

Abstract
The adsorption and insertion kinetics for the association of two 34-residue cyclic peptides with phosphocholine membranes have been studied using circular and linear dichroism approaches. The two peptides studied are identical with the exception of two residues, which are both tyrosine in one of the peptides and tryptophan in the other. Both peptides adopt random coil conformations in solution in the absence of membranes and do not aggregate at concentrations below 20 μM. After addition to liposome dispersions, circular dichroism spectroscopy indicated that both peptides undergo an extremely rapid transformation to a β-conformation that remains unchanged throughout the remainder of the experiment. Linear dichroism (LD) spectroscopy was used to study the kinetics of membrane adsorption and insertion. The data were analyzed by nonlinear least squares approaches, leading to identification of a number of bound states and their corresponding LD spectra. Two pseudo-first order processes could be identified that were common to both peptides. The first occurred with a time constant of the order of 1 min and led to a bound state characterized by weak LD signals, with significant bands corresponding to the transitions of aromatic side chains. The second process occurred with an unusually long time constant of between 75 and 100 min, forming a state with considerably stronger positive LD absorbance in the far-ultraviolet region of the spectrum. For the tyrosine-substituted peptide, a third slow process with a long time constant (76 min) could also be delineated and was attributed to rearrangements of the peptide within the membrane.
Introduction
There has been much speculation in recent years concerning the roles of aromatic amino acids in the interactions of proteins and peptides with lipid bilayers (1–4). Recent studies have suggested that tryptophan in particular, has significant interactions with lipids, using the hydrogen bonding donor potential of the indole ring combined with a possible role for cation-π interactions of the large aromatic ring system (5,6). Tyrosine has also been found to have significant interactions with phospholipid headgroups (5,1,2).
From data produced by studies on model transmembrane peptides (7,8) it is clear that tryptophan plays a key role in membrane anchoring, with NMR evidence suggesting that the indole group of the tryptophan side chain interacts with lipid acyl carbonyl groups (9); recent experiments with ether lipids have shown similar peptide behaviors to those observed with acyl lipids (10). These experiments, however, have been generally restricted to transmembrane species that are helical in nature. Systematic studies on model transmembrane β-peptides have been conducted less frequently, despite the relative abundance of stable membrane proteins and antimicrobial peptides rich in β-structure. Experiments on short tryptophan- and leucine-rich β-sheet peptides have highlighted the particular importance of hydrogen bond formation in the low dielectric membrane environment (11,12), and shown that significant changes in behavior can occur in response to single amino acid changes, particularly concerning substitutions for tryptophan. Similar modifications to the membrane activities of β-sheet peptides after single amino acid substitutions for tryptophan have been shown elsewhere (13), raising questions with regard to the mechanisms that control the membrane insertion and stability of peptides.
To obtain mechanistic information, it is desirable to have access to both kinetic and structural information during the insertion process itself. Circular dichroism (CD) spectroscopy has proved to be a useful tool for monitoring the conformations of peptides in membrane-associated and free forms, but does not show information concerning the location of the peptide within the membrane. Oriented CD (14–18) and linear dichroism (LD) approaches have been used to address this concern. LD shows the orientation of electronic transitions with respect to the experimental frame of reference and can therefore furnish information on the orientation of peptides with respect to membranes, provided that the latter can be aligned. The development of a low volume Couette cell (19,20) has allowed facile measurement of LD spectra from membrane-bound species in shear-aligned liposomes (21,22). This has been used to study the alignments of cell-penetrating peptides and was able to correlate reduced membrane penetrating ability with α-helix alignment in the plane of the membrane (23). Combined CD and LD studies on gramicidin have enabled probing of the tryptophan side chains in the β6.3 conformation of the peptide, with the axis of the peptide found to align along the bilayer normal and the tryptophan side chains to adopt orientations away from the plane of the membrane (21). This approach therefore has the potential to allow both peptide structure and membrane alignment to be probed as a function of time, enabling insertion kinetics to be determined with greater certainty.
In previous work (24), we described a cyclic peptide system that was able to porate liposomal membranes with high efficacy. In this study, we describe spectroscopic experiments concerning this and a related peptide (Fig. 1) that show that these peptides undergo a range of kinetic processes after binding to membranes, some of which exhibit much slower kinetics than anticipated. The data further highlight the differential effects of tyrosine and tryptophan in relation to membrane anchoring and shows the potential for deconvoluting LD data to reveal details of intermediate states in the insertion process.
Figure 1.

Structures of the cyclic peptides described in this study.
Materials and Methods
Materials
All amino acids, benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBOP) and the solid support (Novasyn TGR) were purchased from Novabiochem (Nottingham, UK). 1,1,1,3,3,3-Hexafluoroisopropanol (HFIP) and 2,2,2-trifluoroethanol (TFE) were purchased from Fluorochem (Glossop, UK) and distilled before use. All other solvents and reagents, including N,N-dimethylformamide (DMF), 1-hydroxybenzotriazole (HOBt), N,N-diisopropylethylamine (DIEA), N-methylmorpholine (NMM), N,N-diethyldithiocarbamate, sodium salt (DEDC), trifluoroacetic acid (TFA), 1,2-ethanedithiol (EDT), triisopropylsilane (TIS) and 2-(4-hydroxyphenylazo)benzoic acid (HABA) were purchased from Aldrich (Gillingham, UK), Fisher Scientific (Loughborough, UK), or Lancaster Synthesis (Heysham, UK) and used without further purification.
For liposome preparation, egg phosphatidylcholine (EPC; 100 mg/mL in chloroform) was purchased from Sigma (Gillingham, UK) and 100 nm laser-etched polycarbonate membranes from Whatman (Maidstone, UK).
Peptide chain assembly
Peptides 1 and 2 were synthesized by batch solid-phase peptide synthesis under a nitrogen atmosphere at room temperature on an Advanced Chemtech 348Ω synthesizer using a Novasyn TGR solid support (0.29 mmol/g; 0.10 g, 0.03 mmol) and Fmoc protection strategies. Coupling of amino acids was carried out for 1 h in DMF (2.5 mL) using PyBOP (5 eq) in the presence of HOBt (5 eq) and DIEA (10 eq). Deprotection between cycles was conducted with 20% v/v piperidine in DMF (2 × 2 mL). The solid support was washed with DMF after each coupling and Fmoc-deprotection step (2 × 2.5 mL and 4 × 2.5 mL respectively). In the first cycle, N-α-Fmoc-l-aspartic acid α-allyl ester (Fmoc-Asp-OAll) was double-coupled to the solid support. Valine, leucine, tryptophan, and tyrosine residues were added as pseudoproline dipeptides (Fmoc-Val-Ser(ψMe,MePro)-OH, Fmoc-Leu-Ser(ψMe,MePro)-OH, Fmoc-Trp(Boc)-Ser(ψMe,MePro)-OH, Fmoc-Tyr(tBu)-Ser(ψMe,MePro)-OH) (25). After peptide assembly, a small portion (∼5 mg) of the resin was removed for analysis.
Peptide cyclization
The resin was washed with CH2Cl2 (3 × 2 mL) and then treated with CH2Cl2 (2 mL) and a freshly prepared solution of tetrakis(triphenylphosphine)palladium(0) in CHCl3/AcOH/NMM (37:2:1; 30 mM, 1.5 mL) (26). The mixture was agitated for 2 h at room temperature before being washed successively with DIEA (0.5 M in DMF, 3.3 mL), DEDC (0.3 M in DMF, 2.5 mL), and DMF (2 × 3 mL). The resin was then treated with a solution of DIEA (0.026 g, 0.15 mmol), HOBt (0.023 g, 0.15 mmol), and PyBOP (0.078 g, 0.15 mmol) in DMF (3 mL) for 5 h at room temperature before being washed successively with DMF (4 × 1.5 mL), MeOH (5 × 2 mL), and CH2Cl2 (5 × 2 mL) and dried under a stream of nitrogen for 30 min. This yielded 0.19 g and 0.18 g of peptide-bound resin for peptides 1 and 2 respectively.
Cleavage of peptides from the solid support
Cleavage of the peptide was conducted when necessary by treating portions of the resin (50 mg) with a mixture of TFA/water/EDT/TIS (9.5:0.25:0.25:0.1; 1 mL) for 4 h at room temperature. The mixture was then filtered and the resin washed with TFA (3 × 0.5 mL). The combined filtrants were treated with dry diethyl ether (20 mL) and cooled to 0°C. The precipitate was pelleted by centrifugation at 14,000 × g for 10 min and triturated with diethyl ether (20 mL) before repeating the centrifugation/trituration process a further two times and drying the pellet in vacuo to give the crude product.
Peptide purification and analysis
The crude product was dissolved in TFE (1.5 mL) for purification. Three eluants were used for HPLC purification: 80% HFIP/TFE (10% v/v; eluant A), TFA/MeCN (0.1% v/v; eluant B), and 20% TFA/water (0.1% v/v; eluant C). HFIP and TFE were obtained from Fluorochem and distilled before use. Other solvents were of HPLC grade from Fisher Scientific. Chromatography was carried out on a Supelcosil LC-8 (C8) column (Supelco; 25 cm × 1 cm) using isocratic elution between 0 and 6 min with an eluant composition of 80% A and 20% C, followed by a linear gradient between 6–9 min of 80%–50% A, 0%–50% B, and 20%–0% C. The flow rate was 2 mL/min throughout. Retention times were 7.23 min (1) and 6.56 min (2). Fractions corresponding to the top of peaks in the elution profile were collected manually, pooled and concentrated in vacuo before being rechromatographed. Fractions from the base of peaks were pooled separately and subjected to a further two rounds of chromatography, producing final yields of 0.6 μmol (19%) for 1 and 0.2 μmol (12%) for 2, (based on the maximum theoretical yield for 50 mg of resin). Peptides were analyzed by MALDI mass spectrometry on a Voyager-DE STR apparatus using a stainless steel target. Peptides and matrix (HABA) were codissolved in HFIP at a molar ratio of 1:1400 to 1:3000 and 1 μL of the solution spotted on to the MALDI target. Calculated for linear allyl ester of 1: C161H236N38O59Na requires m/z 3670.81; found 3670.54, [M + Na]+. Calculated for linear allyl ester of 2: C165H238N40O57Na requires m/z 3716.88; found 3716.35, [M + Na]+. Calculated for 1: C158H228N38O58Na3 requires m/z 3656.69; found 3656.73, [M + 3Na − 2H]+. Calculated for 2: C162H230N40O56Na3 requires m/z 3702.76; found 3702.77, [M + 3Na − 2H]+.
Stock solutions of the peptides were prepared in TFE and their concentrations determined through absorbance measurements at 280 nm after dilution of small aliquots into water (extinction coefficients were calculated as ɛ = 8940 mol−1 cm−1 and ɛ = 16,960 mol−1 cm−1 at 280 nm for peptides 1 and 2 respectively) (27).
Liposome preparation
A solution of egg phosphatidylcholine (100 μL, 10 mg) was concentrated in vacuo in a 25 mL round bottomed flask to form a thin film that was dried in vacuo overnight. The lipid film was hydrated with 1 mL of aqueous medium containing 150 mM NaCl buffered with 10 mM tris at pH 7.4 and mixed thoroughly before being subjected to five freeze thaw cycles and extruded 10 times through 100 nm laser-etched polycarbonate membranes at 30°C using a thermobarrel extruder (Northern Lipids, Burnaby, Canada).
Circular and LD spectroscopy
Experiments were carried out on Jasco J-810 (CD) or Jasco J-715 (LD) spectropolarimeters and processed using the Spectra Analysis program (version 1.50, Jasco, Great Dunmow, UK). All experiments were carried out in 10 mM tris buffer at pH 7.4 containing 150 mM NaCl using a 1 cm quartz cell (CD) or a purpose made 50 μL quartz Couette cell (LD) (19) at 25°C. A portion of the stock solution of the peptide in TFE was added to the aqueous medium containing liposomes at concentrations in the range 1.5–1.82 mg/mL (∼2 mM) and mixed by gentle shaking, producing peptide concentrations in the range 0.34–10.1 μM. Volumes were selected such that the proportion of TFE was never >10% v/v. Time series spectra were acquired between 350–190 nm at a scan rate of 10 nm/min, with a response time of 16 s and a bandwidth of 2 nm. Blank spectra were recorded after the addition of identical quantities of pure TFE to the aqueous medium and subtracted from the raw time series spectra. LD spectra were further processed using 15-point binomial smoothing, before analysis. Data from time series acquisitions were fitted to two- or three-state models (Fig. 2) using Eq. 1 ([P]3 and LD3 = 0 for the 2-state model).
| (1) |
where [P]1, [P]2, and [P]3 are the concentrations of peptide in states 1, 2, and 3 respectively, and LD1, LD2, and LD3 are the LD signals corresponding to these states. [P]T is the total concentration of peptide. LDcalc is the calculated LD signal.
Figure 2.

The kinetics model used for analysis of LD data.
Concentrations of the peptides in each state were calculated according to Eqs. 2–4:
| (2) |
| (3) |
| (4) |
where k1, k2, and k3 are the rate constants for the formation of states 1–3 and t is time. With k3 = 0, Eqs. 3 and 4 revert to those for a 2-state model.
For the purposes of these calculations, time t at which the instrument was at a given wavelength (λt) on each independently collected scan could be related to the scan rate using Eq. 5:
| (5) |
where λs is the starting wavelength of each scan, λe is the end wavelength of the scan, λt is the wavelength of the monochromator at time t, s is the scan rate in nm/min, and n is an integer corresponding to the number of completed scans (n = 0, 1, 2…). Global least squares fitting to shared rate constants (k1, k2, and k3) was carried out using the data from four wavelengths (193 nm, 228 nm, 245 nm, and 254 nm) to calculate LD evolution profiles (LDcalc). Nonlinear least squares curve fitting procedures were carried out using the Solver function in the program Excel (Microsoft, Redmond, WA). The rate constants determined from the above process were then used to evaluate the LD parameters at all wavelengths by nonlinear least squares fitting, to generate the full LD spectrum for each of the bound states.
Results
Peptide synthesis
Peptides 1 and 2 were synthesized in moderate yield using an approach based on coupling the majority of the amino acids as pseudoproline dipeptides. This approach was expected to enable assembly of the peptide chains while minimizing the formation of nonproductive conformations and favor the final cyclization process. To further facilitate cyclization, the syntheses were carried out on a solid support with a polyethylene glycol spacer to ensure that the reaction occurred in more solution-like conditions. The use of an allyl group as orthogonal protection for the first amino acid proved successful, although it was necessary to prepare fresh Pd(PPh3)4 in-house (see Supporting Material) for the deprotection to proceed to completion. Analysis of small portions of cleaved resin by mass spectrometry before cyclization indicated that the chain had been assembled successfully. Minor peaks at lower mass than the target peptide were consistent with small amounts of peptide for which coupling of Fmoc-Val-Ser(ψMe,MePro)-OH had not gone to completion, but at levels that were not detrimental to isolation of the target peptides. Cyclization and cleavage proceeded without difficulties, to yield the crude peptides. As anticipated, these were not trivial to manipulate due to the lack of charged residues and high amphiphilicity. Nevertheless, successful purification to ≥90% was reproducibly achieved through the use of fluorinated alcohols as the principal solvents. Once solubilized, peptide solutions could be diluted in to aqueous media at solvent compositions ≤10% alcohol by volume and remained stable over several hours.
Liposome binding experiments
Both peptides exhibited CD spectra in solution in the absence of membranes that were consistent with random coil conformations (see Supporting Material). Furthermore, at the concentrations used for these experiments there was no evidence of aggregation. After addition to liposomes, CD spectra were produced that were consistent with rapid association to the liposomal membranes (Fig. 3, A and B). Single minima at 213 nm and 216 nm were observed for 1 and 2 respectively, similar to those observed for β-barrel membrane proteins (28) and consistent with the adoption of β-structure. Due to the high salt conditions and path lengths used for CD experiments, data could not be recorded at wavelengths <210 nm. The profiles of the CD spectra exhibited by both peptides did not change significantly during the course of the experiments. LD measurements over the same time period (Fig. 3, C and D) yielded spectra indicative of changes in membrane alignment occurring on a timescale of minutes to hours. During these measurements, the LD signal remained positive at all wavelengths, with notable shoulders observed in the regions of 190–195 nm and 245–250 nm for both peptides and additional shoulders at 227 nm and 291 nm for peptide 2.
Figure 3.
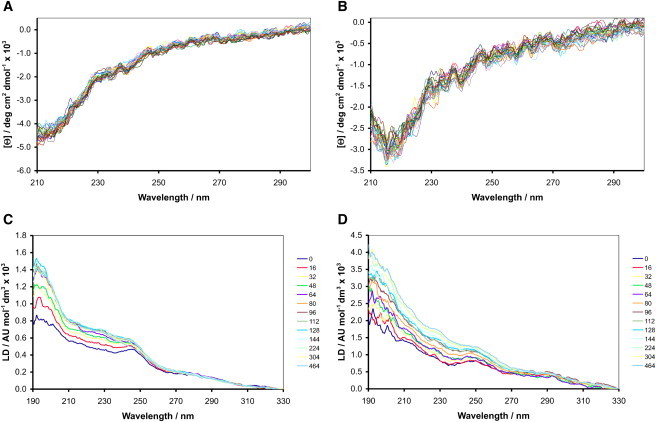
Time series spectra acquired at 20°C over an 8-h period after the addition of peptides to 100 nm EPC liposomes in 10 mM tris/150 mM NaCl at pH 7.4. CD spectra for 1 (A) and 2 (B) at respective peptide concentrations of 10.1 μM and 3.23 μM and an EPC concentration of 1.82 mg/mL (2 mM); LD spectra for 1 (C) and 2 (D) at respective peptide concentrations of 1.05 μM and 0.34 μM and an EPC concentration of 1.5 mg/mL (2 mM). The legend associated with each LD spectrum indicates the time (min) after which acquisition of the corresponding spectrum commenced.
Least squares fitting of the experimental data
LD data were analyzed using kinetic models that assumed pseudo-first order kinetics for consecutive processes. This was reasonable, given the combination of the excess lipid used in the experiments and the likelihood that the early phases in the binding of peptides to membranes do not involve aggregates. Examination of semi-log plots for the evolution of the LD signal (Fig. 4, A and B) support these models, particularly for peptide 2, where it is evident that the semi-log plot consists of two processes with strikingly linear rate profiles. For peptide 1, the data could be modeled adequately using a triple exponential model (Eqs. 1–5). Applying a double exponential model for peptide 1 yielded inferior fits to the experimental data. Data from four wavelengths corresponding to regions with the major shoulders in the LD spectra were used for the analysis of both peptides. Global least-squares fitting to shared rate constant values yielded good fits to the experimental data for both peptides (Table 1).
Figure 4.

The evolution of LD signals after the addition of peptides 1 and 2 at respective concentrations of 1.05 μM and 0.34 μM to EPC liposomes (1.5 mg/mL; 2 mM) in 10 mM tris/150 mM NaCl at pH 7.4: semi-log plots for peptide 1 at 193 nm (A) and 2 at 228 nm (B); experimental and calculated profiles for peptides 1 (C) and 2 (D) at 193 nm (■), 228 nm (▴), 245 nm (□) and 254 nm (○). Experimental data are presented as points and the calculated profiles from nonlinear regression as lines.
Table 1.
Parameters obtained from nonlinear least squares fitting of the model described by Eqs. 1-5 to the experimental data for the binding of peptides 1 and 2 at respective concentrations of 1.05 μM and 0.34 μM to EPC liposomes (1.5 mg/mL; 2 mM) in 10 mM tris/150 mM NaCl at pH 7.4
| Parameter | Peptide | |
|---|---|---|
| 1∗ | 2∗ | |
| k1/min−1 | 1.01 ± 0.20 | 1.62 ± 0.47 |
| k2/min−1 | 0.013 ± 0.001 | 0.010 ± 0.001 |
| k3/min−1 | 0.013 ± 0.001 | — |
| τ1/min | 0.99 ± 0.19 | 0.62 ± 0.18 |
| τ2/min | 76.3 ± 5.9 | 98.6 ± 9.7 |
| τ3/min | 76.4 ± 5.9 | — |
| LD/AU mol−1 dm3 | ||
| 193 nm | ||
| LD1 | 570 ± 13 | 1649 ± 9 |
| LD2 | 2130 ± 79 | 3874 ± 4 |
| LD3 | 1340 ± 20 | — |
| 228 nm | ||
| LD1 | 377 ± 2 | 706 ± 7 |
| LD2 | 909 ± 37 | 1286 ± 5 |
| LD3 | 659 ± 10 | — |
| 245 nm | ||
| LD1 | 423 ± 9 | 662 ± 2 |
| LD2 | 739 ± 12 | 1715 ± 1 |
| LD3 | 505 ± 4 | — |
| 254 nm | ||
| LD1 | 300 ± 3 | 631 ± 4 |
| LD2 | 482 ± 7 | 1142 ± 2 |
| LD3 | 352 + 4 | — |
Errors are reported to 95% confidence limits and were estimated by repeating the nonlinear regression ten times from random estimates of the variables.
The binding of peptide 1 (Fig. 4 C) was characterized by three processes, with k1 > k2 ≈ k3. The calculated LD values for each of the bound states are indicative of an initially weakly-oriented state (P1, formed with rate constant k1) with low LD. The peptide then converts to a considerably more oriented state (P2, formed with rate constant k2) before relaxing to a state with intermediate LD values (LD3, formed with rate constant k3). For peptide 2 (Fig. 4 D), two processes were characterized, with k1 > k2. As with peptide 1, a state with low orientation forms with rapid kinetics before converting to a state that is considerably more ordered.
Calculation of the LD parameters at all wavelengths using the kinetic parameters in Table 1 enabled the LD spectra corresponding to each of the bound states to be determined (Fig. 5). In the case of peptide 1 (Fig. 5 A), the initial bound state (P1) is characterized by an LD spectrum (LD1) that is relatively weak across the whole spectrum, but has a pronounced maximum at 246 nm and a shoulder at ∼275 nm. This gives way to spectra (LD2 and LD3, Fig. 5 A) exhibiting significantly higher LD values in the far-ultraviolet (UV) range, with a significant maximum at 193 nm. Interestingly, changes in the near-UV spectrum are considerably smaller than those of the far-UV for both of these bound states. The spectrum of the initial bound state of peptide 2 (LD1, Fig. 5 B) shares similar features with that of peptide 1, with a maximum at 249 nm and a shoulder at 292 nm. This also gives way to a spectrum (LD2, Fig. 5 B) with higher LD values in the far-UV range and smaller changes in the near-UV, but with a shoulder at 228 nm in addition to the maximum at 192 nm.
Figure 5.

LD spectra for each binding state described by Eq. 1, calculated by regression analysis of the binding profiles for the association of peptides 1 (A) and 2 (B) with EPC liposomes in 10 mM tris/150 mM NaCl at pH 7.4. The identity of the binding state is indicated by the text associated with each spectrum.
One feature enabled by the nonlinear regression using this model is the ability to calculate the concentration of all intermediates throughout the experiment. This gives rise to the concentration profiles in Fig. 6. For both peptides, the initially formed state (P1) forms rapidly and then decays exponentially to zero during the period of the experiments. In the case of peptide 2, the final state (P2) forms completely during the same period. With peptide 1, this state exists as a transient intermediate, decaying to zero and being replaced by the final state (P3).
Figure 6.

Concentrations of each of the liposome-bound states after addition of peptides 1 (A) and 2 (B) at respective concentrations of 1.05 μM and 0.34 μM to EPC liposomes (1.5 mg/mL; 2 mM) in 10 mM tris/150 mM NaCl at pH 7.4, calculated by fitting of Eq. 1 to the experimental data by nonlinear regression. (Dashed line) [P]1; (solid line) [P]2; (dotted line) [P]3.
Discussion
Peptides 1 and 2 were designed following bioinformatics exercises on β-barrel membrane proteins on the basis that, due to their hydrophobicity and amphiphilicity, membrane association would be favored (24). Their cyclic nature was intended to promote the formation of secondary structure after membrane binding and render analysis of insertion processes more straightforward, as the issue of which end of the peptide inserts into the membrane first would not be a consideration.
Both peptides adopt random coil conformations in aqueous solution at concentrations <20 μM in the absence of membranes. However, after addition to membranes, secondary structure forms rapidly and once formed, remains unchanged for the duration of the experiment. As a consequence, LD data can be interpreted with the certainty that they reflect only changes in alignment with respect to the experimental frame of reference and do not contain contributions from secondary structure changes. It further enables LD profiles at all wavelengths to be simultaneously fitted to the same kinetics parameters, improving the reliability of data analysis. The purpose of this study was to explore the possibilities for using LD to probe the membrane-association kinetics of two peptides designed to undergo insertion, by assaying relative changes in alignment. As such, it was not necessary to use an internal reference to define the experimental frame of reference, as has been described elsewhere (29). For this work, the semiquantitative assessment made here enables us to extract the mechanistic information required.
Nonlinear fitting of the model described by Eqs. 1–5 produced good fits to the experimental data for both peptides. Two processes are common to both peptides. The first (k1) is a rapid process that leads to a bound state (P1) characterized by a weak positive LD spectrum with distinct maxima and shoulders in the near-UV that correspond closest to the La and Lb transitions of tyrosine and tryptophan (20). The Lb transition of tyrosine is oriented normal to the Cβ-C1-C4-O axis and is usually found in the region of 260 nm, although transitions in the region of 250 nm have been reported for β-rich proteins with a high aromatic amino acid content, such as retinol binding protein (30). Tryptophan has an La transition occurring at ∼265 nm that lies on an axis that bisects the indole N1-C4 atoms and an Lb transition at 287 nm oriented orthogonally to the La transition. The deconvoluted LD spectrum for P1 is consistent with a state that is weakly bound to the membrane through the side chains of aromatic residues, leading to alignment of these side chains, with the peptide backbone either poorly aligned or aligned close to 54.7° with respect to the membrane normal (that would also give a weak signal) (22). As the wavelengths corresponding to these bands are red-shifted by ∼5 nm in peptide 2 with respect to the corresponding bands of peptide 1, indicating that they arise from the tryptophan residues of the former, it would seem that the residues near the ends of the peptides as depicted in Fig. 1 are those that are primarily participating in interactions, although it does not rule out contributions from the others. Positive LD bands for both La and Lb of tryptophan have been observed in other systems (23) and are indicative of the indole side chain being oriented perpendicular to the membrane normal. In these studies, the same rationale can be applied to the phenolic side chain of tyrosine. The observation of a spectrum that is positive across the wavelength range suggests that the alignment of the peptide as a whole is nearer to the membrane normal than the plane of the membrane, as the main amide transitions in β-hairpins that contribute to the far-UV region have a cross strand orientation. Taken together, these observations are consistent with an anchoring interaction of a small number of residues with the membrane surface. It is noteworthy that the signals corresponding to the Lb transitions of tyrosine and tryptophan do not change significantly throughout the duration of the experiment, which suggests that some of the side chain interactions are relatively strong and implies that changes in alignment of the peptide backbone are accommodated by changes in the side chain torsional angles.
The second process (k2) is characterized by surprisingly slow kinetics, which was not anticipated at the outset of this work. The spectrum of the bound state (P2) has significantly more positive LD values across the spectrum, particularly for the 190 nm region (corresponding to the π-π∗ transition of the backbone), indicative of an alignment that is closer to the membrane normal than P1. Accounts of kinetic processes for the binding of peptides to membranes with half-lives of the order of ∼1 h are rare. White et al. (16) reported conditions under which a designed helical peptide underwent slow membrane insertion kinetics, on a timescale of hours, although this phenomenon was only observed at nonphysiological pH values (≤pH 5.3). It will be of interest to determine whether peptide rearrangements on these timescales are a general phenomenon or a property particular to these peptides. Earlier studies on marker release by peptide 1 indicated that apparent poration of EPC membranes occurs on a timescale of seconds to minutes (24), which is more consistent with the kinetics of k1 than k2, raising the possibility that membrane disruption occurs primarily during the initial binding process, rather than by specific poration. An explanation for the slow kinetics lies in the lack of charge complementaity between the peptides and the membrane, both of which are neutral in the physiological pH range. Given the low molar peptide/lipid ratios (typically 1:1000) and that the peptides do not promote significant changes in lipid morphology at higher ratios (24), the changes in LD can be assigned to changes in peptide orientation.
A third kinetic process is observed for peptide 1, but not peptide 2. This also occurs on a slow timescale, and leads to a diminution of positive LD values, consistent with the adoption of a conformation that lies further away from the membrane normal. One possibility is that this represents a realignment of peptide 1 to minimize the effects of mismatch (31,32) between the peptide and the membrane or promote peptide aggregation, whereas peptide 2 does not realign due to the known anchoring effects of tryptophan in the interfacial region of lipid membranes (9), which would favor lipid redistribution as a means of overcoming mismatch.
One study of the refolding of a β-barrel outer membrane protein reported three processes with kinetics on a similar timescale to those reported here (33). Of these, a moderately slow process with a timescale of minutes was attributed to the formation of a partially folded state within the membrane, with the slower process on a timescale of hours attributed to adoption of the folded state. These are broadly similar to the processes observed here, indicating that these peptides may prove to be good models for transmembrane β-barrels. A model for the behavior of these peptides is shown schematically in Fig. 7.
Figure 7.

Peptide binding conformations consistent with the calculated LD spectra for binding states of peptides 1 and 2. The membrane is represented by the hatched boxes and the peptide by open rectangles.
The peptides form secondary structure on a timescale that is more rapid than k1. The full description of binding therefore requires at least one additional rate constant to be included in the process depicted in Fig. 7. An advantage of the rapid adoption of secondary structure, however, is that changes in LD relate only to changes in peptide orientation, enabling us to interpret the LD data more definitively than if folding and insertion were coupled. The data illustrate that the LD methodology is a useful tool for probing membrane adorbtion kinetics that is of general applicability to systems where confomational changes during adsorbtion can be assayed.
Acknowledgments
This study was supported by grant RSRG 24281 from the Royal Society (J.M.S.) and the Engineering and Physical Sciences Research Council (GR/T09224/01).
Supporting Material
References
- 1.Ulmschneider M.B., Sansom M.S. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta. 2001;1512:1–14. doi: 10.1016/s0005-2736(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 2.Landolt-Marticorena C., Williams K.A., Deber C.M., Reithmeier R.A. Non-random distribution of amino acids in the transmembrane segments of human type I single span membrane proteins. J. Mol. Biol. 1993;229:602–608. doi: 10.1006/jmbi.1993.1066. [DOI] [PubMed] [Google Scholar]
- 3.Liu W., Caffrey M. Interactions of tryptophan, tryptophan peptides, and tryptophan alkyl esters at curved membrane interfaces. Biochemistry. 2006;45:11713–11726. doi: 10.1021/bi0608414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanderson J.M. Peptide-lipid interactions: insights and perspectives. Org. Biomol. Chem. 2005;3:201–212. doi: 10.1039/b415499a. [DOI] [PubMed] [Google Scholar]
- 5.Sanderson J.M., Whelan E.J. Characterization of the interactions of aromatic amino acids with diacetyl phosphatidylcholine. Phys. Chem. Chem. Phys. 2004;6:1012–1017. [Google Scholar]
- 6.Sanderson J.M. Refined models for the preferential interactions of tryptophan with phosphocholines. Org. Biomol. Chem. 2007;5:3276–3286. doi: 10.1039/b707502b. [DOI] [PubMed] [Google Scholar]
- 7.Killian J.A. Synthetic peptides as models for intrinsic membrane proteins. FEBS Lett. 2003;555:134–138. doi: 10.1016/s0014-5793(03)01154-2. [DOI] [PubMed] [Google Scholar]
- 8.Wimley W.C., White S.H. Designing transmembrane alpha-helices that insert spontaneously. Biochemistry. 2000;39:4432–4442. doi: 10.1021/bi992746j. [DOI] [PubMed] [Google Scholar]
- 9.de Planque M.R.R., Bonev B.B., Demmers J.A.A., Greathouse D.V., Koeppe R.E. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 2003;42:5341–5348. doi: 10.1021/bi027000r. [DOI] [PubMed] [Google Scholar]
- 10.van der Wel P.C.A., Reed N.D., Greathouse D.V., Koeppe R.E., II Orientation and motion of tryptophan interfacial anchors in membrane-spanning peptides. Biochemistry. 2007;46:7514–7524. doi: 10.1021/bi700082v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wimley W.C., Hristova K., Ladokhin A.S., Silvestro L., Axelsen P.H. Folding of beta-sheet membrane proteins: A hydrophobic hexapeptide model. J. Mol. Biol. 1998;277:1091–1110. doi: 10.1006/jmbi.1998.1640. [DOI] [PubMed] [Google Scholar]
- 12.Wimley W.C., White S.H. Reversible unfolding of beta-sheets in membranes: a calorimetric study. J. Mol. Biol. 2004;342:703–711. doi: 10.1016/j.jmb.2004.06.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Y., Mozsolits H., Hammer J., Zmuda E., Zhu F. Influence of tryptophan on lipid binding of linear amphipathic cationic antimicrobial peptides. Biochemistry. 2003;42:9395–9405. doi: 10.1021/bi034338s. [DOI] [PubMed] [Google Scholar]
- 14.Pan Y.-L., Cheng J.T.-J., Hale J., Pan J., Hancock R.E.W. Characterization of the structure and membrane interaction of the antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J. 2007;92:2854–2864. doi: 10.1529/biophysj.106.097238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clayton A.H.A., Sawyer W.H. Oriented circular dichroism of a class a amphipathic helix in aligned phospholipid multilayers. Biochim. Biophys. Acta. 2000;1467:124–130. doi: 10.1016/s0005-2736(00)00208-x. [DOI] [PubMed] [Google Scholar]
- 16.Ladokhin A.S., White S.H. Interfacial folding and membrane insertion of a designed helical peptide. Biochemistry. 2004;43:5782–5791. doi: 10.1021/bi0361259. [DOI] [PubMed] [Google Scholar]
- 17.Wu Y., Huang H.W., Olah G.A. Method of oriented circular dichroism. Biophys. J. 1990;57:797–806. doi: 10.1016/S0006-3495(90)82599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen F.-Y., Lee M.-T., Huang H.W. Evidence for membrane thinning effect as the mechanism for peptide-induced pore formation. Biophys. J. 2003;84:3751–3758. doi: 10.1016/S0006-3495(03)75103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marrington R., Dafforn T.R., Halsall D.J., MacDonald J.I., Hicks M. Validation of new microvolume Couette flow linear dichroism cells. Analyst. 2005;130:1608–1616. doi: 10.1039/b506149k. [DOI] [PubMed] [Google Scholar]
- 20.Dafforn T.R., Rajendra J., Halsall D.J., Serpell L.C., Rodger A. Protein fiber linear dichroism for structure determination and kinetics in a low-volume, low-wavelength Couette flow cell. Biophys. J. 2004;86:404–410. doi: 10.1016/S0006-3495(04)74116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodger A., Rajendra J., Marrington R., Ardhammar M., Nordén B. Flow oriented linear dichroism to probe protein orientation in membrane environments. Phys. Chem. Chem. Phys. 2002;4:4051–4057. [Google Scholar]
- 22.Rodger A., Marrington R., Geeves M.A., Hicks M., de Alwis L. Looking at long molecules in solution: what happens when they are subjected to Couette flow? Phys. Chem. Chem. Phys. 2006;8:3161–3171. doi: 10.1039/b604810m. [DOI] [PubMed] [Google Scholar]
- 23.Caesar C.E.B., Esbjorner E.K., Lincoln P., Nordén B. Membrane interactions of cell-penetrating peptides probed by tryptophan fluorescence and dichroism techniques: correlations of structure to cellular uptake. Biochemistry. 2006;45:7682–7692. doi: 10.1021/bi052095t. [DOI] [PubMed] [Google Scholar]
- 24.Sanderson J.M., Yazdani S. The design, synthesis and characterization of channel-forming peptides. Chem. Commun. 2002:1154–1155. doi: 10.1039/b203046b. [DOI] [PubMed] [Google Scholar]
- 25.Wohr T., Wahl F., Nefzi A., Rohwedder B., Sato T. Pseudo-prolines as a solubilizing, structure-disrupting protection technique in peptide synthesis. J. Am. Chem. Soc. 1996;118:9218–9227. [Google Scholar]
- 26.Kates S.A., Sole N.A., Johnson C.R., Hudson D., Barany G. A novel, convenient, three-dimensional orthogonal strategy for solid-phase synthesis of cyclic peptides. Tetrahedron Lett. 1993;34:1549–1552. [Google Scholar]
- 27.Pace C.N., Vajdos F., Fee L., Grimsley G., Gray T. How to measure and predict the molar absorbtion coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamm L.K., Hong H., Liang B. Folding and assembly of beta-barrel membrane proteins. Biochim. Biophys. Acta. 2004;1666:250–263. doi: 10.1016/j.bbamem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Ardhammar M., Lincoln P., Nordén B. Ligand substituents of ruthenium dipyridophenazine complexes sensitively determine orientation in liposome membrane. J. Phys. Chem. B. 2001;105:11363–11368. [Google Scholar]
- 30.Greene L.H., Chrysina E.D., Irons L.I., Papageorgiou A.C., Acharya K.R. Role of conserved residues in structure and stability: tryptophans of human serum retinol-binding protein, a model for the lipocalin superfamily. Protein Sci. 2001;10:2301–2316. doi: 10.1110/ps.22901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harzer U., Bechinger B. Alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: a CD, 15N and 31P solid-state NMR spectroscopy investigation. Biochemistry. 2000;39:13106–13114. doi: 10.1021/bi000770n. [DOI] [PubMed] [Google Scholar]
- 32.de Planque M.R.R., Boots J.-W.P., Rijkers D.T.S., Liskamp R.M.J., Greathouse D.V. The effects of hydrophobic mismatch between phosphatidylcholine bilayers and transmembrane alpha-helical peptides depend on the nature of interfacially exposed aromatic and charged residues. Biochemistry. 2002;41:8396–8404. doi: 10.1021/bi0257686. [DOI] [PubMed] [Google Scholar]
- 33.Surrey T., Jähnig F. Kinetics of folding and membrane insertion of a beta-barrel membrane protein. J. Biol. Chem. 1995;270:28199–28203. doi: 10.1074/jbc.270.47.28199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.