

Abstract
Degranulation of bone marrow-derived mast cells (BMMCs) triggered by antigens (e.g., 2,4-dinitrophenylated bovine serum albumin (DNP-BSA) and secretagogues (e.g., poly-L-lysine) was investigated by combined atomic force microscopy (AFM) and laser scanning confocal microscopy (LSCM). This combination enables the simultaneous visualization and correlation of membrane morphology with cytoskeletal actin arrangement and intracellular granules. Two degranulation mechanisms and detailed membrane structures that directly corresponded to the two stimuli were revealed. In DNP-BSA triggered activation, characteristic membrane ridges formed in accordance with the rearrangement of underlying F-actin networks. Individual granules were visualized after they released their contents, indicating a “kiss-and-run” pathway. In BMMCs stimulated by poly-L-lysine, lamellopodia and filopodia were observed in association with the F-actin assemblies at and near the cell periphery, whereas craters were observed on the central membrane lacking F-actin. These craters represent a new membrane feature resulting from the “kiss-and-merge” granule fusion. This work provides what we believe is important new insight into the local membrane structures in correlation with the cytoskeleton arrangement and detailed degranulation processes.
Introduction
The morphology and local structures of cellular membranes are sensitive and characteristic indications of cellular viability and signaling processes (1–3). For instance, membrane ridges appear upon antibody-mediated activation of rat basophilic leukemia cells (RBL-2H3), whereas a high density of microvilli indicates resting cells (4). Lamellopodia and filopodia correspond to cellular spreading and locomotion (5). Membrane blebs occur during cell apoptosis (6). Throughout these processes, the change of membrane structure is caused by cross-linking of receptors by external antigens (7), the redistribution of actin filaments (8), or the decoupling of the cytoskeleton from the plasma membrane (9). In the case of mast cells, degranulation is also implicated in membrane morphological changes (10,11), although the detailed correlations between the membrane structures and the specific degranulation processes are still not clear. This is mainly due to the lack of high-resolution structural characterization tools that are capable of visualizing cellular membranes, cytoskeletons, and intracellular structures for individual cells in their living and hydrated states.
In this investigation we demonstrate that a combined laser scanning confocal microscopy (LSCM) and atomic force microscopy (AFM) technique shows great promise to fill the technical void discussed above. A primary cell type, bone marrow-derived mast cells (BMMCs), was used. The culturing and activation protocols for this cell type are well established (12,13), but structural characterizations have not been extensively studied, mainly because of their nonadherent nature (14), as well as the long time required to attain primary cells. Systematic investigations were conducted to compare the resting cells with cells activated by antigens or secretagogues. Two activation pathways were previously reported in the investigation of mast cells (15–18). The antibody-mediated pathway, also known as the FcɛRI-dependent pathway, involves the preliminary binding of immunoglobulin-E (IgE) to specific high-affinity IgE receptors (FcɛRI) and the subsequent cross-linking of bound IgE molecules by multivalent antigens. The cross-linking process leads to receptor aggregations, which further triggers the signaling cascade, resulting in degranulation. The characteristic cell morphology associated with this pathway is the presence of ridges and disappearance of villi (4,19). Stimulation with basic secretagogues, such as anaphylatoxins, neuropeptides, compound 48/80, and poly-L-lysine, results in a distinct FcɛRI-independent pathway for the activation of mast cells (20,21). These stimuli share a common characteristic of being polycations (22–24), which are able to internalize into or possibly penetrate halfway through the plasma membrane and stimulate the guanine nucleotide-binding proteins (G-proteins) (25,26), ultimately leading to degranulation. A previous structural study of the RBL-2H3 cells activated by this pathway indicates an intermediate surface morphology between the resting cells and antigen-activated cells (11). A systematic comparison of membrane structures in three dimensions and in high resolution for both activation pathways has not been performed, and less is known about the membrane-cytoskelton-granule correlation.
Taking advantage of the capabilities of LSCM in three-dimensional (3D) imaging, in conjunction with AFM in high-resolution imaging of the cell membrane, we focus on studying the impact of the rearrangement of F-actin and degranulation on membrane structures as a result of various external activation stimuli, i.e., antigens and secretagogues. The results from this investigation provide a high-resolution view of BMMCs' membrane morphology and local structures; establish correlations among membrane morphology, cytoskeletal F-actin, and intracellular granules; and shed what we believe is new light on degranulation mechanisms, i.e., the fate of granules and cytoskeleton-membrane interactions.
Materials and Methods
Preparation of BMMCs
The animal study was approved by the Institutional Animal Care and Use Committee of the University of California, Davis. Bone marrow cells were collected from the femurs of C57BL/6 mice and cultured following our previously established procedures (27) for 4–10 weeks at 37 °C and 5% CO2 in RPMI 1640 medium containing 2 mM L-glutamine, 10% fetal bovine serum, 100 U/mL penicillin, and 20% conditioned medium from Interleukin 3 (IL-3) producing cell line WEHI-3B. The purity of mast cells exceeded 95%, as determined by the expression of FcɛRI on the cell surface (13).
Activation and stimulation of BMMCs
BMMCs were activated with the use of established protocols via antibody-mediated FcɛRI clustering. The purpose was to examine whether combined AFM and LSCM is able to probe characteristic membrane and cytoskeletal structures associated with mast cell activation, and to obtain new insights into mast cell degranulation. Two forms of 2,4-dinitrophenylated bovine serum albumin (DNP-BSA, A23018; Invitrogen, Carlsbad, CA), surface-bond and solubilized, were utilized as antigens. Cells were first sensitized with anti-DNP IgE (0.5 μg/mL, prepared following our previously established procedures (28)) overnight, and then washed by phosphate-buffered saline (PBS, D8662; Sigma-Aldrich, St. Louis, MO) to remove excess IgE. Afterward, activation was triggered by adding DNP-BSA to the culture medium or centrifuging (450 × g) the cells onto DNP-BSA-coated coverslips, made by incubating the coverslips in PBS containing 20 μg/mL DNP-BSA for 30 min. Based on the antibody sensitization, antigen stimulation, and surface coating used in this protocol, we use BMMCsIgE/DNP(s)/− and BMMCsIgE/DNP(c)/PL to represent this category of cells, as summarized in Table 1.
Table 1.
Summary of activation conditions for BMMCs and the corresponding abbreviations
| Abbreviation | Description |
|---|---|
| BMMCs−/−/− | Control, BMMCs were spun onto clean/bare glass coverslips. |
| BMMCsIgE/DNP(s)/− | BMMCs were sensitized with anti-DNP IgE overnight and then stimulated by DNP-BSA immobilized on coverslips. |
| BMMCs−/−/PL | BMMCs were spun onto poly-L-lysine-coated coverslips. |
| BMMCsIgE/DNP(c)/PL | BMMCs were sensitized with anti-DNP IgE overnight and then spun onto poly-L-lysine-coated coverslips in culture medium containing soluble DNP-BSA. |
As an alternative stimulation approach, poly-L-lysine (152689, MW 1500–8000; MP Biomedicals, Solon, OH) coated onto coverslips was used. The degranulation of mast cells triggered by poly-L-lysine has been reported by previous studies (29–31). Our goal was to demonstrate the sensitivity of combined AFM and LSCM to the variations in cell signaling, and to capture the characteristic membrane and cytoskeletal structures of BMMCs under this protocol. In these cases, cells were directly spun onto poly-L-lysine-coated coverslips (BMMCs−/−/PL). The concentration of poly-L-lysine coating solution was 1 mg/mL.
For comparison, BMMCs spun on glass surfaces (BMMCs−/−/−) were used as control. All of the cell samples were incubated for 30 min under 5% CO2 and 37 °C before fixatives were added to terminate the stimulation. The 30-min incubation time was selected to ensure that most of the cells would reach the activation stage at which dramatic morphological changes would occur (5).
Combined AFM and LSCM
A combined LSCM and AFM system was used to image the cell samples. The AFM system (MFP-3D-CF, Asylum Research, Santa Barbara, CA) has a scan range of 90, 90, and 16 μm in the X, Y, and Z directions, respectively. The X, Y, Z scanner has a closed-loop configuration with the precision of 0.6, 0.6, and 0.3 nm, respectively. The AFM's capabilities in wide scanning range in 3D, high movement precision, and versatility of imaging in various environments enable investigation of cellular systems, as well as its integration with confocal microscopy.
The LSCM is a FluoView 1000 system (Olympus America, Center Valley, PA) with spectral detection on an IX81 stand, in which a fully automated internal stepper motor with Z-resolution of 0.01 μm is integrated. The XY scanning is performed with a pair of galvanometric mirrors, yielding a wide scanning range that images both individual cells and cell colonies. All confocal images were acquired using a Plan Apo N (60×, N.A. = 1.42) oil immersion bright-field objective with a working distance of 0.15 mm.
The integration of AFM and LSCM is achieved by the modification of the sample stage of the LSCM to accommodate the AFM scanner. The AFM scanner is designed in such a way that it has a window in the center to allow the alignment of the AFM tip with the laser beam. This enables concurrent imaging of desired cells by both microscopes. An in-house-constructed sample holder, which enables the mounting of coverslips, was utilized for simultaneous AFM and LSCM imaging in the same medium.
For AFM imaging, the cells were incubated on coverslips and then fixed with 3.7% formaldehyde for 30 min and maintained in PBS. Unless otherwise specified, contact-mode AFM was employed to probe the cell morphology. In some cases when the cell surface was very sticky, tapping-mode AFM was employed to minimize the disturbance of the AFM tip to the membrane and to achieve a high-resolution view of local membrane structures. In contact mode, silicon cantilevers with a spring constant of 0.03 N/m (CSC38 lever B; MikroMasch, San Jose, CA) were used. The force constant was measured by the thermal noise method (32). The imaging force was controlled to be <1 nN as determined by the force-distance curve. In tapping mode, silicon nitride cantilevers with a spring constant of 0.03 N/m (Biolever B, Olympus, Japan) were used. The driving frequency was set at the fundamental resonance of the cantilever, typically 6–8 kHz. The imaging set point was adjusted to 60% damping of the free amplitude.
For LSCM imaging, parallel cell samples were labeled with two dyes (each sample with one dye). To visualize the cytoskeletal actin, phalloidin staining was used. BMMCs were first fixed with 3.7% formaldehyde for 10 min and washed with PBS. Then, with or without the permeabilization of cell membrane with 1 mL of 0.1% Triton-X 100 (21123; Invitrogen, Carlsbad, CA) for 5 min at room temperature (RT), the cells (after washing with PBS twice) were stained with 0.1 μM of phalloidin (A12380; Invitrogen, Carlsbad, CA) for 20 min at RT, rinsed, and stored in PBS before imaging. The LSCM images were collected with the emission between 555 nm and 630 nm under an excitation of 543 nm.
To reveal the intracellular structural changes upon cell stimulation, carboxyfluorescein diacetate succinimidyl ester (CFSE, V12883; Invitrogen, Carlsbad, CA) was used to highlight intracellular homogeneity or heterogeneity. Cell pellets were resuspended in 37 °C PBS containing 10 μM of the CFSE dye and then incubated for 15 min. The dye solution was then removed by centrifuging and resuspending the cells in the fresh medium. To ensure complete uptake and binding of the dye, the cells were incubated for another 30 min and then washed again with PBS. The LSCM images were collected with emissions between 497 and 531 nm under an excitation of 488 nm.
Image display and analysis
All of the AFM images were acquired and analyzed using Asylum MFP3D software developed on the Igor Pro 5.04 platform. Because of the large dynamic range of cells (2–10 μm high), the display of AFM topographic images for cells is not trivial. In this study, deflection images (collected simultaneously) are displayed in conjunction with the topographic images. The deflection images allow clear visualization of structural features on the cell membrane in a wide dynamic range, and quantitative 3D measurements can be made from the corresponding topographic image. Calculation of cell-spreading areas from AFM images was done using the ImageJ software (National Institutes of Health, Bethesda, MD).
All of the confocal fluorescence images were acquired and analyzed using the Olympus Fluoview 1.5 software. To visualize chromophores that are only membrane bound, such as phalloidin, 3D stacking images were displayed using Z-projection to present the 3D spatial distribution of dye in one frame. To visualize the intracellular structure (e.g., CFSE-stained cells), 3D stacking images were displayed in a three-panel view, where cross sections of the XY, XZ, and YZ planes were selected to emphasize important features. The color of the confocal images was assigned to represent the real emission wavelength of the chromophores.
The overlay of confocal images with AFM topographic images was accomplished using Igor Pro. First, at least three different landmarks were selected from the AFM and confocal images. Then, both images were aligned by matching all of the landmarks using the transform tool in Adobe Photoshop (Adobe Systems Inc., San Jose, CA). Afterward, the AFM topograph (Z as a function of X and Y) was displayed in three dimensions. Finally, the 3D AFM image was colored according to the aligned fluorescence image using MFP-3D software as described previously (33). The overlay enables a direct correlation of cytoskeletal and intracellular structures captured by LSCM with the membrane structures revealed in AFM topographs.
Results
AFM enables high-resolution visualization of the 3D landscape of the cell membrane in buffer
High-resolution AFM images were acquired in PBS for the resting and stimulated BMMCs under the four sample conditions summarized in Table 1. Fig. 1 shows representative cell images to reveal the overall cellular morphology and close-range views on the top of the cell body. From left to right in Fig. 1, each column displays the AFM topograph, deflection, and 5 × 5 μm2 zoom-in images, respectively. From the overall view of cell morphology acquired by the AFM contact mode, the control cells (BMMCs−/−/−) exhibit a “hat” shape with low degree of spreading. The typical contact diameter and height of BMMCs−/−/− (e.g., the cell in Fig. 1 A) measures 9 ± 1 and 7.5 ± 0.5 μm, respectively (cursor profile 1). Under an AFM tapping mode and by zooming onto the top of the cell (Fig. 1 C), clear domains of fine membrane structures are revealed. From their morphology and distribution, these fine structures are consistent with microvilli and small protrusions with apparent height ranging from 200 to 500 nm, as shown in cursor profile 2. A more detailed survey of structural diversity and statistics of the fine structures, including villi and protrusions, was reported in our previous study (34) and is of little relevance to the focus of this work.
Figure 1.
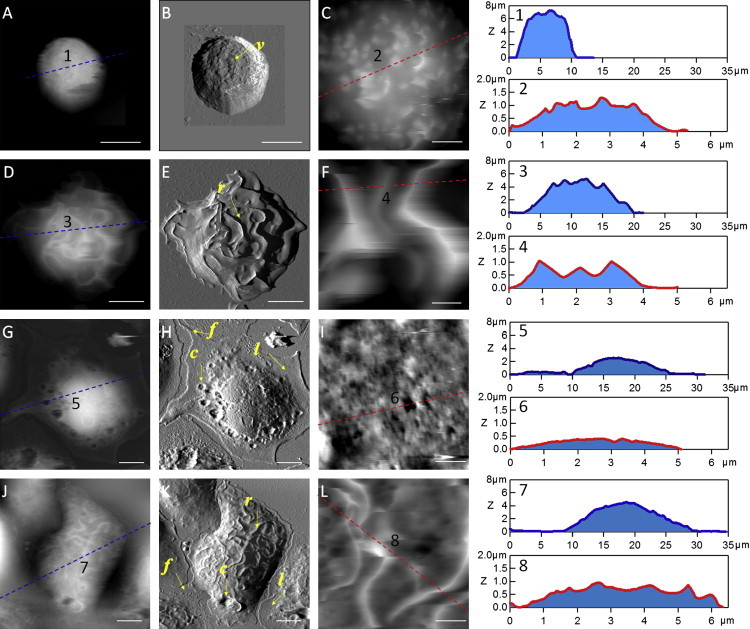
AFM images reveal characteristic 3D membrane morphologies for BMMCs under various sample preparation conditions. (A) AFM topograph of a BMMC−/−/−. (B) The corresponding deflection image of A. (C) A 5 μm × 5 μm zoom-in image from the top of the cell. Cursor profiles as indicated in A and C are shown on the right. The layout for the rest of rows is the same as for the top, except that the subject cells are a BMMCIgE/DNP(s)/−, BMMC−/−/PL, and BMMCIgE/DNP(c)/PL, respectively. Scale bars: (A, B, D, E, G, H, J, and K) 5 μm; and (C, F, I, and L) 1 μm. Local structures (villi, ridge, lamellopodia, filopodia, and crater) are indicated by letters (v, r, l, f, and c, respectively).
Characteristic membrane ridges appear on BMMCs upon activation by DNP-BSA immobilized on a glass surface (BMMCsIgE/DNP(s)/−) as shown in Fig. 1, D–F. The overall shape of BMMCsIgE/DNP(s)/− varies due to cell spreading. The spreading also results in the decrease in cell height, e.g., the height of the cell shown in Fig. 1 D is 5.2 μm—2 μm lower than the cell in Fig. 1 A (cursor profile 1 vs. 3). Membrane ridges are the most prominent features of this activated BMMC. From the AFM topograph and deflection images in Fig. 1, D and E, one can count all 25 ridges on this cell. The length of these membrane ridges varies from 2 to 10 μm, and their height measures 0.2–2.0 μm. By taking cursor profile measurements on BMMCsIgE/DNP(s)/− with different spreading levels, as exemplified by cursor profile 4, we noticed that the ridge height decreased with increased cell spreading. The presence of membrane ridges on activated BMMCs is consistent with a previous study using scanning electron microscopy (SEM), in which the formation of membrane ridges was associated with a later stage of degranulation (5). In contrast to SEM, AFM enables 3D measurements of membrane morphology in hydrated cells.
Using surface-bound poly-L-lysine instead of DNP-BSA as coatings led to a different morphology, as exemplified by Fig. 1, G–I. Compared to the heights of BMMCs−/−/− and BMMCsIgE/DNP(s)/−, the cells exhibit a much higher level of spreading, as seen from the AFM images and the corresponding cursor profile measurements. Characteristic membrane features associated with cell spreading, such as lamellopodia and filopodia, are clearly visible. The cursor profile measurements (e.g., cursor profile 5) reveal that the thickness of the lamellopodia is as thin as 65 nm, whereas the filopodia are as long as 4 μm. The evolution of lamellopodia and filopodia during cell spreading has been well reported by previous microscopic studies (35,36). A new observed feature associated with poly-L-lysine stimulation is the membrane crater. For instance, in Fig. 1, G and H, craters can be seen at the membrane region between the cell bulk and the lamellopodia. The opening of these craters ranges from 0.5 to 4.8 μm, and the depth varies from 0.1 to 0.7 μm. Upon zooming into the top of the cell, as shown in Fig. 1 I, fine membrane features, such as domains and pits, are visible, whereas ridges are not present.
Coexisting morphology was observed on the BMMCs when stimulated by both DNP-BSA and poly-L-lysine (BMMCsIgE/DNP(c)/PL) as shown in Fig. 1, J–L. From the overall view of these cells, it can be seen that BMMCsIgE/DNP(c)/PL contain both characteristic features of BMMCsIgE/DNP(s)/− and BMMCs−/−/PL. Membrane ridges, lamellopodia, filopodia, and craters were observed, as highlighted in the AFM deflection image of Fig. 1 K. Furthermore, from the quantification of these structures, we can conclude that the morphology of BMMCsIgE/DNP(c)/PL is intermediate between that of BMMCsIgE/DNP(s)/− and BMMCs−/−/PL. For example, the average spreading area for BMMCsIgE/DNP(c)/PL is 355 ± 60 μm2 (n = 81), which is smaller than that of BMMCs−/−/PL (473 ± 46 μm2, n = 86) but higher than that of BMMCsIgE/DNP(s)/− (298 ± 76 μm2, n = 168). In addition, the area of lamellopodia, the number of craters, and the height of ridges for BMMCsIgE/DNP(c)/PL all fall between those for BMMCsIgE/DNP(s)/− and BMMCs−/−/PL.
The results shown in Fig. 1 demonstrate that the “fix-and-look” methodology using AFM enables 3D and high-resolution characterization of cell morphology and fine membrane features for hydrated cells. Well-known features, such as ridges, are identified in association with antigen activation of BMMCs, which validates our approach. In addition, the high-resolution imaging reveals a new type of structural feature (craters) that is a characteristic of poly-L-lysine stimulation. Further, the 3D information of characteristic structural features correlates closely with stimulation protocols and cellular spreading behaviors. The nanoscopic information sheds light on detailed local structure changes of membrane during cell activation.
F-actin redistribution is directly responsible for the membrane features of ridges, filopodia, and lamellopodia
Combined AFM and LSCM investigations of BMMCs labeled with phalloidin enable simultaneous and in situ observations of cytoskeletal F-actin and membrane morphologies. Fig. 2, A–C, show the LSCM Z-projection image and the AFM topograph, as well as the overlay images taken on the same cell after antigen activation. The Z-projection images in Fig. 2 A involve only the Z slices taken on the cortical side of the cell. In these images one can see that the BMMCIgE/DNP(s)/− exhibits a heterogeneous network arrangement of F-actin, whereas the resting cell in the inset of Fig. 2 A (BMMC−/−/−) shows a more homogeneous fluorescence contrast. If these confocal images were displayed via a 2D cross-sectional view, each individual slice of the activated BMMC would exhibit a broken circle, whereas that of the resting cell would display a continuous ring. This observation is consistent with a previous study by Nishida et al. (37) using confocal microscopy, in which the cytoskeletal F-actin was found to be fragmentized upon antibody-mediated activation. Our results with the complete Z stacks contribute further spatial distribution data regarding this rearrangement of F-actin.
Figure 2.
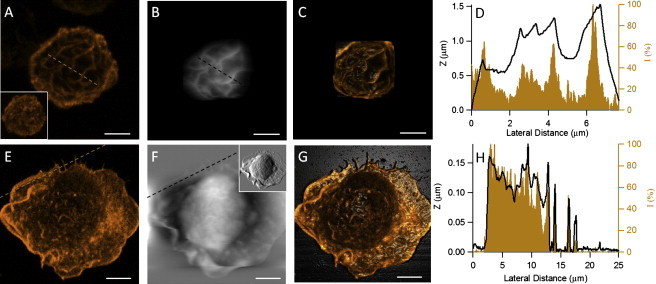
Correlation of membrane and cytoskeletal features is achieved by the overlay of AFM topographic images with LSCM images of F-actin. (A) LSCM Z-projection image. (B) AFM topographic image of a BMMCIgE/DNP(s)/− labeled with phalloidin. (C) Overlay image of A and B. (D) Comparison of the AFM cursor profile drawn in B with the fluorescence intensity profile drawn in A. The layout for the second row (E–H) is the same as for the top, except that the subject cell is a BMMC−/−/PL without surfactant treatment. The inset in A shows the Z-projection image of a BMMC−/−/− labeled with phalloidin, and the inset in F is the corresponding AFM deflection image of F. Scale bars, 5 μm.
In experiments shown in Fig. 2, we modified the commonly used phalloidin assay by excluding surfactants during labeling to preserve membrane integrity. Fig. 2 B displays the membrane topograph of the same cell in Fig. 2 A acquired by tapping-mode AFM, where ridge structures are clearly visible. Because we aimed to acquire high-resolution images of the top part of the cell with minimum tip disturbance, a soft AFM probe with a short tip (5 μm high) was selected for this topographic imaging. According to the measurements made on the topograph image, the height of the ridges ranges from 200 nm to 2 μm, and the spacing between them ranges from 0.5 μm to 3 μm. The overlay of the confocal and AFM images is shown in Fig. 2 C, where the three-point method (see Materials and Methods) was applied to ensure the accurate overlay of these two images. From this overlay image, one can see that the membrane ridges are superimposed over the F-actin network. This colocalization is also shown in the comparison of the AFM height profile with the fluorescence intensity profile in Fig. 2 D. To the best of our knowledge, this is the first direct evidence that membrane ridges are due to the lipid bilayer decoration of the F-actin network underneath.
As discussed in the previous section, poly-L-lysine prompts cell spreading on surfaces with membrane features such as lamellopodia and filopodia. Using combined AFM and LSCM, we further investigated the correlations between these membrane features and cytoskeletal F-actin. Fig. 2 E displays the Z-projection of all confocal slices taken on a BMMC−/−PL, including both cortical and ventral sides, from which one can see the fluorescence intensity is much stronger at the cell periphery where lamellopodia and filopodia reside. On the top of the cell, some small features are visible, indicating a few villi structures, but their dye intensity is much weaker than that of the peripheral area. The AFM topograph of the same cell is shown in Fig. 2 F, in which a large area of lamellopodia and a few filopodia can be seen. In contrast to the confocal image in Fig. 2 E, some of the lamellopodia on the leading edge in the AFM image fold up as induced by the AFM probe during scanning. On the top of the cell, the membrane is relatively smooth compared to Fig. 2 B, indicating that the F-actin signal seen on the top of cell in Fig. 2 E derives from villi structures that faded into the membrane due to cell spreading. No craters were found on this cell; however, several membrane protrusions can be found in the region between the cell bulk and lamellopodia. As shown in Fig. 2 F, one of the largest protrusions has an elliptical shape with long and short axes of 3.5 and 2.5 μm, respectively. The overlay image in Fig. 2 G shows that the lamellopodia and filopodia correlate with an increased fluorescence signal, and the comparison of cursor profiles in Fig. 2 H shows an almost exact match between the membrane topograph and F-actin for the lamellopodia and filopodia. Therefore, the observed membrane features at the leading and trailing edges of BMMCs are the lipid bilayers decorating over cytoskeletal F-actin.
Much interest and effort have been devoted to the investigation of local membrane structures, such as budding, villi, and ridges, resulting from protein or cytoskeleton regulation (3,4,38). The combination of AFM and LSCM enables in situ and simultaneous observations of cellular membrane and cytoskeletal structures underneath, such as F-actin assemblies. This investigation offers direct proof that the formation of membrane ridges, filopodia, and lamellopodia is due to the decoration of lipid bilayers over the F-actin network underneath.
The F-actin network and the lack thereof are associated with the presence of postsecretion granules and membrane craters, respectively
With the use of CFSE to highlight the intracellular homogeneity and heterogeneity, “ghost” granules upon exocytosis were captured under the ridged membrane for BMMCsIgE/DNP(s)/−, as shown in Fig. 3. Fig. 3, A and B, present the AFM topograph and deflection images, respectively, of a BMMCIgE/DNP(s)/− labeled with CFSE. A large number of membrane ridges with various lengths can be seen in these AFM images, indicating the antigen-driven activation of this cell. As discussed in previous sections, these ridges correspond to lipid bilayers and membrane composites decorating over the F-actin network underneath.
Figure 3.
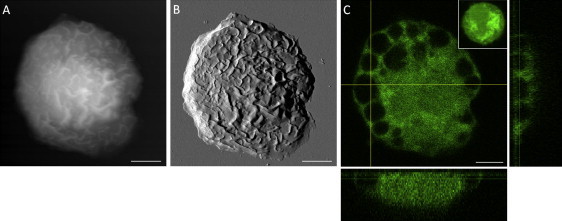
Combined AFM and LSCM images of a BMMCIgE/DNP(s)/− labeled with CFSE. (A) The AFM topographic image. (B) The corresponding deflection image of A. (C) The Z-stack LSCM images displayed in a three-panel view with the XY plane at 1.6 ± 0.1 μm above the glass surface. Scale bars, 5 μm.
Fig. 3 C displays the 3D Z-stacking confocal images in the form of an orthogonal three-panel view for a BMMCIgE/DNP(s)/− labeled with CFSE. The CFSE dye contains acetate groups, which yield highly fluorescent amine-reactive carboxyfluorescein succinimidyl ester upon cleavage by intracellular esterases. The succinimidyl ester group then further reacts with intracellular amine-containing proteins, lighting up the entire cell body (39). The BMMCIgE/DNP(s)/− in Fig. 3 C shows an overall decreased CFSE intensity and a large number of intracellular cavities. The diameter of these cavities measures 0.8–5.0 μm. In contrast, the inset image in Fig. 3 C shows a control cell (BMMC−/−/−) with a higher overall intensity of fluorescence. There is some degree of heterogeneity in dye intensity, indicating heterogeneity in the cytoplasmic protein distribution. Numerous villi structures stained by CFSE can be seen at the periphery of this nonactivated cell. Since the cell in Fig. 3 C differs from the one in the inset only because of exocytosis by antigen activation, we infer that these cavities arise from granules after releasing their contents.
The CFSE-void intracellular cavities were found in large numbers, almost one next to another, at the periphery of the cell. Further inside the cell, only two large (4.5 μm, 3.0 μm in the longest diameter) and two small (0.8, 1.0 μm) intracellular cavities were found. No cavities were observed inside the nucleus from a thorough analysis of all confocal Z slices. The lack of cavities near the central region is consistent with the fact that cavities are granules upon exocytosis, because the dye diffuses with time from the periphery to center, and thus the loss of dye inside the granules is the aftermath of dye uptake.
By comparing Fig. 3 A with Fig. 3 C, one can see that the AFM images show characteristic ridge structures decorating all over the cortical membrane on the top of the cavities, indicating the formation of F-actin network at the cytoskeleton between the plasma membrane and intracellular granules. An overlay of the AFM topogaph and confocal images reveals no obvious correlation between the membrane ridges and the cavities underneath, and no corresponding membrane features for these intracellular cavities.
In some cases of antigen-mediated activation, as exemplified in Fig. 4, we were able to capture pore structures on the membrane in between ridges from the AFM topograph. Fig. 4 shows a series of zoom-in images taken on a BMMCIgE/DNP(c)/PL, from 17 × 17 μm2 in Fig. 4 A, to 5 × 5 μm2 and 0.5 × 0.5 μm2 in Fig. 4, B and C, respectively, as marked by the squares in these images. In Fig. 5 C, two pore structures are visible: a larger one with an opening of 150 ± 10 nm in diameter and >75 nm in depth, and a small one with an opening of 50 ± 10 nm in diameter and >25 nm in depth. Both are located at the “valley” between two membrane ridges as clearly seen from these serial zoom-in images. A closer look at the overlay image in Fig. 4 E reveals the correspondence of these pore structures with a cavity underneath, as pointed to by the arrow in the confocal image of Fig. 4 D. The diameter of this cavity is ∼1.1 ± 0.1 μm, equal to the topographic height, 1.15 ± 0.05 μm, at the specific site where the pore structures are located. The observed dimensions for these membrane indentations are consistent with those reported for fusion pores (50–150 nm) (2,40) and porosomes (41) associated with the functions of exocytosis during mast cell activation. Our data indicate the depletion of CFSE intensity in the cavities through the fusion pores, which is consistent with the transient fusion model of exocytosis (2,42), where granules temporally dock onto the cell membrane to release their contents.
Figure 4.
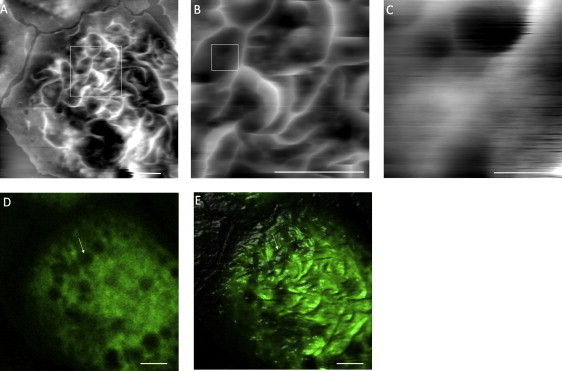
Combined AFM and LSCM images reveal pore structures located in between membrane ridges. (A) A 17 μm × 17 μm AFM topographic image of a BMMC−/−/PL. (B) A 5μm × 5 μm scan on the membrane patch drawn by the square in A. (C) A 500 nm × 500 nm scan on the membrane patch drawn by the square in B. (D) The cross-section confocal image taken from the same cell in A. (E) The overlay of A and D. The arrows point to the location of the membrane pores shown in the systematic AFM zoom-in images. Scale bars: (A, B, D, and E) 2.5 μm; and (C) 200 nm.
Figure 5.
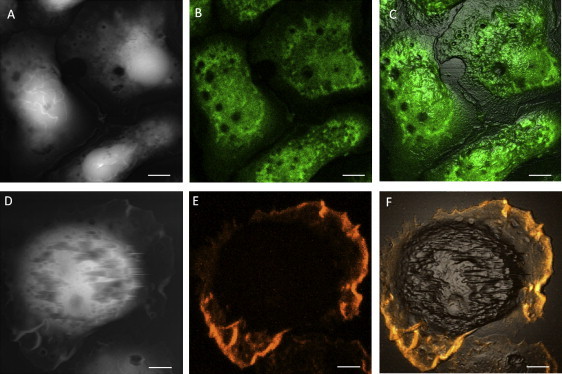
(A) AFM topographic image, and (B) the LSCM cross-section image of BMMCs−/−/PL labeled with CFSE. (C) The overlay image of A and B. (D) The AFM topographic image, and (E) the LSCM Z-projection image of a BMMC−/−/PL treated with Triton-X 100 during phalloidin labeling. (F) The overlay image of D and E. Scale bars, 5 μm.
BMMCs stimulated by poly-L-lysine coated on the surface (BMMCs−/−/PL) exhibit a different degranulation behavior, as revealed by the combined AFM and confocal images shown in Fig. 5. In cells dyed with CFSE (Fig. 5, A–C), the AFM topographic images reveal craters at the cell membrane, whereas the confocal contrast shows dark pits at the same locations. As shown by the overlay image in Fig. 5 C, the membrane craters in the AFM topograph (Fig. 5 A) are accurately superposed with the dark pits of fluorescence in Fig. 5 B. The diameters of these dark pits range from 0.4 to 5.2 μm, consistent with the opening size of craters measured in the AFM images.
By combining AFM images with confocal images taken on BMMCs−/−/PL dyed with phalloidin, we were able to investigate the role played by cytoskeletal F-actin in crater formation. We found little correlation between the two. First, craters are mostly located at the membrane region, where F-actin network is lacking. Second, individual craters are not colocalized with the F-actin network. Fig. 5, D–F, show a BMMC−/−/PL with surfactant treatment during phalloidin labeling. Similarly to the cell in Fig. 2 E, the Z-projection confocal image in Fig. 5 E shows that F-actin is enriched in the lamellopodia and filopodia located at the edges of cells. In contrast to its role in crater formation, the F-actin network does provide support to maintain cells' integrity. The AFM topographic image in Fig. 5 D shows that the cell is partly torn at the center, where there is little phalloidin intensity in the confocal image. Similar tearing is not seen where the F-actin network is present.
Taken collectively, these results suggest that craters are formed by the fusion of granules into the plasma membrane (i.e., permanent fusion), where cytoskeletal F-actin network is lacking. Unlike immunofluorescence or granule-labeling approaches, CFSE induces least perturbation to the BMMC's degranulation process, since CFSE is a cell tracing dye and is known to be inert in cells (43). This combined AFM and confocal imaging methodology provides direct evidence regarding the fate of granules upon transient (in the case of DNP-BSA activation) and permanent (in the case of poly-L-lysine stimulation) fusion. The ability to quantify the cavities and craters, and to correlate the cytoskeleton with membrane and intracellular structures paves the way for understanding granule-membrane interactions.
Discussion
Prior studies have yielded two models of regulated exocytosis for mast cells based on the fate of the granules after they release their contents inside: 1), the permanent fusion (kiss-and-merge) model (44); and 2), the transient fusion (kiss-and-run) model (2,45). The ratio of transient fusions versus permanent fusions was reported to be 3:2 in 70% of total granules for RBL-2H3 cells upon IgE-mediated activation (46). In this investigation, we found that the dominant degranulation events were kiss-and-run in antigen-activated BMMCs, and kiss-and-merge in poly-L-lysine stimulated BMMCs. These two degranulation scenarios are illustrated in Fig. 6 and described below.
Figure 6.
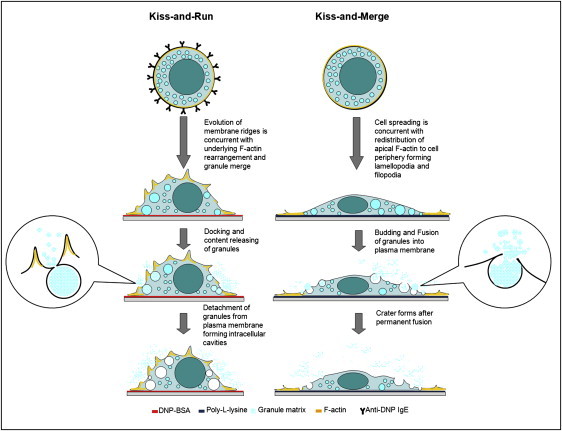
Schematic diagram of the two degranulation mechanisms for BMMCs stimulated by surface coatings of DNP-BSA (left) and poly-L-lysine (right), respectively.
In the kiss-and-run process shown in the left column of Fig. 6, ridges form due to the rearrangement of cytoskeletal F-actin upon the cross-linking of the IgE-FcɛRI complexes by surface-bond antigen. In contact with the interior of the plasma membrane, granules dock onto membrane regions in between ridges and then open the fusion pores to release the granule contents. The contact ends when most of the granule matrix is depleted and the granules retreat to the cytoplasm (2). The overall theme of this kiss-and-run model was proposed previously in a study using patch-clamp amperometry (47) and further confirmed by two-photon microscopy (46). The new (to our knowledge) insights gained from our investigation include 1), the observation and origination of membrane ridges, as well as the 3D characterization of their dimensions; 2), the direct observation of postdegranulation granules manifesting as cavities via a gentle cell-tracing dye; and 3), the capture of transient fusion pores in an AFM topograph of hydrated cells.
In the kiss-and-merge process shown in the right column of Fig. 6, craters form as a result of granules merging into the plasma membrane. Upon contact with the poly-L-lysine coated on glass coverslips, the cells spread quickly. This process involves the formation of extensive lamellopodia and filopodia, which are mainly constructed by cytoskeletal F-actin under the plasma membrane (36). However, de novo synthesis is not able to provide the necessary cytoskeletal F-actin in such a short period to compensate for the quick expansion of the cell (48). Thus, the main pool of the cellular actin moves to and near the periphery of the cell. The spreading of the cell is likely to trap the granules toward the periphery of the cell bulk, where the gap between the cortical and ventral membrane is narrow. The granules then bud out from the cortical membrane and merge with the plasma membrane after releasing their contents inside. The budding and merging of granules into the plasma membrane has been reported by previous studies using transmission electron microscopy (49). The new insights gained by our investigation include 1), the capture and quantification of the intermediate structures of permanent fusion (i.e., craters); and 2), the observation of noncolocalization between these craters and F-actin, indicating the lack of F-actin participation in degranulation and crater formation.
In addition to the new information regarding the two mechanisms of degranulation, this investigation elucidates the role of cytoskeletal F-actin in determining the fate of granules, as well as the local membrane structures. When the cell is in its resting state, F-actin underlying the lipids of plasma membranes serves as a physical barrier to the access of secretory vesicles to the plasma membrane, and thus blocks the membrane fusion. Upon the cross-linking of FcɛRI-bound IgE by antigen, the F-actin molecules, originally distributed evenly under plasma membrane, reorganize into a more heterogeneous network structure. This effectively breaks the original F-actin barrier and allows temporary fusion to occur. Meanwhile, this newly formed F-actin network still serves as confinement to hinder the complete fusion between granules and the plasma membrane. Thus, ridges and pores are the primary membrane features for BMMCs upon antigen activation. When the cell is spreading on a poly-L-lysine-coated surface, most of the cytoskeletal F-actin, which originally was evenly distributed on the cell surface, relocates to and near the periphery of the spreading cells. This disassembly of F-actin on the cortical and ventral membranes removes the barrier for granules to contact the plasma membrane, and thus favors permanent fusion and crater formation on the cell surface.
Membrane morphological changes induced by the underlying F-actin further affect the fate of fusions between granules and the plasma membrane. As discussed in the previous sections, the BMMC membrane exhibits ridged morphology as a result of the lipid bilayers' decoration of F-actin network. This wrinkled membrane structure is more relaxed, due to cytoskeleton confinement (left inset of Fig. 6), compared to the stretched membrane of BMMCs spreading on poly-L-lysine (right inset of Fig. 6) (50). As such, lack of F-actin and high lateral tension promote permanent fusion between granules and the plasma membrane (kiss-and-merge) in the poly-L-lysine stimulated cells, and the presence of F-actin network and small lateral tension favors transient fusion (kiss-and-run) in the antigen-activated BMMCs.
Conclusions
This work represents our initial effort to apply combined AFM and LSCM to cellular biology research. This combination provides a powerful tool for characterizing 3D structures of cellular membranes, from nanometer to single-cell (tens of micrometers) levels, as well as for imaging cytoskeleton structures and intracellular features under the cellular membrane. The high-resolution images acquired provide important insights into local membrane structures in correlation with the cytoskeleton underneath: 1), membrane ridges are formed during antigen activation of BMMCs, due to the follow of lipid bilayer to F-actin network underneath and 2), craters appear during the merging of granules into the cellular membrane, where F-actin is lacking. In conjunction with the direct visualization of post-release granules intracellularly, we are able to rationalize the role of F-actin and local membrane structures in terms of determining the fate of granules after releasing their contents. Future work will include correlations among the membrane morphology, cytoskeletal structure, and cellular mechanics at nanometer scales (51) to further our understanding of the local structures of membrane and cytoskeleton in correlation with the cellular property and function. Investigations are in progress to apply the combined microscopy techniques in live-cell studies and to explore the contribution of other types of cytoskeletal machinery, such as tubulin, myosin, and caveolin, to reveal characteristic morphologies and nanostructures associated with the activation of mast cells.
Acknowledgments
We thank Mr. Alan Hicklin at the University of California, Davis, and Dr. Kevin Jia at Olympus American for their assistance in using the combined LSCM and AFM. Dr. Christopher Koehler is acknowledged for his help with manuscript preparation.
This work was supported by the University of California, Davis; the National Science Foundation (DMR0421521 and DMR0723118); and the National Institutes of Health (R21 GM77850-01 and R01 AI20958).
References
- 1.Horber J.K.H., Miles M.J. Scanning probe evolution in biology. Science. 2003;302:1002–1005. doi: 10.1126/science.1067410. [DOI] [PubMed] [Google Scholar]
- 2.Schneider S.W. Kiss and run mechanism in exocytosis. J. Membr. Biol. 2001;181:67–76. [PubMed] [Google Scholar]
- 3.Frankel D.J., Pfeiffer J.R., Surviladze Z., Johnson A.E., Oliver J.M. Revealing the topography of cellular membrane domains by combined atomic force microscopy/fluorescence imaging. Biophys. J. 2006;90:2404–2413. doi: 10.1529/biophysj.105.073692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfeiffer J.R., Seagrave J.C., Davis B.H., Deanin G.G., Oliver J.M. Membrane and cytoskeletal changes associated with Ige-mediated serotonin release from rat basophilic leukemia-cells. J. Cell Biol. 1985;101:2145–2155. doi: 10.1083/jcb.101.6.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zou X., Block M., Lofman C., Nilsson G. IgE-mediated mast cell degranulation and recovery monitored by time-lapse photography. J. Allergy Clin. Immunol. 2001;108:116–121. doi: 10.1067/mai.2001.116124. [DOI] [PubMed] [Google Scholar]
- 6.Fackler O.T., Grosse R. Cell motility through plasma membrane blebbing. J. Cell Biol. 2008;181:879–884. doi: 10.1083/jcb.200802081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seagrave J., Pfeiffer J.R., Wofsy C., Oliver J.M. Relationship of Ige receptor topography to secretion in Rbl-2h3 mast-cells. J. Cell. Physiol. 1991;148:139–151. doi: 10.1002/jcp.1041480117. [DOI] [PubMed] [Google Scholar]
- 8.Ludowyke R.I., Kawasugi K., French P.W. Pma and calcium ionophore induce myosin and F-actin rearrangement during histamine-secretion from Rbl-2h3 cells. Cell Motil. Cytoskeleton. 1994;29:354–365. doi: 10.1002/cm.970290408. [DOI] [PubMed] [Google Scholar]
- 9.Charras G.T., Coughlin M., Mitchison T.J., Mahadevan L. Life and times of a cellular bleb. Biophys. J. 2008;94:1836–1853. doi: 10.1529/biophysj.107.113605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burwen S.J., Satir B.H. Plasma-membrane folds on mast-cell surface and their relationship to secretory activity. J. Cell Biol. 1977;74:690–697. doi: 10.1083/jcb.74.3.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sahara N., Siraganian R.P., Oliver C. Morphological-changes induced by the calcium ionophore-A23187 in rat basophilic leukemia (2h3) cells. J. Histochem. Cytochem. 1990;38:975–983. doi: 10.1177/38.7.1693935. [DOI] [PubMed] [Google Scholar]
- 12.Sharma B.B., Apgar J.R., Liu F.T. Mast cells—receptors, secretagogues, and signaling. Clin. Rev. Allergy Immunol. 2002;22:119–148. doi: 10.1385/CRIAI:22:2:119. [DOI] [PubMed] [Google Scholar]
- 13.Chen H.Y., Sharma B.B., Yu L., Zuberi R., Weng I.C. Role of galectin-3 in mast cell functions: galectin-3-deficient mast cells exhibit impaired mediator release and defective JNK expression. J. Immunol. 2006;177:4991–4997. doi: 10.4049/jimmunol.177.8.4991. [DOI] [PubMed] [Google Scholar]
- 14.Lebduska P., Korb J., Tumova M., Heneberg P., Draber P. Topography of signaling molecules as detected by electron microscopy on plasma membrane sheets isolated from non-adherent mast cells. J. Immunol. Methods. 2007;328:139–151. doi: 10.1016/j.jim.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 15.Gilfillan A.M., Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat. Rev. Immunol. 2006;6:218–230. doi: 10.1038/nri1782. [DOI] [PubMed] [Google Scholar]
- 16.Rivera J., Gilfillan A.M. Molecular regulation of mast cell activation. J. Allergy Clin. Immunol. 2006;117:1214–1225. doi: 10.1016/j.jaci.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 17.Mousli M., Hugli T.E., Landry Y., Bronner C. Peptidergic pathway in human skin and rat peritoneal mast-cell activation. Immunopharmacology. 1994;27:1–11. doi: 10.1016/0162-3109(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 18.Berlin G. The dynamics of mast-cell secretion mediated by Ige or polyamines. Agents Actions. 1984;15:482–487. doi: 10.1007/BF01966760. [DOI] [PubMed] [Google Scholar]
- 19.Edgar A.J., Davies G.R., Anwar M.A., Bennett J.P. Loss of cell surface microvilli on rat basophilic leukaemia cells precedes secretion and can be mimicked using the calmodulin antagonist trifluoperazine. Inflamm. Res. 1997;46:354–360. doi: 10.1007/s000110050201. [DOI] [PubMed] [Google Scholar]
- 20.Lagunoff D., Martin T.W., Read G. Agents that release histamine from mast-cells. Annu. Rev. Pharmacol. Toxicol. 1983;23:331–351. doi: 10.1146/annurev.pa.23.040183.001555. [DOI] [PubMed] [Google Scholar]
- 21.Baxter J.H., Adamik R. Differences in requirements and actions of various histamine-releasing agents. Biochem. Pharmacol. 1978;27:497–503. doi: 10.1016/0006-2952(78)90384-2. [DOI] [PubMed] [Google Scholar]
- 22.Foreman J.C., Lichtenstein L.M. Induction of histamine-secretion by polycations. Biochim. Biophys. Acta. 1980;629:587–603. doi: 10.1016/0304-4165(80)90164-6. [DOI] [PubMed] [Google Scholar]
- 23.Dufton M.J., Cherry R.J., Coleman J.W., Stanworth D.R. The capacity of basic peptides to trigger exocytosis from mast-cells correlates with their capacity to immobilize band-3 proteins in erythrocyte-membranes. Biochem. J. 1984;223:67–71. doi: 10.1042/bj2230067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ialenti A., Ianaro A., Brignola G., Marotta P., Dirosa M. Hyaluronic-acid inhibits polycation-induced cellular responses. Mediators Inflamm. 1994;3:287–289. doi: 10.1155/S0962935194000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ortner M.J., Chignell C.F. Spectroscopic studies of rat mast-cells, mouse mastocytoma-cells, and compound 48-80. 3. Evidence for a protein-binding site for compound 48-80. Biochem. Pharmacol. 1981;30:1587–1594. doi: 10.1016/0006-2952(81)90385-3. [DOI] [PubMed] [Google Scholar]
- 26.Bueb J.L., Dasilva A., Mousli M., Landry Y. Natural polyamines stimulate G-proteins. Biochem. J. 1992;282:545–550. doi: 10.1042/bj2820545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Razin E., Menciahuerta J.M., Stevens R.L., Lewis R.A., Liu F.T. Ige-mediated release of leukotriene C-4, chondroitin sulfate E-proteoglycan, β-hexosaminidase, and histamine from cultured bone marrow-derived mouse mast-cells. J. Exp. Med. 1983;157:189–201. doi: 10.1084/jem.157.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu F.T., Bohn J.W., Ferry E.L., Yamamoto H., Molinaro C.A. Monoclonal dinitrophenyl-specific murine Ige antibody—preparation, isolation, and characterization. J. Immunol. 1980;124:2728–2737. [PubMed] [Google Scholar]
- 29.Ennis M., Pearce F.L., Weston P.M. Some studies on the release of histamine from mast-cells stimulated with polylysine. Br. J. Pharmacol. 1980;70:329–334. doi: 10.1111/j.1476-5381.1980.tb07940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Courtoy R., Boniver J., Simar L.J. Cyto-chemistry of mouse mast-cell reaction to polylysine. Histochemistry. 1980;66:49–58. doi: 10.1007/BF00493245. [DOI] [PubMed] [Google Scholar]
- 31.Padawer J. Reaction of rat mast cells to polylysine. J. Cell Biol. 1970;47:352–372. doi: 10.1083/jcb.47.2.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hutter J.L., Bechhoefer J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 1993;64:1868–1873. [Google Scholar]
- 33.Proksch R. Multifrequency, repulsive-mode amplitude-modulated atomic force microscopy. Appl. Phys. Lett. 2006;89:113121. [Google Scholar]
- 34.Zink T., Deng Z., Chen H.-Y., Yu L., Liu F.-T. High-resolution three-dimensional imaging of the rich membrane structures of bone marrow-derived mast cells. Ultramicroscopy. 2008;109:22–31. doi: 10.1016/j.ultramic.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mattila P.K., Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008;9:446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- 36.Mejillano M.R., Kojima S., Applewhite D.A., Gertler F.B., Svitkina T.M. Lamellipodial versus filopodial mode of the actin nanomachinery: pivotal role of the filament barbed end. Cell. 2004;118:363–373. doi: 10.1016/j.cell.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 37.Nishida K., Yamasaki S., Ito Y., Kabu K., Hattori K. Fcɛri-mediated mast cell degranulation requires calcium-independent microtubule-dependent translation of granules to the plasma membrane. J. Cell Biol. 2005;170:115–126. doi: 10.1083/jcb.200501111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dvorak A.M. Ultrastructural studies of human basophils and mast cells. J. Histochem. Cytochem. 2005;53:1043–1070. doi: 10.1369/jhc.5R6647.2005. [DOI] [PubMed] [Google Scholar]
- 39.Parish C.R. Fluorescent dyes for lymphocyte migration and proliferation studies. Immunol. Cell Biol. 1999;77:499–508. doi: 10.1046/j.1440-1711.1999.00877.x. [DOI] [PubMed] [Google Scholar]
- 40.Chandler D.E., Heuser J.E. Arrest of membrane fusion events in mast cells by quick-freezing. J. Cell Biol. 1980;86:666–674. doi: 10.1083/jcb.86.2.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jena B.P. Discovery of the porosome: revealing the molecular mechanism of secretion and membrane fusion in cells. J. Cell. Mol. Med. 2004;8:1–21. doi: 10.1111/j.1582-4934.2004.tb00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeremic A., Kelly M., Cho S.J., Stromer M.H., Jena B.P. Reconstituted fusion pore. Biophys. J. 2003;85:2035–2043. doi: 10.1016/S0006-3495(03)74631-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hawkins E.D., Hommel M., Turner M.L., Battye F.L., Markham J.F. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat. Protocols. 2007;2:2057–2067. doi: 10.1038/nprot.2007.297. [DOI] [PubMed] [Google Scholar]
- 44.Rohlich P. Electron microscope observations on compound 48/80-induced degranulation in rat mast cells—evidence for sequential exocytosis of storage granules. J. Cell Biol. 1971;51:465–483. doi: 10.1083/jcb.51.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Breckenridge L.J., Almers W. Currents through the fusion pore that forms during exocytosis of a secretory vesicle. Nature. 1987;328:814–817. doi: 10.1038/328814a0. [DOI] [PubMed] [Google Scholar]
- 46.Williams R.M., Webb W.W. Single granule pH cycling in antigen-induced mast cell secretion. J. Cell Sci. 2000;113:3839–3850. doi: 10.1242/jcs.113.21.3839. [DOI] [PubMed] [Google Scholar]
- 47.Albillos A., Dernick G., Horstmann H., Almers W., deToledo G.A. The exocytotic event in chromaffin cells revealed by patch amperometry. Nature. 1997;389:509–512. doi: 10.1038/39081. [DOI] [PubMed] [Google Scholar]
- 48.Tobacman L.S., Korn E.D. The kinetics of actin nucleation and polymerization. J. Biol. Chem. 1983;258:3207–3214. [PubMed] [Google Scholar]
- 49.Dvorak A.M. Similarities in the ultrastructural morphology and developmental and secretory mechanisms of human basophils and eosinophils. J. Allergy Clin. Immunol. 1994;94:1103–1134. doi: 10.1016/0091-6749(94)90320-4. [DOI] [PubMed] [Google Scholar]
- 50.Sackmann E., Duwe H.P., Engelhardt H. Membrane bending elasticity and its role for shape fluctuations and shape transformations of cells and vescicles. Faraday Discuss. 1986;81:281–290. doi: 10.1039/dc9868100281. [DOI] [PubMed] [Google Scholar]
- 51.Schutz G.J., Sonnleitner M., Hinterdorfer P., Schindler H. Single molecule microscopy of biomembranes. Mol. Membr. Biol. 2000;17:17–29. doi: 10.1080/096876800294452. (review) [DOI] [PubMed] [Google Scholar]