

Abstract
Molecular flexibility and rigidity are required to determine the function and specificity of protein molecules. Some psychrophilic enzymes demonstrate a higher catalytic efficiency at low temperatures, compared to the efficiency demonstrated by their meso/thermophilic homologous. The emerging picture suggests that such enzymes have an improved flexibility of the structural catalytic components, whereas other protein regions far from functional sites may be even more rigid than those of their mesophilic counterparts. To gain a deeper insight in the analysis of the activity-flexibility/rigidity relationship in protein structure, psychrophilic carbonic anhydrase of the Antarctic teleost Chionodraco hamatus has been compared with carbonic anhydrase II of Bos taurus through fluorescence studies, three-dimensional modeling, and activity analyses. Data demonstrated that the cold-adapted enzyme exhibits an increased catalytic efficiency at low and moderate temperatures and, more interestingly, a local flexibility in the region that controls the correct folding of the catalytic architecture, as well as a rigidity in the hydrophobic core. The opposite result was observed in the mesophilic counterpart. These results suggest a clear relationship between the activity and the presence of flexible and rigid protein substructures that may be useful in rational molecular and drug design of a class of enzymes playing a key role in pathologic processes.
Introduction
Molecular flexibility and the complementary concept of rigidity are important characteristics for protein activity. A protein can be considered a mechanically heterogeneous 3D structure composed of locally rigid and flexible substructures. Enzymes isolated from cold-adapted organisms are generally characterized by a higher plasticity or flexibility of their molecular structure to compensate for the lower thermal energy provided by the low-temperature habitat (1–4). High structural flexibility of the psychrophilic enzymes could allow better interaction with substrates and could explain their higher catalytic rate (kcat), lower thermostability, and lower activation energy (Ea) requirements, compared with requirements for mesophilic and thermophilic counterparts (2,4–6). Psychrophilic enzymes have been shown to use different structural adaptation strategies to increase molecular flexibility, such as weakening intramolecular hydrogen bonds, increasing the number of hydrophobic side chains exposed to the solvent, and reducing the number of salt bridges (4,7). However, each protein family adopts its own structural strategy (2,4). As a consequence of their high structural flexibility, most of these enzymes usually improve kcat at the expense of affinity for substrates (Km), with a consequent loss of catalytic efficiency (kcat/Km) (8).
Conversely, some psychrophilic intracellular enzymes, operating at subsaturating substrate concentrations, showed Km values as much as 10 times lower than that of their mesophilic homologs at their respective physiologic temperatures (7–9), keeping the characteristic high-reaction rate (4). The decrease in Km, crucial to maintain catalytic efficiency values compatible with the cell metabolism, is achieved by acquiring rigidity in substructures that restrict the number of conformational states available for the enzyme-substrate complex (8). As a matter of fact, all cold-adapted enzymes with catalytic efficiency greater than that of their mesophilic counterparts display local flexibility/rigidity (5,8). Flexibility and rigidity are likely to “work” together, each acting on specific areas of the enzyme structure. Although flexibility and rigidity are properties difficult to quantify for a small and anisotropic material such as a protein molecule, the study of a cold-adapted enzyme compared with a closely related mesophilic counterpart may allow an interesting analysis at the molecular level (10–12).
To elucidate how local flexibility and rigidity are exploited in enzymes and to clarify the connections between structure, dynamics, and function of enzymes, we have investigated two cytoplasmatic enzymes, i.e., the cold-adapted gill carbonic anhydrase from the Antarctic icefish Chionodraco hamatus (Ice-CA) and its mesophilic counterpart bovine carbonic anhydrase II (BCAII).
Structural features of Ice-CA and BCAII
Ice-CA consists of 259 residues with an acetylated N-terminus; it has been shown to have a shift of the optimal temperature toward low values, an increased catalytic rate, and relatively low activation energy (13). Because of its cytoplasmic localization, its high sensitivity to sulphonamides, and the analysis of the amino acids within 10 Å from the catalytic core, the cold-adapted Ice-CA isoform can be considered closely related to the mammalian CAII isoform (13–15). However, similarly to CAIII, Ice-CA is also S-glutathionylated, suggesting a protective action of S-glutathionylation against oxidative stress (13). For these reasons, we chose, as the mesophilic reference, BCAII, an enzyme that, along with the human counterpart (HCAII), has been extensively studied as model characterize protein folding (16–22), catalytic features (23,24), protein-chaperone interactions (25,26), and protein-surface adsorption mechanisms (27).
BCAII consists of a single polypeptide chain with 260 amino acid residues. The central fold consists of 12 strands of parallel or antiparallel β-sheets, forming a core structure with seven α-helices surrounding the β-sheets (28). The overall structure of the molecule is roughly spherical, also a good model for its human homolog. (The root mean-square deviation for backbone atom positions between HCA II and BCA II is in fact 0.58 Å (28)). The active site of BCAII forms a funnel-shaped channel with an outer diameter of 13 Å that extends from the molecular surface to the center, with the zinc ion located at the bottom of the channel at a depth of 10 Å from the surface. Three direct ligands from the protein (His-94, His-96, His-119) and a water molecule placed in the middle of the β-sheets coordinate to Zn2+ ion in a tetrahedral geometry. The indirect ligands (Gln-92, Glu-117, Thr-199, Asn-243) enhance the basicity of direct histidine ligands and also help the positioning of the direct ligands for optimal metal coordination (29).
The proton transfer between zinc-bound water and bulk-solvent is mediated by the active site “shuttle” residue His-64; this residue, along with Glu-106, Thr-199, and zinc-hydroxide, forms a hydrogen bond network (30) that maintains the orientation and reactivity of the zinc hydroxide group, catalyzes proton transfer, and discriminates inhibitor (31–33). The aromatic residues Phe-93, Phe-95, Trp-97, and Phe-97 form a large hydrophobic cluster that anchors the β-strand containing the direct ligands His-94 and His-96 in the hydrophobic core of the enzyme and stabilizes its conformation (34). A binding site, 3.4 Å away from the zinc, is formed by the side chains of Val-121, Val-143, Leu-197, and Trp-208 (35). The C-terminal region of BCAII, as well as that of HCAII, is characterized by a unique “knot” topology, in which the polypeptide chain crosses itself in such a manner that one chain segment goes “below-above-below” the other segment. The “knot” topology of BCA II is tighter and more stable than that of HCAII (28).
BCAII and Ice-CA are of similar size, share all the properties common to carbonic anhydrases, but are adapted to different temperatures, therefore constituting an adequate series of homologous enzymes for adaptation studies.
Materials and Methods
Protein purification
Cytoplasmic carbonic anhydrase of Ice-CA was obtained as previously described (14). Ice-CA was purified by fast protein liquid chromatography affinity chromatography on p-aminomethylbenzene-sulphonamide immobilized on cyanogenbromide-activated agarose gel (13,14). The gel column (1.6 × 20 cm), fitted to an AKTA-FPLC system (Amersham Pharmacia Biotech, Uppsala, Sweden), was equilibrated with: Tris 25 mM, Na2SO4 100 mM, adjusted to pH 8.7 with HCl, and rinsed with Tris 25 mM, NaClO4 300 mM, adjusted to pH 8.7 with HCl; then the enzyme was eluted at 8 mL h−1 by CH3COOH 100 mM, NaClO4 500 mM, pH 5.6 at 4°C. Protein elution was monitored by measuring the eluate absorbance at 280 nm, and all fractions containing CA activity, measured by an electrometric method (14), were pooled and concentrated by ultra filtration with YM10 membrane (Amicon, Lexington, MA), under nitrogen pressure (7 × 105 Pa). All purification steps were carried out at 4°C.
Purified BCAII was purchased from Sigma (St. Louis, MO).
Sequence analysis and homology modeling
The sequence of the cold-adapted Ice-CA was retrieved from the Swiss-Prot database (http://www.expasy.org/; accession number P83299); bovine (Bos taurus) CAII was retrieved from the Protein Data Bank (PDB) structural database (http://www.rcsb.org/) with the PDB ID 1v9e. These sequences were aligned with ClustalW (version 1.9; http://www.ebi.ac.uk/clustalw). For the modeling procedure, the sequence of Ice-CA was submitted to the Swiss Model method, using the first approach mode (http://swissmodel.expasy.org//SWISS-MODEL.html) in combination with the DeepView 3.7 program (http://swissmodel.expasy.org/spdbv/). The protein structures were analyzed using the PyMOL program (http://pymol.sourceforge.net/); the accessible solvent area (ASA) was calculated using the NOC 1.5.2.10 program (http://www.noch.sourceforge.net/).
Quality validation of the Ice-CA model
The Protein Structure Quality Score (PSQS) (http://www1.jcsg.org/psqs), ERRAT, Verify3D, and ProCheck (http://nihserver.mbi.ucla.edu/SAVS/) programs were used to validate the quality of the Ice-CA model. The PSQS program uses an energy-like measure of the quality of the protein structure. The energy is calculated based on statistical potentials of mean force, which describes the interactions between residues pairs and between single residues and the solvent. The Verify3D program uses a potential based on the relative burial of residues and local secondary structure. The ERRAT program analyzes the statistics of nonbonded interactions between different atom types. By comparison with statistics from high-quality structures, the error values have been calibrated to give confidence limits. The ProCheck geometry validation program compares the geometry (standard Ramachandran plot) of the model with that of high-quality x-ray structures.
Carbonic anhydrase activity
Ice-CA and BCAII activities were measured by a radioactive method described earlier (13,14). Kinetic parameters of Ice-CA and BCAII (substrate affinity, Km, and maximum reaction rate, Vmax) were obtained by measuring their activity at 2, 18, and 35°C, using increasing concentration of NaH14CO3 (0.1–700 mM) and 500 ng of purified enzyme for each determination. Experimental data were fitted to a Michaelis-Menten equation by a curve-fitting subroutine in the Graph-Pad software package.
CA thermodependence was investigated by measuring the enzymatic activity in the 2–70°C range in a saturating concentration of substrate (100 mM NaH14CO3) and by recording the maximum velocity of bicarbonate dehydration.
Fluorescence measurements
All fluorescence measurements were recorded in photon counting mode, by using a 450-W Xenon lamp (Ushio, Tokyo, Japan) as the source of excitation and double monochromators both in excitation and emission (2 nm bandwidths). The emitted light was observed at right angles to the excitation radiation. Photoluminescence spectra of control samples, without protein, were recorded and subtracted from the experimental samples to correct for background interference. All of the spectra were recorded in 20 mM phosphate buffer, pH 7.2 at room temperature (18°C). For 8-anilino-1-naphtalenesulfonic acid (ANS) experiments, dye/enzyme molar ratio ([ANS]/[CA]) was ∼50.
Thermal unfolding of the two enzymes was monitored by recording the fluorescence intensity change at 350 nm for BCAII (1.7 μM) and Ice-CA (3.5 μM) in 20 mM phosphate buffer, pH 7.2 (typical scan rate: 1°C/min). Data were normalized using the pre- and post-transition baseline slopes, as previously described (36).
Time-resolved photoluminescence measurements were performed by time-correlated single photon counting at 18°C, using a protein concentration of 4.4 μM in 20 mM phosphate buffer, pH 7.2. Sample solution was excited at 280 or 295 nm by high-frequency light emitting diodes (LEDs) (∼10 nm bandwidth; ∼1 ns pulse duration; 1 MHz repetition rate). Protein fluorescence was detected at 330, 310, and 380 nm for BCAII and at 321 nm for Ice-CA (see Results and Discussion for details), using a standard right angle geometry (emission band width: 4 nm).
Fluorescence-quenching experiments were performed by analyzing the effect of acrylamide (0–0.6 M) on the emission of the two enzymes (4.4 μM, 20 mM phosphate buffer, pH 7.2). The fluorescence from the two carbonic acid species was quenched by progressive addition of small aliquots of a stock solution (5 M) of the quencher to the fluorimetric cuvette. After correction for dilution of the sample and for inner-filter effects, arising from light absorption by acrylamide, the decrease of fluorescence (calculated as the area under the emission curves) was analyzed according to the Stern-Volmer equation (37).
The above experiments were also performed in time-resolved mode at 18°C, using excitation at 280 nm with fluorescence emission set at 330, 310, or 380 nm for BCAII, and at 321 nm for Ice-CA (emission band width: 4 nm).
Dynamic light-scattering measurements
Protein size measurements were performed on a Zetasizer Nano-S Dynamic Light Scattering (DLS) system (Malvern Instruments Ltd., Malvern, UK), using a 633 nm laser as the excitation source and a back-scattering signal collection. Samples were prepared in 20 mM phosphate buffer, pH 7.2, and the protein concentrations were 2.45 mg/mL for BCAII and 3.1 mg/mL for Ice-CA.
Thermal unfolding experiments were also performed by DLS technique. Data were recorded in the 10–60°C temperature range for Ice-CA and in the 30–80°C range for BCAII to obtain the protein unfolding pattern in size and to calculate the protein melting point.
Statistics
For statistical considerations, the experimental results are the average of five independent measurements. The data shown in the figures throughout the manuscript are the mean values of the data from experiments, and the error bars indicate the standard deviations.
Results and Discussion
Sequence analysis and homology modeling
Sequence alignment between Ice-CA and BCAII revealed a 63% amino acid similarity and highlighted that the psychrophilic enzyme preserves all residues contributing to its activity (Fig. 1): direct zinc ligands (His-94, His-96, His-119), indirect zinc ligands (Gln-92, Glu-117, Thr-199, Asn-243), residues involved in hydrogen-bond-network (His-64, Glu-106, Thr-199), proton shuttling (Gly-63, His-64, Ser-65, and Phe-66), substrate-binding hydrophobic sites (Val-121, Val-143, Leu-198, and Trp-209), and aromatic clusters (Phe-93, Phe-95, Trp-97). Compared with the sequence of BCAII, the Ice-CA sequence shows an increased content of serines (9.7% vs. 6.2%) and glycine (8.5% vs. 7.7%) and a decreased amount of major hydrophobic residues Leu, Val, Ile, Phe, and Met (23.6% vs. 25.4%), positive-charged residues Lys and Arg (8.9% vs. 10.4%), Asn (3.5% vs. 5.0%) and Gln (3.1% vs. 4.6%). Similar features have been observed for other psychrophilic enzymes (4,7).
Figure 1.
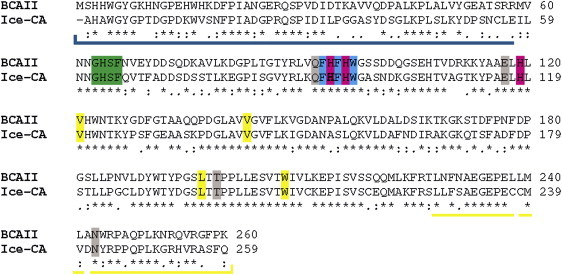
Pairwise sequence alignment of Ice-CA and BCAII. The identical (asterisk) and similar (double points) residues are highlighted. In particular, the direct ligands His-94–His-96–His-119 (magenta), the proton shuttling residues Gly-63–His-64–Ser-65–Phe-66 (green), the binding site Val-121–Val-143–Leu-197–Trp-208 (yellow), the indirect zinc ligands Gln-92–Glu-117–Thr-199–Asn-243 (gray), and the aromatic cluster Phe-93–Phe-95–Trp-97 (cyan) are spotlighted (numeration valid for BCAII). The N-term and C-term regions that form the upper part of the enzyme (“knot” topology) are underlined in blue and yellow, respectively.
The homology modeling of Ice-CA elaborated by SwissModel utilized BCAII as a template, because of the high homology between the two sequences and the high resolution of BCAII structure (1.95 Å) (28). We utilized two classes of verification methods, one mainly focusing on the local environment (PSQS and Verify3D) and the second focusing on the local geometry (ERRAT and ProCheck). The environment-oriented methods PSQS and Verify3D gave scoring values for the model that were higher than those of the template (Table 1). In the geometry-oriented methods the scoring differences between the model and the template were small (Table 1). The Ramachandran plot (not shown) showed that 85.8% of the amino acids in the Ice-CA model were in the most favored regions and 12.8% were in additional allowed regions, whereas 1.4% of the amino acids were in generously allowed regions and no residues were in disallowed regions. All of these tests indicated a high quality of the model, which is thus suitable as a tool for analyzing the 3D structure of Ice-CA.
Table 1.
Quality scores of the Ice-CA model using different quality control programs
| Structure | Verify 3D∗ | PSQS† | ERRAT‡ | ProCheck§ |
|---|---|---|---|---|
| model (Ice-CA) | 91.05 | −0.31 | 92.74 | 85.8 |
| Template (BCAII) | 88.85 | −0.33 | 94.42 | 85.6 |
Data accessed from http://nihserver.mbi.ucla.edu/SAVS and http://www1.jcsg.org/psqs/psqs.cgi.
Percentage of residues with average 3D–1D score 0.2.
The average value for the PSQS scores of a representative set of PDB structures is −0.27.
Percentage of residues with scoring below the 95% limit.
Percentage of residues found in the core region of the Ramachandran plot.
As expected from the high homology between BCA II and Ice-CA, the two- and three-dimensional structures of the two enzymes are highly similar. Ice-CA folds like BCAII, with 12 strands of parallel or antiparallel β-sheet forming a core structure and with 7 α-helices surrounding the β-sheets (Fig. 2). Also, akin to BCAII (19), the Ice-CA molecule can be divided in two distinct domains: the upper part of the protein consists of the N-terminal and C-terminal regions, which wrap around each other (“knot” topology); the lower part comprises the β-sheets core that forms the bottom of the catalytic hole (Fig. 3). In this latter region we found the highest homology degree between Ice-CA and BCAII (Fig. 1). The upper domain of the two enzymes shows the lower homology degree (45% and 47% in the N-terminal and C-terminal regions, respectively). As shown in Fig. 3, this domain closes as a lid the overall catalytic architecture that is kept inside the lower part of the enzyme.
Figure 2.

Schematic view of the three-dimensional structures of Ice-CA (a) and BCAII (b). Dots representation shows the direct ligand His-94–His-96—His-119 (magenta), the main proton shuttling residue His-64 (green), and the binding site Val-121–Val-143–Leu-197–Trp-208 (yellow) (numeration valid for BCAII). The zinc ion is shown as a gray sphere.
Figure 3.
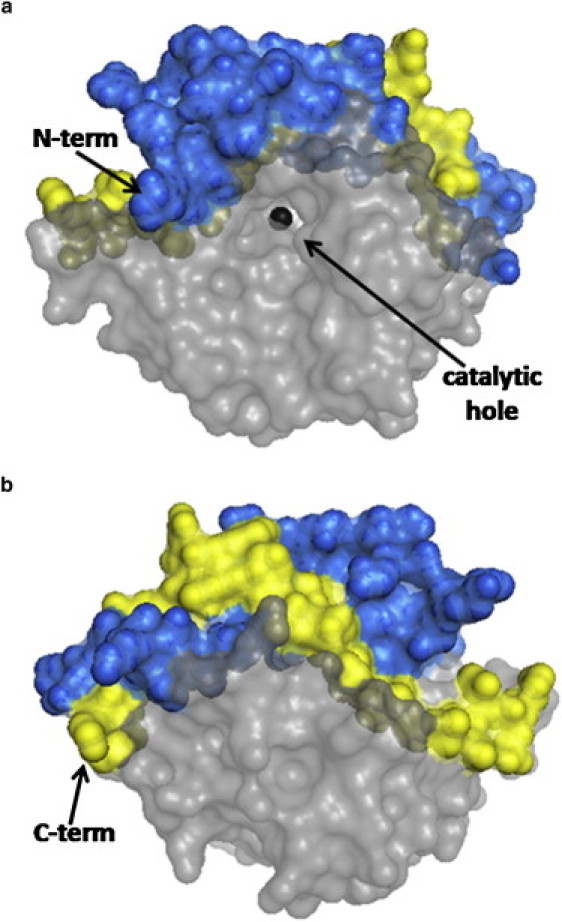
BCAII molecular surface highlighting the upper part of the molecule consisting of the N-terminal (blue spheres) and the C-terminal (yellow spheres) regions. (a) Frontal view of the funnel shaped catalytic hole where the zinc ion is shown. (b) Back view with respect to the catalytic hole. The same features are present in Ice-CA (not shown).
The ASA of Ice-CA and BCAII was then calculated (Table 2). In Ice-CA there is an increase of negatively charged ASA (22.57% vs. 19.99%) and a decrease of positively charged ASA (16.12% vs. 20.64%), accounting for the significantly lower Ice-CA isoelectric point (13,15). The hydrophobic ASAs in Ice-CA and BCAII are quite similar (12.11% vs. 12.69%, respectively).
Table 2.
ASA values of Ice-CA and BCAII
| Total ASA | Positively charged ASA (%) | Negatively charged ASA (%) | Hydrophobic ASA (%) | |
|---|---|---|---|---|
| BCAII | 11421.39 Å2 | 20.64 | 19.99 | 12.69 |
| Ice-CA | 11246.24 Å2 | 16.12 | 22.57 | 12.11 |
Kinetic parameters of Ice-CA and BCAII
The determination of the kinetic parameters for Ice-CA and BCAII revealed that at 2°C the psychrophilic enzyme is more active than its mesophilic counterpart (Table 3). At 2°C Ice-Ca is characterized by a Vmax value comparable to that exhibited by BCAII at 35°C, therefore demonstrating identical enzymatic activities at their respective physiologic temperatures. In contrast, the Km value of Ice-CA at 2°C is lower than that measured for BCAII at 35°C (Table 3) and, in particular, the psychrophilic enzyme Km value increased from 14.53 to 26.55 mM from 2–20°C, whereas BCAII showed an opposite trend, with Km value lower at 35°C than at 2°C (Table 3). Hence, Ice-CA shows a greater catalytic efficiency than that of BCAII at their respective physiologic temperatures. It may be argued that the Ice-CA retains some stable domains that restrict the number of conformational states available for the enzyme-substrate complex, improving the affinity for the substrate. These assumptions, related to the concept of local flexibility/rigidity in psychrophilic enzymes, are consistent with previous observation of these features in intracellular cold-adapted enzymes relying on Km to regulate their activity (7–9).
Table 3.
Kinetic parameters of Ice-CA and BCAII at different temperatures
| Temperature (°C) | Km (mM) | Vmax (μmol CO2min−1 mg−1 protein) | |
|---|---|---|---|
| Ice-CA | 2 | 14.53 ± 2.51 | 6310.0 ± 466.3 |
| BCAII | 2 | 30.71 ± 3.35 | 4880.0 ± 258.4 |
| Ice-CA | 20 | 26.55 ± 3.23 | 7946.0 ± 495.0 |
| BCAII | 20 | 27.97 ± 2.92 | 5803.0 ± 253.1 |
| BCAII | 35 | 23.13 ± 3.87 | 7174.0 ± 401.9 |
Photophysics of Ice-CA and BCAII
Although an in-depth study of BCAII photophysics has not been yet performed (and thus comparable data are not yet directly available), it can be assumed that the high degree of structural similarity between BCAII and HCAII (28) produces the same fluorescence pattern (38). HCAII and its fluorescence properties have been extensively characterized, thanks to its crystal structure determination (30) and by means of detailed site-directed mutagenesis experiments, in which the individual photoluminescence contributions of the seven Trp residues were strongly elucidated (39). This latter study showed that several energy transfer processes among different Trps and strong quenching mechanisms by nearby amino acid residues account for the complex photophysics underlying the HCAII fluorescence emission. In particular, it has been demonstrated that such intramolecular interactions result in a quasi-complete quenching of five of the protein fluorophores, with the major fluorescence emitters Trp-97 (located in the lower part of the enzyme) and Trp-244 (placed in the upper part of the enzyme, in particular in the “knot” topology) in the native state. We have, therefore, utilized such comprehensive data to address the specific contribution of the different fluorescing residues in the case of the psychrophilic enzyme, thus also providing interesting information about its conformational properties. This was obtained by examining the microenvironment of each of the seven Trp residues (considering all the surrounding amino acids contributing to the microenvironment) and then directly comparing the results from the modeling of Ice-CA with those from BCAII (not shown). We found nearly identical structural features for all of the Trp residues, except for Trp-243, which, in the icefish enzyme, is replaced by a Tyr residue. Because this latter, along with all the other Tyr residues, provides negligible contribution to Ice-CA fluorescence (see below), a single emitter, Trp-96, is expected in principle to account for the overall Ice-CA intrinsic fluorescence.
We found experimentally that the Ice-CA photoluminescence spectrum is centered around 321 nm and is quite broad (spectral width = 51 nm) (Fig. 4 a), accounting for the conformational heterogeneity of the enzyme, in agreement with other studies on cold-adapted enzymes (10). Also, consistent with the photophysics of HCAII (39), the photoluminescence emission of Ice-CA was found to be mostly due to Trp residues, with a negligible contribution from the Tyr residues. (No differences in the emission spectrum were recorded by exciting the protein at 270 or 295 nm, in terms of line-shape, emission maximum, and spectral width.) On the other hand, BCAII showed a broad band emission centered around 330 nm, which can be ascribed to Trp-97 and Trp-244 residues (38): Trp-97 is deeply buried in the lower part of the protein interior, whereas Trp-244 is largely exposed to the solvent in the “knot” topology of the upper part of the enzyme (Fig. 3), similar to HCAII protein. Because Ice-CA lacks the second fluorescence emitter (Trp-243), Trp-96 alone accounts for the overall emission of the psychrophilic enzyme. This Trp exhibits a very “blue” emission (λmax = 321 nm), indicating a rather apolar environment, consistent with its function as active site organizer in the protein hydrophobic core (34). In Fig. 4 a, we report the BCAII and Ice-CA fluorescence spectra, together with their difference spectrum to further characterize their photophysical behavior. This difference spectrum shows an emission maximum (λmax = 343 nm) typical of a Trp exposed to the solvent, strongly suggesting that it may account for the fluorescence of Trp-244 (BCAII). This hypothesis was experimentally verified by time-resolved experiments performed on the two enzymes (see below); also, the deconvolution of the BCAII emission spectrum resulted in two bands peaking at 321 and 343 nm (data not shown), in close agreement with the above assumption. This analysis confirmed our predictions, indicating that Trp-97 of BCAII has the same spectral properties as those of the single Ice-CA emitters (Trp-96).
Figure 4.
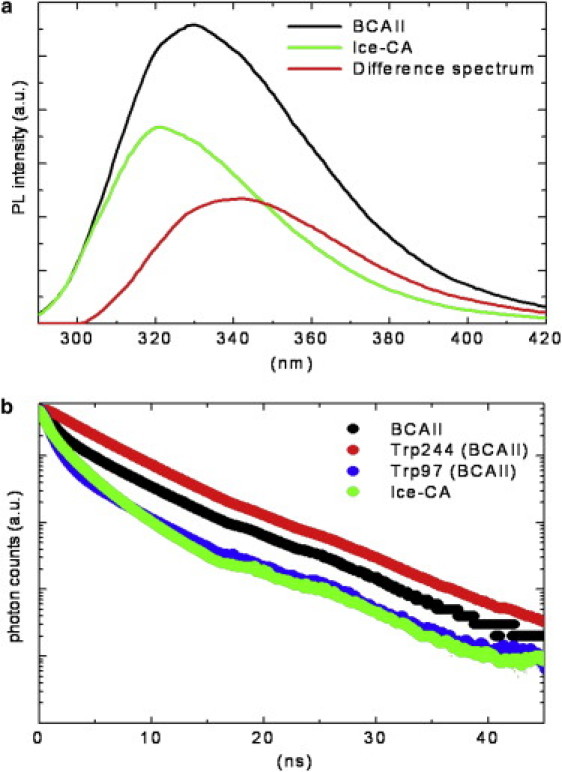
(a) BCAII and Ice-CA photoluminescence spectra obtained by exciting protein samples (2 μM in phosphate buffer, pH 7.2, at 18°C) at 280 nm wavelength, plotted together with their difference spectrum. (b) Time-resolved fluorescence emission registered at 18°C for BCAII, its Trps residues (Trp-244 and Trp-97), and Ice-CA, setting the emission wavelengths at 330, 380, 310, and 321 nm, respectively. 380- and 310-nm emission wavelengths were selected to account for the individual BCAII Trps residues (see text). λexc was 280 nm or 295 nm with no appreciable differences in the observed fluorescence decays. Ordinate scale is logarithmic.
Time-resolved fluorescence measurements were carried out on the two proteins by collecting the emission signals at their fluorescence maximum (λem = 321 and 330 nm for Ice-CA and BCAII, respectively). Moreover, in the case of the bovine enzyme we examined the lifetime decays at two additional emission wavelengths (310 nm and 380 nm) to spectrally separate the contributions of the two fluorescing Trps (at 310 nm most of the BCAII fluorescence signal is due to the effect of Trp-97) (Fig. 4 b). All the fluorescence decays were found to be composed of three exponentials, accounting for the conformational heterogeneity of the two enzymes. BCAII showed a longer fluorescence lifetime (3.4 ± 0.1 ns) than did Ice-CA (2.2 ± 0.1 ns). The fluorescence decay of Trp-244 of the bovine enzyme is slower (4.4 ± 0.1 ns) than its overall emission, whereas Trp-97 is more rapid (2.1 ± 0.1 ns) and, importantly, quite similar to the Ice-CA decay. This confirms the previous assumptions and the modeling data, indicating that the Trp exhibits the same spectral properties in the two enzymes.
Fluorescence quenching
The hydrophobic cluster of Trp-97, in the lower part of the enzyme, has a crucial role in the stability of the molecule, because it serves as an anchor that helps the preorientation of the direct metal ligands and second-shell metal ligands to optimize the tetrahedral geometry for zinc and destabilize alternative geometries (34). For this reason, we further investigated the microenvironment (and the physicochemical properties) of Trp-97 and Trp-96 residues, in BCAII and Ice-CA respectively, selecting all the amino acids within a 6 Å radius sphere centered on the specific Trps (these two structures are superimposed in Fig. 5). These Trps are in contact with the amino acids that integrally belong to the catalytic architecture, in particular with the solvent exposed loop Asn-61→Phe-66, containing the proton shuttling residue His-64, and with the β-sheets core shown in Fig. 3: these structures border the lower contour of the catalytic cavity. The overall microenvironment of the two enzymes is quite similar (Fig. 5), although some noncatalytic amino acidic variation occurs. To probe the dynamic properties of this region, we performed fluorescence quenching experiments, using acrylamide. Such experiments were carried out by both steady-state and time-resolved fluorescence measurements. As reported in Fig. 6, the fluorescence intensities of the two enzymes decreased markedly with increasing quencher concentration. In particular, the luminescence of Ice-CA (Fig. 6 a) was less affected by acrylamide than by its mesophilic counterpart (Fig. 6 b); moreover, in the case of BCAII, a significant blue-shift in the emission band can be observed. A more careful analysis of the quenching process (Fig. 7, a and b) clearly revealed an increasing shift of the emission maximum and spectral width in BCAII toward lower values, at variance with values for Ice-CA. In particular, BCAII emission peak ranged from 330 nm (in absence of acrylamide) to 324 nm (at 0.6 M acrylamide concentration); similarly, the spectral width showed a significant reduction. Importantly, both of these spectral features seem to approach the Ice-CA values, consistent with a higher quenching efficiency of the exposed Trp-244 residue by acrylamide, compared to the values of buried “blue” Trp-97. The Stern-Volmer plots of the two enzymes are reported in Fig. 7 c, revealing a higher slope of BCAII with respect to Ice-CA. The calculated values (1.91 M−1 for BCAII vs. 0.93 M−1 for Ice-CA) clearly suggest an overall accessibility of Trp residues higher in BCAII compared to accessibility of the residues with Ice-CA. On the other hand, a deeper insight in the quenching analysis may be obtained by directly comparing the behavior of Trp96 and Trp97 (along with their environment) in the presence of the quencher. Analogous to time-resolved experiments (see above), we collected the lifetime signals at three different emission wavelengths for BCAII (330, 380, 310 nm) to monitor how acrylamide affected the fluorescence decays of the single emitters (Trp-244 and Trp-97). This analysis revealed that the normalized fluorescence lifetime (τ/τ0) of the different contributions decreased with increasing quencher concentration (Fig. 8), in line with steady-state data and the Stern-Volmer plot. Trp-244 of BCAII is the most quenched emitter, as expected from the three-dimensional structural analysis of the protein. However, it is interesting to observe that the behavior of Trp-97 is quite different from that of Trp-96 (Ice-CA), because the latter is less quenched. Although the environments of such Trps residues in the two CA structures are very similar, the distinct quenching efficiency indicates that the Ice-CA structure near Trp-96 impedes more strongly acrylamide-emitter interaction with respect to the BCAII case. This may be ascribed to a more rigid local domain in Ice-CA close to the aromatic cluster. Such local rigidity in the lower part of Ice-CA, compared to rigidity with BCAII, is likely to compensate the negative effects of flexibility on the catalytic efficiency; acquiring rigidity in regions that organize the active site, but that do not participate in catalytic processes, might allow the enzyme to maintain the Km parameter in a physiologic range.
Figure 5.
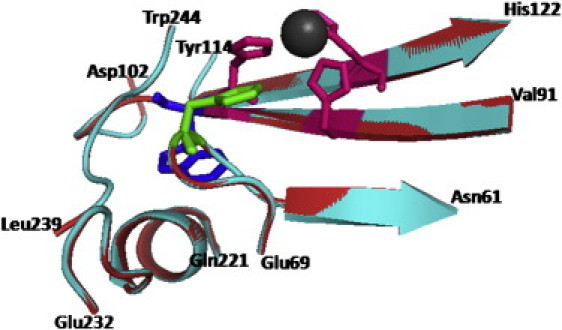
Schematic representation of Ice-CA (cyan) and BCAII (red) lower parts alignment near Trp-96 (Ice-CA) and Trp-97 (BCAII), shown as blue sticks. Additionally, the zinc ion (gray sphere), zinc direct ligands (magenta sticks), and the main proton shuttling residue (green sticks) are shown.
Figure 6.
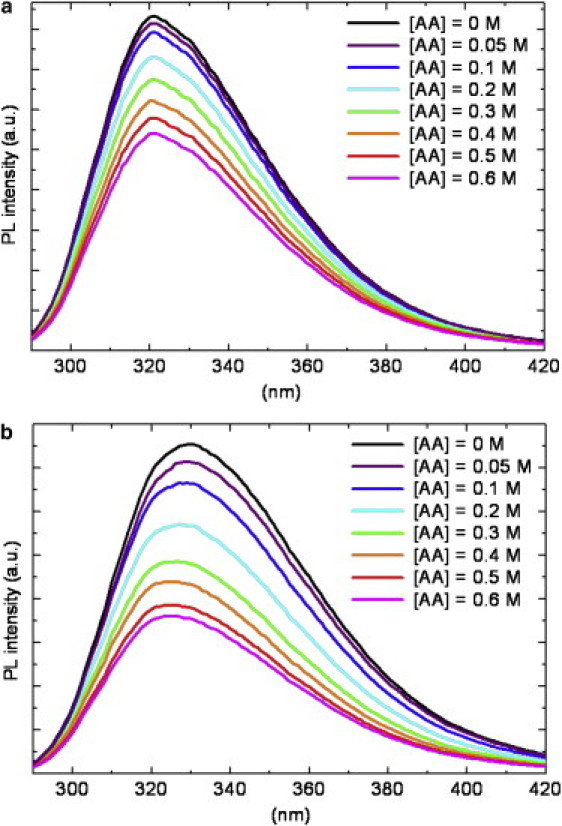
Intrinsic fluorescence quenching by acrylamide (AA): decrease of Ice-CA (a) and BCAII (b) photoluminescence spectra at increasing quencher concentration (λexc = 280 nm).
Figure 7.
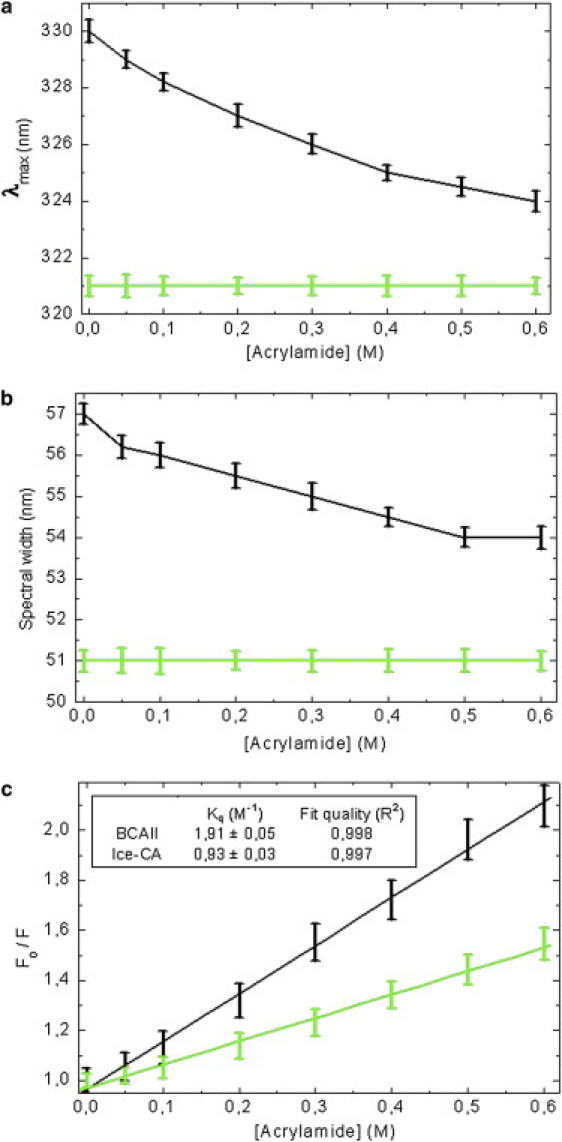
Analysis of the fluorescence quenching process for BCAII (black line) and Ice-CA (green line). Spectral modifications recorded as a function of acrylamide concentration: (a) emission maximum (λmax), (b) spectral width, (c) Stern-Volmer plots and (inset) calculated values of the quenching constant (Kq) for the two enzymes.
Figure 8.
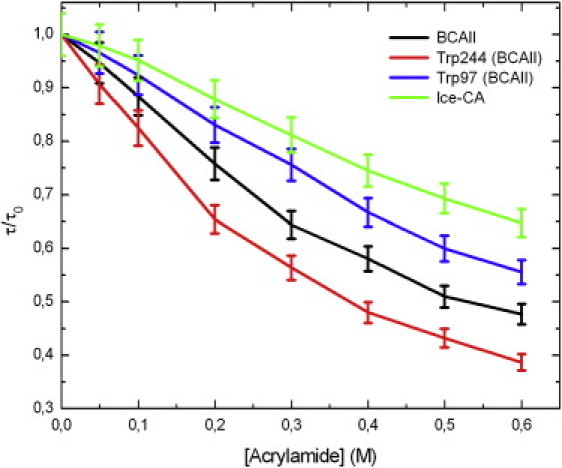
Decrease of the normalized fluorescence lifetime (τ/τ0) with increasing quencher concentration. Data were extracted from fluorescence decays collected at each acrylamide concentration for Ice-CA (λem = 321 nm) and for BCAII and its Trps residues (λem = 330, 380, 310 nm, respectively).
ANS fluorescence
The fluorescent hydrophobic dye ANS is widely used to probe the presence of hydrophobic residue clusters on the surfaces of proteins. Upon binding to these clusters, there is a considerable increase in the fluorescence intensity of the dye, as well as a blue shift of its emission maximum (40,41).
The ASA values for the solvent accessible hydrophobic area in BCAII and Ice-CA were calculated to be quite similar (Table 2); these residues are not organized in patches both in BCAII (42) and in Ice-CA surfaces. The only hydrophobic cluster for ANS interaction, apart from the protein core, is the hydrophobic half of the active-site cavity Val-121, Val-143, Leu-198, and Trp-209 that forms the binding site on the frontal side of the central β-sheet (35). The ASA analysis for this pocket in Ice-CA and BCAII gave comparable values (0.55% and 0.54%, respectively). However, as revealed by ANS experiments (Fig. 9), BCAII exhibits a weak binding of the dye, suggesting a minimum interaction between ANS and the BCAII binding site, whereas a strong enhancement of ANS fluorescence was recorded for Ice-CA, underlining a significant ANS accessibility to the binding site in the cold-adapted enzyme. In particular, we found a BCAII/Ice-CA ANS intensity ratio of 0.22, meaning that ANS binding to the psycrophilic enzyme is 4.5 times more effective than ANS binding to BCAII.
Figure 9.
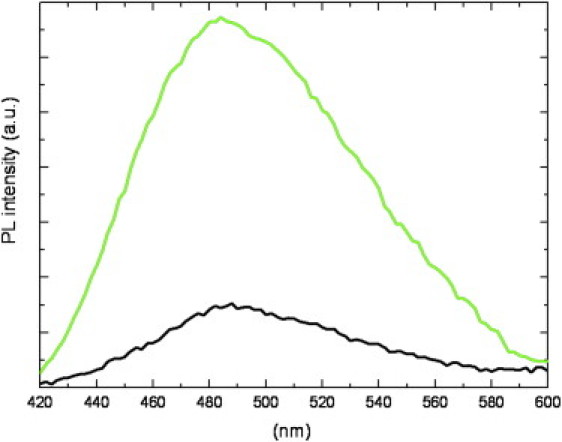
ANS fluorescence spectra (λexc = 390 nm) in the presence of Ice-CA (green) and BCAII (black) in the native state. ANS/enzyme molar ratio ([ANS]/[CA]) was ∼50.
The high solvent exposure of the substrate binding site in Ice-CA is achieved by protein backbone motions around the catalytic hole (flexibility). Because we observed a more rigid domain in the lower part of the enzyme with respect to its mesophilic counterpart, local flexibility allowing an increase of substrate binding site exposure in Ice-CA is expected to occur in the “knot” structure of the N-terminal and C-terminal regions that compose the upper part of the enzyme (Figs. 2 and 3). Such a domain borders the upper part of the catalytic hole and contains vital information for the correct folding of the enzyme, leading to the formation of the active site (21); in BCA II, this domain is significantly more rigid than that of Ice-CA. This structural feature is strengthened by the evidence that the “knot topology” of BCAII is tighter and more stable compared to that of another mesophilic enzyme such as HCA II (28).
Thermal unfolding and thermodependence of activity
Generally, an increase of the conformational flexibility of cold-adapted enzymes causes a low thermal stability of the molecular structure (11,12,43). To investigate this behavior in Ice-CA, we performed thermal unfolding experiments and thermodependence studies of enzyme activities, because these features are in close connection (Fig. 10). Thermal unfolding measurements were carried out by intrinsic fluorescence. We found the BCAII melting point (Tm) to be near 66°C, consistent with results in previous studies (44,45), and the maximum of its activity to be near 35°C. On the other hand, in Ice-CA we recorded a Tm of ∼30°C and a maximum activity of near 20°C, reflecting its weak stability and highlighting a clear link in psychrophilic enzymes between temperature adaptation of optimal activity and decreased stability. Thermally induced aggregation of the two enzymes was also monitored by a dynamic light-scattering technique, revealing a trend very similar to that in fluorescence studies.
Figure 10.
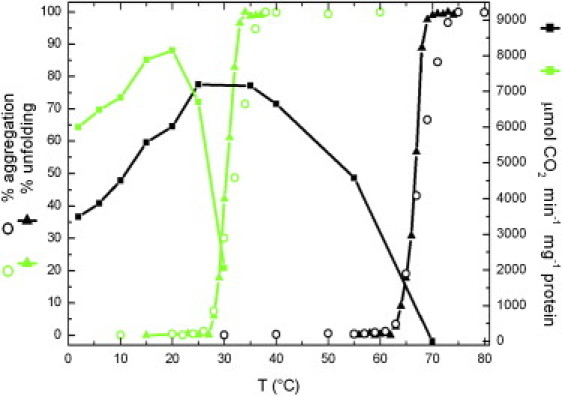
Unfolding, aggregation, and activity dependence of Ice-CA (green) and BCAII (black) as a function of temperature. Thermal unfolding (left axis) was recorded by intrinsic fluorescence for Ice-CA and BCAII. Thermally induced aggregation (left axis) was monitored by DLS. The specific activity of the enzymatic reaction, expressed as μmol CO2 min−1 mg−1 protein (right axis), was measured in saturating substrate conditions.
In Ice-CA, the temperature corresponding to 50% of activity loss was reached only 3°C before its Tm (Fig. 10), whereas, for BCAII, this shift was higher (∼13°C). In line with our photophysical data (see above), this suggests that, in Ice-CA, the lower part of the enzyme is the most stable domain, its unfolding leading to a break of catalytic architectures with activity loss. In BCAII, although the lower domain is more flexible than that of its psycrophilic counterpart, the unfolding of the lower part (Trp-97) and of the upper part (Trp-244) occur long after enzyme inactivation. This may be explained by considering the upper part of BCAII the most rigid domain found in this mesophilic enzyme: while some thermolabile active site regions are inactivated, the “knot topology” continues to keep the entire structure extremely packed, causing a shift in Tm.
Conclusions
Mesophilic carbonic anhydrases II are some of the most active of the known enzymes, with a catalytic efficiency approaching the limit of diffusion-controlled processes (46). Psychrophilic organisms have evolved several strategies to overcome the effect of low temperatures on catalytic efficiency, including producing enzymes in which the reaction rate tends to become temperature-independent and approaches diffusion-control. This latter strategy is achieved in Ice-CA, where kcat/Km ratio increases by means of a fine balance between local flexibility/rigidity. We have explored this issue in Ice-CA in a comparative manner with respect to BCAII, finding in the psychrophilic enzyme a higher exposure to solvent of the substrate binding site kept inside the catalytic funnel-shaped channel. This enzymatic region has a crucial role in catalysis: interaction with this cluster is assumed to lead to an electrophilic activation of the carbon dioxide for the subsequent nucleophilic attack of the zinc-bound hydroxide. A part of the binding energy of the in situ-formed carboxylate in this hydrophobic cavity might then be utilized to lower the activation energy for dissociation of the product (30,35). In Ice-CA, we demonstrated a gain in flexibility in the upper part of the enzyme that controls the correct folding of the catalytic architecture, and an increased rigidity in the lower part of the enzyme that anchors the catalytic β-strands into the hydrophobic core, stabilizing their conformation when compared to results with the mesophilic counterpart. In particular, the upper domain of the two enzymes shows the lower homology degree. This strategy is in agreement with hypotheses suggesting that a local rigidity can be beneficial for keeping Km values in a physiologic range (4).
The results obtained for the two carbonic anhydrases, adapted to different thermal habitats, clearly establish a link between activity, flexibility, and rigidity that has different possible implications. The first pertains to adaptation studies of psycrophilic organisms to different life conditions. The second, pertaining to protein engineering, is the idea that desired patterns of flexibility might be designed and inserted into a protein by specific mutation, as a means of either selecting or regulating a given function. The third is related to rational design of novel therapeutic inhibitors and activators on a class of enzymes involved in a range of disorders, including edema, glaucoma, obesity, cancer, epilepsy, osteoporosis, and, recently, Alzheimer's disease, aging, and infection (46). If dynamics are important for enzyme function, and if the protein regions most responsible for the motion can be identified, they might represent a potential target for drug binding and inhibition.
Acknowledgments
The authors gratefully acknowledge E. D'Amone and V. Fiorelli for their expert technical assistance. This work was supported by the Italian Ministry of Research through MIUR “FIRB” projects (RBLA03ER38_001 and RBNE03FMCJ_003). This work is also in the framework of the Italian National Program for Antarctic Research.
References
- 1.Siddiqui K.S., Cavicchioli R. Cold-adapted enzymes. Annu. Rev. Biochem. 2006;75:403–433. doi: 10.1146/annurev.biochem.75.103004.142723. [DOI] [PubMed] [Google Scholar]
- 2.Fields P.A. Protein function at thermal extremes: balancing stability and flexibility. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2001;129:417–431. doi: 10.1016/s1095-6433(00)00359-7. [DOI] [PubMed] [Google Scholar]
- 3.Feller G. Molecular adaptations to cold in psychrophilic enzymes. Cell. Mol. Life Sci. 2003;60:648–662. doi: 10.1007/s00018-003-2155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Georlette D., Blaise V., Collins T., D'Amico S., Gratia E. Some like it cold: biocatalysis at low temperatures. FEMS Microbiol. Rev. 2004;28:25–42. doi: 10.1016/j.femsre.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Papaleo E., Riccardi L., Villa C., Fantucci P., De Gioia L. Flexibility and enzymatic cold-adaptation: a comparative molecular dynamics investigation of the elastase family. Biochim. Biophys. Acta. 2006;1764:1397–1406. doi: 10.1016/j.bbapap.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Feller G., Gerday C. Psychrophilic enzymes: molecular basis of cold adaptation. Cell. Mol. Life Sci. 1997;53:830–841. doi: 10.1007/s000180050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smalås A.O., Leiros H.K., Os V., Willassen N.P. Cold adapted enzymes. Biotechnol. Annu. Rev. 2000;6:1–57. doi: 10.1016/s1387-2656(00)06018-x. [DOI] [PubMed] [Google Scholar]
- 8.Lonhienne T., Gerday C., Feller G. Psychrophilic enzymes: revisiting the thermodynamic parameters of activation may explain local flexibility. Biochim. Biophys. Acta. 2000;1543:1–10. doi: 10.1016/s0167-4838(00)00210-7. [DOI] [PubMed] [Google Scholar]
- 9.Hoyoux A., Jennes I., Dubois P., Genicot S., Dubail F. Cold-adapted beta-galactosidase from the Antarctic psychrophile Pseudoalteromonas haloplanktis. Appl. Environ. Microbiol. 2001;67:1529–1535. doi: 10.1128/AEM.67.4.1529-1535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D'Amico S., Claverie P., Collins T., Georlette D., Gratia E. Molecular basis of cold adaptation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2002;357:917–925. doi: 10.1098/rstb.2002.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins T., Meuwis M.A., Gerday C., Feller G. Activity, stability and flexibility in glycosidases adapted to extreme thermal environments. J. Mol. Biol. 2003;328:419–428. doi: 10.1016/s0022-2836(03)00287-0. [DOI] [PubMed] [Google Scholar]
- 12.D'Amico S., Marx J.C., Gerday C., Feller G. Activity-stability relationships in extremophilic enzymes. J. Biol. Chem. 2003;278:7891–7896. doi: 10.1074/jbc.M212508200. [DOI] [PubMed] [Google Scholar]
- 13.Rizzello A., Ciardiello M.A., Acierno R., Carratore V., Verri T. Biochemical characterization of a S-glutathionylated carbonic anhydrase isolated from gills of the Antarctic icefish Chionodraco hamatus. Protein J. 2007;26:335–348. doi: 10.1007/s10930-007-9076-1. [DOI] [PubMed] [Google Scholar]
- 14.Maffia M., Rizzello A., Acierno R., Rollo M., Chiloiro R. Carbonic anhydrase activity in tissues of the icefish Chionodraco hamatus and of the red-blooded teleosts Trematomus bernacchii and Anguilla anguilla. J. Exp. Biol. 2001;204:3983–3992. doi: 10.1242/jeb.204.22.3983. [DOI] [PubMed] [Google Scholar]
- 15.Marino S., Hayakawa K., Hatada K., Benfatto M., Rizzello A. Structural features that govern enzymatic activity in carbonic anhydrase from a low-temperature adapted fish, Chionodraco hamatus. Biophys. J. 2007;93:2781–2790. doi: 10.1529/biophysj.107.107540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCoy L.F., Jr., Wong K.P. Renaturation of bovine erythrocyte carbonic anhydrase B denatured by acid, heat, and detergent. Biochemistry. 1981;20:3062–3067. doi: 10.1021/bi00514a012. [DOI] [PubMed] [Google Scholar]
- 17.Cleland J.L., Wang D.I. Refolding and aggregation of bovine carbonic anhydrase B: quasi-elastic light scattering analysis. Biochemistry. 1990;29:11072–11078. doi: 10.1021/bi00502a009. [DOI] [PubMed] [Google Scholar]
- 18.Hammarström P., Kalman B., Jonsson B.H., Carlsson U. Pyrene excimer fluorescence as a proximity probe for investigation of residual structure in the unfolded state of human carbonic anhydrase II. FEBS Lett. 1997;420:63–68. doi: 10.1016/s0014-5793(97)01488-9. [DOI] [PubMed] [Google Scholar]
- 19.Mårtensson L.G., Karlsson M., Carlsson U. Dramatic stabilization of the native state of human carbonic anhydrase II by an engineered disulfide bond. Biochemistry. 2002;41:15867–15875. doi: 10.1021/bi020433+. [DOI] [PubMed] [Google Scholar]
- 20.Carlsson U., Jonsson B.H. Folding and stability of human carbonic anhydrase II. EXS. 2000;90:241–259. doi: 10.1007/978-3-0348-8446-4_13. [DOI] [PubMed] [Google Scholar]
- 21.Hammarström P., Carlsson U. Is the unfolded state the Rosetta Stone of the protein folding problem? Biochem. Biophys. Res. Commun. 2000;276:393–398. doi: 10.1006/bbrc.2000.3360. [DOI] [PubMed] [Google Scholar]
- 22.Ohta S., Alam M.T., Arakawa H., Ikai A. Origin of mechanical strength of bovine carbonic anhydrase studied by molecular dynamics simulation. Biophys. J. 2004;87:4007–4020. doi: 10.1529/biophysj.104.045138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindskog S., Malmström B.G. Metal binding and catalytic activity in bovine carbonic anhydrase. J. Biol. Chem. 1962;237:1129–1137. [PubMed] [Google Scholar]
- 24.Alam M.T., Yamadaa T., Carlsson U., Ikai A. The importance of being knotted: effects of the C-terminal knot structure on enzymatic and mechanical properties of bovine carbonic anhydrase II. FEBS Lett. 2002;519:35–40. doi: 10.1016/s0014-5793(02)02693-5. [DOI] [PubMed] [Google Scholar]
- 25.Persson M., Lindgren M., Hammarström P., Svensson M., Jonsson B.H. EPR mapping of interactions between spin-labeled variants of human carbonic anhydrase II and GroEL: evidence for increased flexibility of the hydrophobic core by the interaction. Biochemistry. 1999;38:432–441. doi: 10.1021/bi981442e. [DOI] [PubMed] [Google Scholar]
- 26.Yazdanparast R., Khodarahmi R. Evaluation of artificial chaperoning behavior of an insoluble cyclodextrin-rich copolymer: solid-phase assisted refolding of carbonic anhydrase. Int. J. Biol. Macromol. 2007;40:319–326. doi: 10.1016/j.ijbiomac.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Karlsson M., Mårtensson L.G., Jonsson B.H., Carlsson U. Adsorption of human carbonic anhydrase II variants to silica nanoparticles occur stepwise: binding is followed by successive conformational changes to a molten-globule-like state. Langmuir. 2000;16:8470–8479. [Google Scholar]
- 28.Saito R., Sato T., Ikai A., Tanaka N. Structure of bovine carbonic anhydrase II at 1.95 A resolution. Acta Crystallogr. D Biol. Crystallogr. 2004;60:792–795. doi: 10.1107/S0907444904003166. [DOI] [PubMed] [Google Scholar]
- 29.Huang C.C., Lesburg C.A., Kiefe L.L., Fierke C.A., Christianson D.W. Reversal of the hydrogen bond to zinc ligand histidine-119 dramatically diminishes catalysis and enhances metal equilibration kinetics in carbonic anhydrase II. Biochemistry. 1996;35:3439–3446. doi: 10.1021/bi9526692. [DOI] [PubMed] [Google Scholar]
- 30.Håkansson K., Carlsson M., Svensson L.A., Liljas A.J. Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J. Mol. Biol. 1992;227:1192–1204. doi: 10.1016/0022-2836(92)90531-n. [DOI] [PubMed] [Google Scholar]
- 31.Merz K.M., Jr. Insights into the function of the zinc hydroxide-Thr-199-Glu-106 hydrogen bonding network in carbonic anhydrases. J. Mol. Biol. 1990;214:799–802. doi: 10.1016/0022-2836(90)90333-H. [DOI] [PubMed] [Google Scholar]
- 32.Krebs J.F., Fierke C.A., Alexander R.S., Christianson D.W. Conformational mobility of His-64 in the Thr-200—Ser mutant of human carbonic anhydrase II. Biochemistry. 1991;30:9153–9160. doi: 10.1021/bi00102a005. [DOI] [PubMed] [Google Scholar]
- 33.Thoms S. Hydrogen bonds and the catalytic mechanism of human carbonic anhydrase II. J. Theor. Biol. 2002;215:399–404. doi: 10.1006/jtbi.2002.2528. [DOI] [PubMed] [Google Scholar]
- 34.Cox J.D., Hunt J.A., Compher K.M., Fierke C.A., Christianson D.W. Structural influence of hydrophobic core residues on metal binding and specificity in carbonic anhydrase II. Biochemistry. 2000;39:13687–13694. doi: 10.1021/bi001649j. [DOI] [PubMed] [Google Scholar]
- 35.Loferer M.J., Tautermann C.S., Loeffler H.H., Liedl K.R. Influence of backbone conformations of human carbonic anhydrase II on carbon dioxide hydration: hydration pathways and binding of bicarbonate. J. Am. Chem. Soc. 2003;125:8921–8927. doi: 10.1021/ja035072f. [DOI] [PubMed] [Google Scholar]
- 36.Pace C.N. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 37.Lakowicz J.R. 2nd ed. Plenum Press; New York: 2001. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 38.Montich G.G. Partly folded states of bovine carbonic anhydrase interact with zwitterionic and anionic lipid membranes. Biochim. Biophys. Acta. 2000;1468:115–126. doi: 10.1016/s0005-2736(00)00250-9. [DOI] [PubMed] [Google Scholar]
- 39.Mårtensson L.G., Jonasson P., Freskgård P.-O., Svensson M., Carlsson U. Contribution of individual tryptophan residues to the fluorescence spectrum of native and denatured forms of human carbonic anhydrase II. Biochemistry. 1995;34:1011–1021. doi: 10.1021/bi00003a036. [DOI] [PubMed] [Google Scholar]
- 40.Stryer L. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J. Mol. Biol. 1965;13:482–495. doi: 10.1016/s0022-2836(65)80111-5. [DOI] [PubMed] [Google Scholar]
- 41.Semisotnov G.V., Radionova N.A., Razgulyaev O.I., Uversky V.N., Gripas A.F. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–128. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 42.Kundu B., Guptasarma P. Use of a hydrophobic dye to indirectly probe the structural organization and conformational plasticity of molecules in amorphous aggregates of carbonic anhydrase. Biochem. Biophys. Res. Commun. 2002;293:572–577. doi: 10.1016/S0006-291X(02)00257-7. [DOI] [PubMed] [Google Scholar]
- 43.Georlette D., Damien B., Blaise V., Depiereux E., Uversky V.N. Structural and functional adaptations to extreme temperatures in psychrophilic, mesophilic, and thermophilic DNA ligases. J. Biol. Chem. 2003;278:37015–37023. doi: 10.1074/jbc.M305142200. [DOI] [PubMed] [Google Scholar]
- 44.Sarraf N.S., Saboury A.A., Ranjbar B., Moosavi-Movahedi A.A. Structural and functional changes of bovine carbonic anhydrase as a consequence of temperature. Acta Biochim. Pol. 2004;51:665–671. [PubMed] [Google Scholar]
- 45.Lavecchia R., Zugaro M. Thermal denaturation of erythrocyte carbonic anhydrase. FEBS Lett. 1991;292:162–164. doi: 10.1016/0014-5793(91)80858-z. [DOI] [PubMed] [Google Scholar]
- 46.Supuran C.T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]