1686(+) cm−1 have not been resolved in temperature-dependent experiments. They are tentatively assigned to turn-structure elements, because the changes in β-sheets led only to weak signals in this spectral region (23). In the same time range as the transition from LOV390-I to LOV390-II, a small volume change or energy release with τ = 9 ms was detected in the same LOV domain without a linker (16,44). The process did not result in any change in diffusion coefficient. An extended construct including the Jα helix exhibited a similar process with τ = 11 ms (44). However, these conformational changes were interpreted as insignificant in terms of signaling. Further time-resolved infrared experiments are needed to bridge the time gap of this study between 5–52 ms.
In summary, three different phases of LOV apoprotein response to adduct formation can be distinguished (Fig. 6). Immediately upon bond formation, a loosening of the α-helical network is detected within the LOV domain. The resulting LOV390-I decays with a time constant of 120 μs, characterized by a relaxation of strain in the apoprotein. In the time range between 5–52 ms, structural changes occur, including a tightening of the β-sheet-network and movements of turn elements, leading to the formation of the final adduct state LOV390-II. A follow-up study using an extended LOV-Jα construct must clarify whether LOV390-I or LOV390-II represents the signaling state. The microsecond process detected in transient lens experiments (44) implies a signal transfer to the Jα initiated by the formation of LOV390-I. However, it is difficult to conceive that the structural changes observed in the transition to LOV390-II do not contribute at all to communication with the other domains of phototropin.
Figure 6.
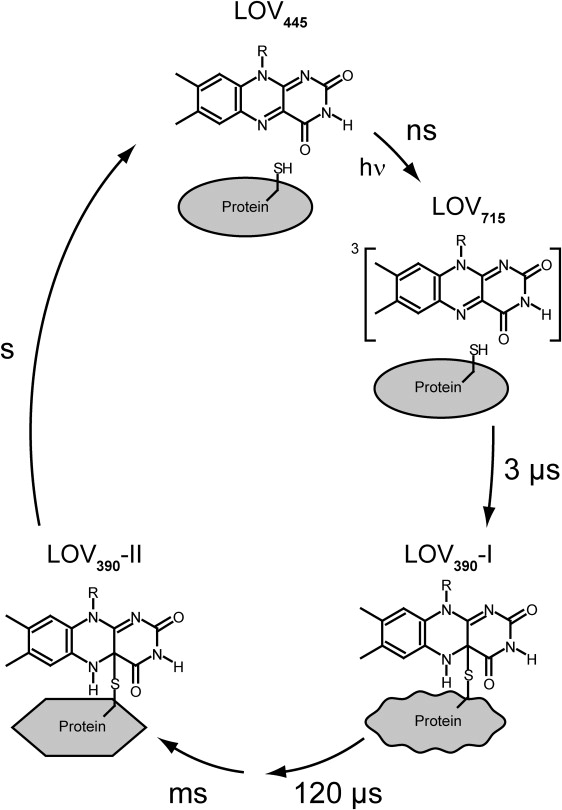
Photoreaction scheme of LOV2 domains without Jα helix, as derived from time-resolved FTIR spectroscopy at 40°C. Upon blue-light illumination, the dark-state LOV445 is converted into triplet excited state LOV715. The latter state is present in its unprotonated form, and decays with τ = 3 μs into the early adduct state LOV390-I. Concomitant differences in secondary structure relax with a time constant of 120 μs. In a subsequent step, structural changes including β-sheets and turn elements of the protein occur, and LOV390-II is formed. Finally, LOV390-II recovers to the dark state within seconds.
Conclusions
This study clarifies two basic features of LOV domain signaling. First, evidence is presented that a neutral flavin triplet state reacts with the cysteine. This finding rules out an ionic mechanism, where the proton of the cysteine is transferred to the flavin well before the sulfur-carbon bond is formed. An ionic mechanism is only conceivable if the proton transfer is rate-limiting. Second, the intermediate LOV390-I shows light-induced changes in helical structure within the LOV domain, even though the Jα helix is missing in the construct. It decays with a time constant of 120 μs, which fits the timescale of processes that were assigned to the outside of the domain. To the best of our knowledge, LOV390-I was not previously characterized because of its transient nature.
Acknowledgments
This work was supported by the Helmholtz Gemeinschaft (grant VH-NG-014) and Deutsche Forschungsgemeinschaft (grant FOR 526).
References
- 1.Christie J.M. Phototropin blue-light receptors. Annu. Rev. Plant Biol. 2007;58:21–45. doi: 10.1146/annurev.arplant.58.032806.103951. [DOI] [PubMed] [Google Scholar]
- 2.Huang K., Beck C.F. Phototropin is the blue-light receptor that controls multiple steps in the sexual life cycle of the green alga Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA. 2003;100:6269–6274. doi: 10.1073/pnas.0931459100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huala E., Oeller P.W., Liscum E., Han I.S., Larsen E. Arabidopsis NPH1: a protein kinase with a putative redox-sensing domain. Science. 1997;278:2120–2123. doi: 10.1126/science.278.5346.2120. [DOI] [PubMed] [Google Scholar]
- 4.Harper S.M., Neil L.C., Gardner K.H. Structural basis of a phototropin light switch. Science. 2003;301:1541–1544. doi: 10.1126/science.1086810. [DOI] [PubMed] [Google Scholar]
- 5.Kennis J.T., Crosson S., Gauden M., van Stokkum I.H., Moffat K. Primary reactions of the LOV2 domain of phototropin, a plant blue-light photoreceptor. Biochemistry. 2003;42:3385–3392. doi: 10.1021/bi034022k. [DOI] [PubMed] [Google Scholar]
- 6.Swartz T.E., Corchnoy S.B., Christie J.M., Lewis J.W., Szundi I. The photocycle of a flavin-binding domain of the blue light photoreceptor phototropin. J. Biol. Chem. 2001;276:36493–36500. doi: 10.1074/jbc.M103114200. [DOI] [PubMed] [Google Scholar]
- 7.Crosson S., Moffat K. Structure of a flavin-binding plant photoreceptor domain: insights into light-mediated signal transduction. Proc. Natl. Acad. Sci. USA. 2001;98:2995–3000. doi: 10.1073/pnas.051520298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salomon M., Christie J.M., Knieb E., Lempert U., Briggs W.R. Photochemical and mutational analysis of the FMN-binding domains of the plant blue light receptor, phototropin. Biochemistry. 2000;39:9401–9410. doi: 10.1021/bi000585+. [DOI] [PubMed] [Google Scholar]
- 9.Schleicher E., Kowalczyk R.M., Kay C.W., Hegemann P., Bacher A. On the reaction mechanism of adduct formation in LOV domains of the plant blue-light receptor phototropin. J. Am. Chem. Soc. 2004;126:11067–11076. doi: 10.1021/ja049553q. [DOI] [PubMed] [Google Scholar]
- 10.Zikihara K., Iwata T., Matsuoka D., Kandori H., Todo T. Photoreaction cycle of the light, oxygen, and voltage domain in FKF1 determined by low-temperature absorption spectroscopy. Biochemistry. 2006;45:10828–10837. doi: 10.1021/bi0607857. [DOI] [PubMed] [Google Scholar]
- 11.Christie J.M., Reymond P., Powell G.K., Bernasconi P., Raibekas A.A. Arabidopsis NPH1: a flavoprotein with the properties of a photoreceptor for phototropism. Science. 1998;282:1698–1701. doi: 10.1126/science.282.5394.1698. [DOI] [PubMed] [Google Scholar]
- 12.Harper S.M., Christie J.M., Gardner K.H. Disruption of the LOV-Jα helix interaction activates phototropin kinase activity. Biochemistry. 2004;43:16184–16192. doi: 10.1021/bi048092i. [DOI] [PubMed] [Google Scholar]
- 13.Crosson S., Moffat K. Photoexcited structure of a plant photoreceptor domain reveals a light-driven molecular switch. Plant Cell. 2002;14:1067–1075. doi: 10.1105/tpc.010475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nozaki D., Iwata T., Ishikawa T., Todo T., Tokutomi S. Role of Gln1029 in the photoactivation processes of the LOV2 domain in Adiantum phytochrome3. Biochemistry. 2004;43:8373–8379. doi: 10.1021/bi0494727. [DOI] [PubMed] [Google Scholar]
- 15.Fedorov R., Schlichting I., Hartmann E., Domratcheva T., Fuhrmann M. Crystal structures and molecular mechanism of a light-induced signaling switch: the Phot-LOV1 domain from Chlamydomonas reinhardtii. Biophys. J. 2003;84:2474–2482. doi: 10.1016/S0006-3495(03)75052-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eitoku T., Nakasone Y., Matsuoka D., Tokutomi S., Terazima M. Conformational dynamics of phototropin 2 LOV2 domain with the linker upon photoexcitation. J. Am. Chem. Soc. 2005;127:13238–13244. doi: 10.1021/ja052523i. [DOI] [PubMed] [Google Scholar]
- 17.Corchnoy S.B., Swartz T.E., Lewis J.W., Szundi I., Briggs W.R. Intramolecular proton transfers and structural changes during the photocycle of the LOV2 domain of phototropin 1. J. Biol. Chem. 2003;278:724–731. doi: 10.1074/jbc.M209119200. [DOI] [PubMed] [Google Scholar]
- 18.Nakasako M., Iwata T., Matsuoka D., Tokutomi S. Light-induced structural changes of LOV domain-containing polypeptides from Arabidopsis phototropin 1 and 2 studied by small-angle x-ray scattering. Biochemistry. 2004;43:14881–14890. doi: 10.1021/bi0485530. [DOI] [PubMed] [Google Scholar]
- 19.Chen E., Swartz T.E., Bogomolni R.A., Kliger D.S. A LOV story: the signaling state of the phot1 LOV2 photocycle involves chromophore-triggered protein structure relaxation, as probed by far-UV time-resolved optical rotatory dispersion spectroscopy. Biochemistry. 2007;46:4619–4624. doi: 10.1021/bi602544n. [DOI] [PubMed] [Google Scholar]
- 20.Nakasone Y., Eitoku T., Matsuoka D., Tokutomi S., Terazima M. Dynamics of conformational changes of Arabidopsis phototropin 1 LOV2 with the linker domain. J. Mol. Biol. 2007;367:432–442. doi: 10.1016/j.jmb.2006.12.074. [DOI] [PubMed] [Google Scholar]
- 21.Iwata T., Nozaki D., Tokutomi S., Kagawa T., Wada M. Light-induced structural changes in the LOV2 domain of Adiantum phytochrome3 studied by low-temperature FTIR and UV-visible spectroscopy. Biochemistry. 2003;42:8183–8191. doi: 10.1021/bi0345135. [DOI] [PubMed] [Google Scholar]
- 22.Barth A., Zscherp C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002;35:369–430. doi: 10.1017/s0033583502003815. [DOI] [PubMed] [Google Scholar]
- 23.Krimm S., Bandekar J. Vibrational spectroscopy and conformations of peptides, polypeptides, and proteins. Adv. Protein Chem. 1986;38:181–364. doi: 10.1016/s0065-3233(08)60528-8. [DOI] [PubMed] [Google Scholar]
- 24.Chizhov I., Chernavskii D.S., Engelhard M., Mueller K.H., Zubov B.V. Spectrally silent transitions in the bacteriorhodopsin photocycle. Biophys. J. 1996;71:2329–2345. doi: 10.1016/S0006-3495(96)79475-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hendler R.W., Shrager R.I. Deconvolutions based on singular value decomposition and the pseudoinverse: a guide for beginners. J. Biochem. Biophys. Methods. 1994;28:1–33. doi: 10.1016/0165-022x(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 26.Kasahara M., Swartz T.E., Olney M.A., Onodera A., Mochizuki N. Photochemical properties of the flavin mononucleotide-binding domains of the phototropins from Arabidopsis, rice, and Chlamydomonas reinhardtii. Plant Physiol. 2002;129:762–773. doi: 10.1104/pp.002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Losi A., Polverini E., Quest B., Gärtner W. First evidence for phototropin-related blue-light receptors in prokaryotes. Biophys. J. 2002;82:2627–2634. doi: 10.1016/S0006-3495(02)75604-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uhmann W., Becker A., Taran C., Siebert F. Time-resolved FTIR absorption spectroscopy using a step-scan interferometer. Appl. Spectrosc. 1991;45:390–397. [Google Scholar]
- 29.Christie J.M., Corchnoy S.B., Swartz T.E., Hokenson M., Han I.S. Steric interactions stabilize the signaling state of the LOV2 domain of phototropin 1. Biochemistry. 2007;46:9310–9319. doi: 10.1021/bi700852w. [DOI] [PubMed] [Google Scholar]
- 30.Ataka K., Hegemann P., Heberle J. Vibrational spectroscopy of an algal Phot-LOV1 domain probes the molecular changes associated with blue-light reception. Biophys. J. 2003;84:466–474. doi: 10.1016/S0006-3495(03)74866-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bednarz T., Losi A., Gärtner W., Hegemann P., Heberle J. Functional variations among LOV domains as revealed by FTIR difference spectroscopy. Photochem. Photobiol. Sci. 2004;3:575–579. doi: 10.1039/b400976b. [DOI] [PubMed] [Google Scholar]
- 32.Swartz T.E., Wenzel P.J., Corchnoy S.B., Briggs W.R., Bogomolni R.A. Vibration spectroscopy reveals light-induced chromophore and protein structural changes in the LOV2 domain of the plant blue-light receptor phototropin 1. Biochemistry. 2002;41:7183–7189. doi: 10.1021/bi025861u. [DOI] [PubMed] [Google Scholar]
- 33.Alexandre M.T., Arents J.C., van Grondelle R., Hellingwerf K.J., Kennis J.T. A base-catalyzed mechanism for dark state recovery in the Avena sativa phototropin-1 LOV2 domain. Biochemistry. 2007;46:3129–3137. doi: 10.1021/bi062074e. [DOI] [PubMed] [Google Scholar]
- 34.Sato Y., Iwata T., Tokutomi S., Kandori H. Reactive cysteine is protonated in the triplet excited state of the LOV2 domain in Adiantum phytochrome3. J. Am. Chem. Soc. 2005;127:1088–1089. doi: 10.1021/ja0436897. [DOI] [PubMed] [Google Scholar]
- 35.Martin C.B., Tsao M.L., Hadad C.M., Platz M.S. The reaction of triplet flavin with indole. A study of the cascade of reactive intermediates using density functional theory and time resolved infrared spectroscopy. J. Am. Chem. Soc. 2002;124:7226–7234. doi: 10.1021/ja0123711. [DOI] [PubMed] [Google Scholar]
- 36.Richard J.P., Williams G., Gao J.L. Experimental and computational determination of the effect of the cyano group on carbon acidity in water. J. Am. Chem. Soc. 1999;121:715–726. [Google Scholar]
- 37.Kottke T., Heberle J., Hehn D., Dick B., Hegemann P. Phot-LOV1: photocycle of a blue-light receptor domain from the green alga Chlamydomonas reinhardtii. Biophys. J. 2003;84:1192–1201. doi: 10.1016/S0006-3495(03)74933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iwata T., Nozaki D., Sato Y., Sato K., Nishina Y. Identification of the C=O stretching vibrations of FMN and peptide backbone by (13)C-labeling of the LOV2 domain of Adiantum phytochrome3. Biochemistry. 2006;45:15384–15391. doi: 10.1021/bi061837v. [DOI] [PubMed] [Google Scholar]
- 39.Majerus T., Kottke T., Laan W., Hellingwerf K., Heberle J. Time-resolved FTIR spectroscopy traces signal relay within the blue-light receptor AppA. Chem. Phys. Chem. 2007;8:1787–1789. doi: 10.1002/cphc.200700248. [DOI] [PubMed] [Google Scholar]
- 40.Crosson S., Rajagopal S., Moffat K. The LOV domain family: photoresponsive signaling modules coupled to diverse output domains. Biochemistry. 2003;42:2–10. doi: 10.1021/bi026978l. [DOI] [PubMed] [Google Scholar]
- 41.Kay C.W., Schleicher E., Kuppig A., Hofner H., Rüdiger W. Blue light perception in plants. Detection and characterization of a light-induced neutral flavin radical in a C450A mutant of phototropin. J. Biol. Chem. 2003;278:10973–10982. doi: 10.1074/jbc.M205509200. [DOI] [PubMed] [Google Scholar]
- 42.Hienerwadel R., Boussac A., Breton J., Diner B.A., Berthomieu C. Fourier transform infrared difference spectroscopy of photosystem II tyrosine D using site-directed mutagenesis and specific isotope labeling. Biochemistry. 1997;36:14712–14723. doi: 10.1021/bi971521a. [DOI] [PubMed] [Google Scholar]
- 43.Imamoto Y., Kataoka M., Tokunaga F. Photoreaction cycle of photoactive yellow protein from Ectothiorhodospira halophila studied by low-temperature spectroscopy. Biochemistry. 1996;35:14047–14053. doi: 10.1021/bi961342d. [DOI] [PubMed] [Google Scholar]
- 44.Eitoku T., Nakasone Y., Zikihara K., Matsuoka D., Tokutomi S. Photochemical intermediates of Arabidopsis phototropin 2 LOV domains associated with conformational changes. J. Mol. Biol. 2007;371:1290–1303. doi: 10.1016/j.jmb.2007.06.035. [DOI] [PubMed] [Google Scholar]