

Abstract
Cholesterol is a major component of biological membranes and is known to affect vesicle fusion. However, the mechanism by which cholesterol modulates SNARE-dependent intracellular fusion is not well understood. Using the fluorescence assay and dye-labeled SNAREs and the fluorescent lipids, we dissected cholesterol effects on individual fusion steps including SNARE complex formation, hemifusion, pore formation, and pore dilation. At physiological high concentrations, cholesterol stimulated hemifusion as much as 30-fold, but its stimulatory effect diminished to 10-fold and three-fold for subsequent pore formation and pore expansion at 40 mol %, respectively. The results show that cholesterol serves as a strong stimulator for hemifusion but acts as mild stimulators for pore opening and expansion. Strong stimulation of hemifusion and mild stimulation of pore formation are consistent with the fusion model based on the intrinsic negative curvature of cholesterol. However, even a milder effect of cholesterol on pore expansion is contradictory to such a simple curvature-based prediction. Thus, we speculate that cholesterol also affects the conformation of the transmembrane domains of SNAREs, which modulates the fusion kinetics.
Introduction
Vital cellular processes such as neurotransmission, fertilization, and trafficking require membrane fusion (1–3). SNAREs are believed to be the core components of the intracellular membrane fusion machinery (4–6). Molecular recognition of a v-SNARE on a cargo vesicle with a t-SNARE on the target membrane leads to formation of a core complex (7–11). This, in turn, brings about the assembly of a supramolecular structure of multiple SNARE complexes that spans two opposing membranes (12,13). There is overwhelming evidence that SNARE-mediated fusion transits through a hemifusion state in which lipids in the proximal leaflets are allowed to be intermixed but those on the distal leaflets are not (14–20).
Cholesterol is a major constituent of the biological membranes, occupying significant fractions of lipids in cellular membranes. In synaptic vesicles, for example, cholesterol is a dominant lipid species whose content reaches as much as 40% of the total lipids (21). Cholesterol and its analogs are shown to play important roles in modulating exocytosis. Depletion of cholesterol out of isolated cortical vesicles from sea urchin eggs (22), or out of a cell-free exocytosis system from PC-12 cells (23), significantly impairs the release, strongly suggesting that cholesterol is essential for Ca2+-regulated exocytosis. Extraction of ergosterol is shown to inhibit plasma membrane fusion for pheromone secretion in mating yeast (24), again indicative of a positive role.
Cholesterol may modulate SNARE-dependent membrane fusion in two different ways. First, cholesterol may regulate the lateral distribution of SNAREs in the membrane (23,25). Recently, it has been hotly debated if the distinctive domains called “lipid rafts”, which are rich in cholesterol and sphingomyelin, exist in cell membranes (26,27). Nevertheless, it was proposed that SNAREs partition into the lipid rafts (28,29). The idea is now disputed by other experimental results (30). Cholesterol is shown to promote crowding of the neuronal t-SNARE Syntaxin via the protein-protein interaction in the isolated plasma membrane of PC-12 cells without involving the rafts (23). Potential relevance of such cholesterol-driven Syntaxin clusters to vesicle fusion was demonstrated experimentally (23). Second, cholesterol is traditionally viewed as a lipid of the negative spontaneous curvature (31,32). Thus, cholesterol can either promote or inhibit the fusion steps depending on the curvature characteristics of the intermediates. For example, cholesterol promotes hemifusion, but inhibits pore formation in a protein-free membrane fusion. For SNARE-mediated fusion, it has been shown that the replacement of cholesterol by other amphiphilic molecules of negative curvature restores the extent of fusion in cortical vesicles (22), suggesting that the curvature plays a role.
Cyclodextrin derivatives have the capacity of extracting cholesterol from the cell membranes (33–35). Until now, information of cholesterol function in exocytosis was based almost entirely on chemical extraction of cholesterol by methyl-β-cyclodextrin. With this method, however, the selective extraction of cholesterol from the desired membrane was nearly impossible. Moreover, the results were often ambiguous because cholesterol depletion may affect other factors, including the regulatory proteins, whereby making the delineation of exact roles of cholesterol in exocytosis is difficult.
In this work, we attempted to analyze cholesterol function in SNARE-mediated fusion in a more defined and controllable setting. We used an in vitro fluorescence fusion assay in which fluorescence-labeled recombinant SNAREs, as well as fluorescent lipids, were used (12). This method made it possible to separately study SNARE complex formation, inner and outer leaflet mixing, and pore dilation (Fig. 1 A). This method allowed us to dissect the effects of cholesterol on individual fusion steps. The results show that cholesterol modulates individual fusion steps differently. The maximal positive effect is on hemifusion, and the negative effect is on pore formation and expansion. The analysis suggests that the cholesterol effects on SNARE-mediated fusion cannot be fully accounted for by its spontaneous curvature. Cholesterol seems to directly modulate the conformation of the transmembrane domains of SNAREs, thereby differentially modulating individual fusion steps.
Figure 1.
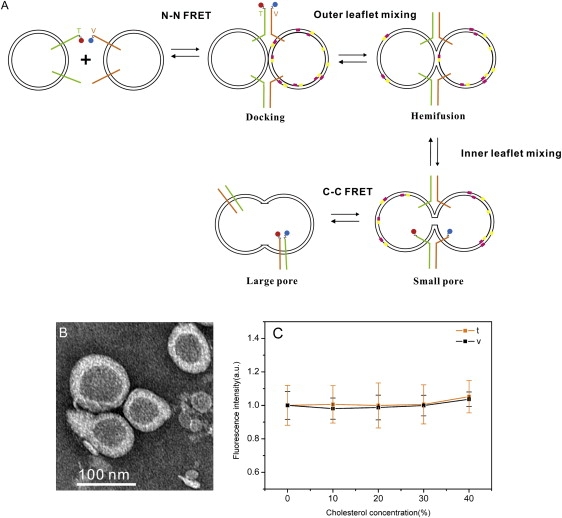
A proposed fusion pathway and the preliminary characterization of SNARE-reconstituted vesicles. (A) A pathway for SNARE-mediated fusion is shown. The green and brown sticks represent v- and t-SNAREs, respectively. The red ball is the fluorescence donor and the blue is the fluorescence acceptor. The pink and yellow dots on the membrane are NBD-PS and rhodamine-PE, respectively. (B) An electron micrograph of the v-SNARE-reconstituted vesicles containing 40% of cholesterol. The vesicles were stained with 1% phosphotungstic acid on the carbon grids. The size of the vesicle averaged over more than 100 individual vesicles was 90 ± 15 nm, which was nearly the same as the vesicles without cholesterol. The small circles on the left were the staining artifacts. (C) The variation of the reconstitution efficiency as a function of cholesterol. After the reconstitution of the dye-labeled SNAREs, the fluorescence intensities were measured. The protein reconstitution efficiency was nearly invariant for all cholesterol concentrations.
Methods
Plasmids and site-directed mutagenesis
DNA sequences encoding Sso1pHT (amino acids 185–290 of Sso1p) and Snc2p (amino acids 1–115) were inserted into the pGEX-KG vector between EcoRI and HINDIII sites as N-terminal glutathione S-transferase (GST) fusion proteins. Sec9c (amino acids 401–651 of Sec9) was inserted into pET-24b(+) between NdeI and XhoI sites as a C-terminal His6-tagged protein. To introduce a unique cysteine residue for the specific dye attachment, native cysteine 266 of Sso1pHT was mutated to alanine. A QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) was used to generate cysteine. DNA sequences were confirmed by the Iowa State University DNA Facility.
Protein expression and purification
Expressions of the yeast SNARE proteins were achieved by using Escherichia coli BL21 (Novagene, San Diego, CA). The cells were incubated at 37°C for ∼3–4 h in LB medium with 100 μg/μL ampicillin. After adding IPTG (isopropyl-β-D-thiogalactopyranoside) for induction, the cells were grown for an additional 5 h at 16°C. Cells were then harvested through centrifugation at 6,000 rpm for 10 min. Purification of the GST fusion proteins was carried out through affinity chromatography using glutathione-agarose beads (Sigma, St. Louis, MO). Cell pellets were resuspended in PBS buffer (phosphate-buffered saline, pH 7.4, with 0.5% Triton X-100 (v/v)) with 2 mM 4-(2-aminoethyl) benzenesulfonyl fluoride (ABESF), and 5 mM dithiothreitol (DTT). Cells were then broken by sonication and centrifuged at 15,000 rpm for 20 min at 4°C. The supernatant was mixed with glutathione-agarose beads in the resuspension buffer and nutated at 4°C for 120 min. The protein-bound GST beads were washed excessively with washing buffer (PBS, pH7.4), and the protein was cleaved by thrombin in cleavage buffer (50 mM Tris-HCl, 150 mM NaCl, pH 8.0). We added 0.8% n-octylglucoside (OG) in the cleavage buffer for Sso1pHT and full-length Snc2p. ABESF was added to the protein after the cleavage (2 mM final concentration).
The His6-tagged protein Sec9c was expressed in E. coli Rosetta (DE3) pLysS. For purification, cells were resuspended in lysis buffer (PBS buffer with 20 mM imidazole, 0.5% Triton X-100, 2 mM AEBSF, pH 7.4). After sonication, the supernatant was mixed with nickel-nitrilotriacetic acid-agarose beads (Qiagen, Valencia, CA) in lysis buffer. The mixture was nutated for binding at 4°C for 120 min. After binding, the beads were washed six times with washing buffer (PBS buffer with 20 mM imidazole, pH 8.0). They were then washed with another washing buffer (PBS buffer with 250 mM imidazole, pH 8.0). All proteins contained 10% glycerol as a cryoprotectant and were kept at −80°C.
Making vesicles and reconstituting SNAREs
A mixture of POPC (1-palmitoyl-2-dioleoyl-sn-glycero-3-phosphatidylcholine), DOPS (1,2-dioleoyl-sn-glycero-3-phosphatidylserine), and cholesterol in chloroform, in an appropriate proportion, was dried in a vacuum and resuspended in a buffer (25 mM HEPES/KOH, 100 mM KCl, pH 7.4). Protein-free large, unilamellar liposomes (∼100 nm in diameter) were prepared by extrusion through polycarbonate filters (Avanti Polar Lipids, Alabaster, AL) after more than 10 iterations of freezing and thawing vesicles to make them big enough. For the lipid-mixing assay, the liposomes containing POPC, DOPS, cholesterol, NBD-PS (1,2-dioleoyl -sn-glycero-3-phosphoserine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl)), and rhodamine-PE (1,2-dioleoyl-snglycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl)) in the molar ratio of 82:15:0:1.5:1.5, 72:15:10:1.5:1.5, 62:15:20:1.5:1.5, 52:15:30:1.5:1.5, and 42:15:40:1.5:1.5 were prepared following the procedure described above. Sso1pHT was reconstituted to the nonfluorescent vesicles whereas Snc2p was reconstituted to the fluorescent vesicles. Proteins were mixed with vesicles at a protein/lipid molar ratio of 1:200 at 4°C for 20 min. The protein/lipid mixture was diluted two times to make the concentration of OG below the critical micelle concentration. The liposomes were then dialyzed overnight against dialysis buffer (25 mM HEPES/KOH, 100 KCl, 5% (w/v) glycerin, pH 7.4) at 4°C. After reconstitution, the samples were centrifuged at 5,000 × g for 10 min to get rid of any protein-lipid aggregates.
Total and inner leaflet fusion assay
Sso1pHT, Snc2p, and Sec9c were used for lipid-fusion assay. In a quartz cuvette, the Sso1pHT-reconstuted vesicles were mixed with Snc2p-reconstituted vesicles. The total lipid concentration was 0.4 mM. The fusion reaction was initiated by adding Sec9c and HEPES buffer (pH7.4, with 5% glycerol (w/v), HEPES, KCl), which made the total volume 100 μL. The fluorescence signal from donor emission frequency was detected by a Varian Cary Eclipse model of a fluorescence spectrophotometer under the temperature of 35°C. To reach the maximum fluorescence intensity (MFI), 1% reduced-Triton solution was applied after 3600 s.
The inner leaflet mixing assay was modified from the method developed by Meers et al. (36). The method is based on the fact that sodium dithionite reacts more rapidly with NBDs in the outer leaflet than those in the inner leaflet. By controlling the time and amount of dithionite the reaction could be limited to the outer leaflet. Small aliquots of 100 mM sodium dithionite in 50 mM Tris buffer (pH 10) were added to the v-SNARE vesicles until a desired reduction of NBD was achieved. The reaction was monitored at 35°C by scanning the fluorescence signal for 15 min from 500 to 700 nm with the excitation at 460 nm. Typically, the reduction was complete in 10 min and no more change of the spectrum was observed. The vesicles with reduced NBDs in the outer leaflets were subjected to the lipid-mixing assay described above. To make sure that the percentage of MFI was independent of the extent of the NBD reduction, the inner leaflet mixing assay was carried out at the level in which 55% and 65% of NBDs were reduced.
The change of the fluorescence intensity in time was fitted with the equation,
| (1) |
where [P] is the concentration of product, C0 is the initial concentration of the reactant, and k1 is the rate constant. The equation represents the second-order kinetics.
N-N and C-C FRET assay
Sso1pHT E185C and Sso1pHT R290C proteins were labeled with Cy3 maleimide, and Snc2p P13C and Snc2p S115C proteins were labeled with Cy5 maleimide (Amersham, Piscataway, NJ) after thrombin cleavage. The free dyes were removed from the proteins by using PD-10 desalting columns (Amersham). These proteins were reconstituted into vesicles with the lipid-to-protein ratio of 200:1. The fusion assay was carried out under identical condition to the procedure described above. The detection was achieved at two channels with the excitation wavelength of 555 nm and emission wavelengths of 570 nm and 668 nm.
Results
In this work, we studied a yeast SNARE family that plays a role in trafficking. Yeast SNAREs share functional and structural similarities with neuronal SNAREs that are involved in the neurotransmitter release. The t-SNARE proteins Sso1p and Sec9 are the counterparts of neuronal Syntaxin 1A and SNAP-25, respectively. Sso1p has a transmembrane domain that anchors the t-SNARE to the membrane. Snc2p is a v-SNARE, a neuronal synaptobrevin (VAMP) analog (2,37,38). We used shorter versions of t-SNAREs Sso1pHT and Sec9c for simplicity (39). Sso1pHT lacks the N-terminal regulatory Habc domain and Sec9c contains the homologous region to SNAP-25.
Cholesterol stimulates lipid mixing
To investigate the effect of cholesterol (Chol) on SNARE-mediated membrane fusion, we incorporated cholesterol into our vesicles. Although ergosterol is a naturally occurring, predominant sterol in yeast membranes (40), we decided to use cholesterol because of the technical difficulties with ergosterol in SNARE reconstitution. The t-SNARE protein Sso1pHT was reconstituted into the 100 nm vesicles of POPC/DOPS/Chol in molar ratios of X:15:Y, where X and Y were adjusted to achieve the desired Chol content (Fig. 1 B). The v-SNARE protein Snc2p was reconstituted into a separate population of vesicles of the same lipid composition. However, this composition contained 1.5 mol % each of NBD-PS and Rhodamine-PE for the fluorescence detection of lipid mixing (Fig. 1 C). The lipid-to-protein ratio was kept at 200:1 for all samples. In the lipid-mixing assay, membrane fusion resulted in the recovery of NBD fluorescence due to the increase of the average distance between the fluorescence donor and the acceptor. When the Sso1p vesicles were mixed with the Snc2p vesicles without the t-SNARE light chain Sec9c, no lipid mixing was observed. This indicated Sec9c was required for fusion. Therefore, membrane fusion can be conveniently initiated by simply adding Sec9c into the mixture of the v- and t-SNARE vesicles.
When Sec9c was added to the mixture, an increase of the NBD fluorescence signal was observed, indicating fusion happened (Fig. 2 A). Lipid mixing was accelerated as the cholesterol content increased. For the quantitative comparisons, we arbitrarily used the equation representing the second-order kinetics to fit the time traces of the fluorescence changes. The analysis showed that the rate of lipid mixing, which was approximated with the inverse of the half time t1/2, increased exponentially as a function of cholesterol (Fig. 2 B). There was only a small increase at 10 mol % cholesterol. But the rate was enhanced 3-, 8-, and even 20-fold at 20, 30, and 40 mol % cholesterol when compared with the rate in the absence of cholesterol. Therefore, the results show that cholesterol is a highly effective stimulator for lipid mixing at the physiological high cholesterol conditions.
Figure 2.
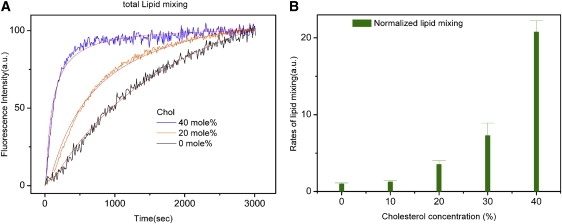
Lipid mixing at various cholesterol concentrations. (A) Fluorescence lipid-mixing assay of SNARE-mediated membrane fusion. Fluorescence change for lipid mixing was normalized with respect to the maximum fluorescence intensity (MFI) obtained by adding 0.1% reduced Triton X-100. The change of the fluorescence intensity in time was fitted with Eq. 1. (B) Rates of lipid mixing at various cholesterol concentrations. The rate was approximated by the inverse of the half-life (t1/2)−1 and the data were normalized with respect to the rate at the 0% cholesterol. The error bars were calculated on the basis of five independent measurements with five separately prepared vesicle samples. Error bars indicate SE.
Inner versus outer leaflet mixing
There is overwhelming evidence that SNARE-mediated membrane fusion transits through a hemifusion state in which outer leaflets are mixed but the inner leaflets remain intact. Because the fluorescent lipids were expected to be distributed equally in the inner and outer leaflets, the observed total lipid mixing represented the average of outer leaflet mixing and inner leaflet mixing. To measure inner leaflet mixing separately, we treated the v-SNARE vesicles with sodium dithionite. Under a controlled condition, sodium dithionite reduces NBD attached to the lipid headgroup in the outer leaflet to a nonfluorescent derivative while leaving NBD in the inner leaflet largely unaffected (14). When Sec9c was added to the mixture of the dithionite-treated v-SNARE vesicles and the Sso1pHT vesicles, an increase of the fluorescence signal was observed, indicating that inner leaflet mixing occurred (Fig. 3 A). This time, cholesterol again increased the rates of inner leaflet mixing but not as drastically as it did for total lipid mixing (Fig. 3 B). Inner leaflet mixing changed little when cholesterol was <20 mol %, but the rates were increased three-fold and 12-fold, respectively, for 30 and 40 mol % cholesterol. Now, the results for outer leaflet mixing can be readily obtained by simple subtraction of the time trace of the inner leaflet mixing from that of total lipid mixing (15). The stimulation of outer leaflet mixing by cholesterol was very strong, reaching astonishing 15- and 30-fold at 30 and 40 mol %. Therefore, the analysis shows that cholesterol enhances outer leaflet mixing much more dramatically than it does inner leaflet mixing.
Figure 3.
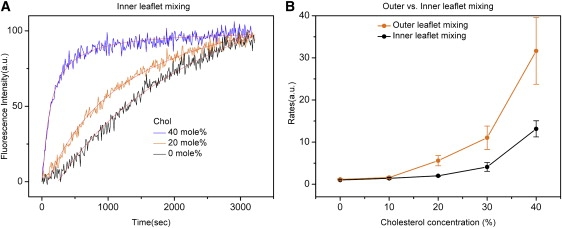
Outer leaflet mixing versus inner leaflet mixing at various cholesterol concentrations. (A) Inner leaflet mixing assay. The fluorescence changes in time were fitted with the second-order kinetics and the best fits are shown as the solid lines. (B) Outer versus inner leaflet mixing. The rates, which are defined as the inverse of the half life, were normalized with respect to the rate at 0% cholesterol. Outer leaflet mixing Po was calculated by using this equation: Po = 2PT − PI, where PT is the percentage for total lipid mixing and PI is the percentage for inner leaflet mixing. The error bars were calculated on the basis of at least five independent measurements.
Effect of cholesterol on pore expansion
The merging of two inner leaflets should be concomitant with formation of the fusion pore. Inner leaflet mixing measured with the dithionite method can therefore be considered a good measure of formation of fusion pore. However, what lipid mixing cannot tell us is the size of the fusion pore. Lipid mixing would be allowed, even if the pore is too small, to permit the passage of peptide or protein cargoes. We assessed the formation of the large functional pore with a FRET method using C termini labeled v- and t-SNARE (12) as an alternative method for content mixing (Fig. 1 A).
To observe FRET between one C-termini-labeled acceptor and donor, one of the dyes must cross the pore so that the acceptor and the donor can come at least within 50–60 Å from each other. Otherwise, the two dyes are separated by at least the thickness of the two bilayers, which would be more than 100 Å. Therefore, observation of FRET here would reflect the presence of a sufficiently wide pore that allows the passage of a few polar amino acids, plus the dyes, at the C-terminal end of the transmembrane domains.
For FRET, cysteine-free versions of Sso1pHT and Snc2p were used to generate two C-terminal mutants: Sso1pHT R290C (Ct) and Snc2p S115C (Cv). The Sso1pHT mutants were derivatized with fluorescence donor Cy3 maleimide. The Snc2p mutants were reacted with acceptor Cy5 maleimide. We first reconstituted the mixture of Cv-Cy5 and unlabeled Snc2p in the ratio of 1:3 into one population of vesicles. These vesicles were then reacted with another population of vesicles containing Ct-Cy3 and unlabeled Sso1pHT in the ratio of 1:3. In this experiment, we diluted the labeled proteins with the corresponding unlabeled wild-types to avoid the potential complications arising from self-quenching (12). When Sec9c was added to the reaction, an increase of the fluorescence intensity in the acceptor channel (Cy5) (Fig. 4 A) and a decrease of the fluorescence intensity in the donor channel (Cy3) were observed (data not shown). The results show that the distance between Cy5 and Cy3 was decreased, due to the colocalization of v- and t-SNARE TMDs in the same aqueous compartment.
Figure 4.
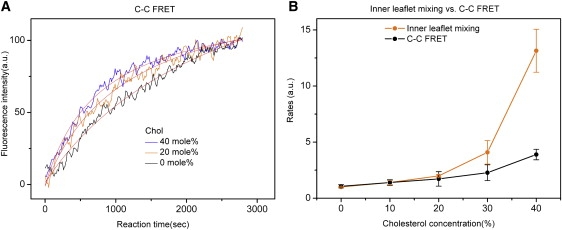
C-to-C FRET at various cholesterol concentrations. (A) The intensity changes of donor fluorescence were fitted with Eq. 1, representing the second-order kinetics. The solid lines were the best fits. (B) The rates of C-C FRET mixing were compared with those of inner leaflet mixing. The data were normalized with respect to the rate of inner leaflet mixing at 0% cholesterol. The error bars were calculated on the basis of at least five independent measurements.
Cholesterol only modestly increased the rate of C-C FRET. The rates were increased only two- and three-fold at 30 and 40 mol % cholesterol, respectively (Fig. 4 B). It is noteworthy that inner leaflet mixing was largely concomitant with C-C FRET when cholesterol content was <20 mol %. However, the latter became slower than the former at higher cholesterol concentrations. Therefore, the results show that cholesterol is not as effective in dilating the pore as it is in opening the small pore.
Effect of cholesterol on SNARE assembly
Cholesterol is thought to influence the lateral distribution of SNAREs. It is also possible that cholesterol alters membrane topologies of the juxtamembrane regions of SNAREs that are proposed to regulate SNARE complex formation (41). Therefore, we asked if SNARE complex formation is affected by cholesterol in the membrane. The kinetics of SNARE assembly was assessed with fluorescence resonance energy transfer (FRET) using dye-labeled SNAREs. The N-terminal residues, amino acid 185 of t-SNARE Sso1pHT and amino acid 13 of full-length v-SNARE Snc2p (amino acids 1–115), were changed to cysteines. The cysteine mutants were labeled with fluorescence donor Cy3 and acceptor Cy5, respectively. Labeled Sso1pHT and labeled Snc2p were reconstituted into two separate populations of the vesicles of POPC, DOPS, and Chol. When the SNARE complex was formed, t-SNARE captured the v-SNARE and the distance between the Cy3 on Sso1pHT and Cy5 on Snc2p decreased. Without Sec9c, the FRET was negligible. An addition of Sec9c increased the donor fluorescence rapidly in time, reporting association of v- and t-SNARE vesicles (Fig. 5 A). The time traces of the fluorescence signal were fitted well with the equation representing the second-order kinetics. An addition of cholesterol did not change the formation time of SNARE assembly much when the cholesterol content was <20 mol % (Fig. 5 B). However, cholesterol promoted the rate by factors of two and six at 30 and 40 mol %, respectively. Therefore, the results show that cholesterol promotes SNARE complex formation as well, although the stimulation was much less than it was for lipid mixing.
Figure 5.
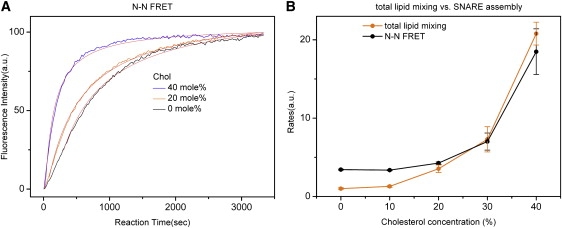
FRET assays of SNARE complex formation. (A) The changes in the fluorescence intensity for the acceptor due to SNARE complex formation at various cholesterol concentrations. The time profiles were fitted with Eq. 1. The solid lines were the best fits. The FRET value (E), calculated with the equation E = IA/(IA + IB), was ∼0.27 and the E values were approximately the same for all cholesterol concentrations. (B) The rates of the SNARE complex formation were compared with those of total mixing. The data were normalized with respect to the minimum rate of total lipid mixing at 0% cholesterol. The error bars were calculated on the basis of at least five independent measurements.
Discussion
The combined fluorescence assays using fluorescent lipids and labeled proteins made it possible to dissect the effects of cholesterol on the individual fusion steps in unprecedented detail. We were able to observe the cholesterol effects on SNARE complex formation, hemifusion, fusion pore opening, and pore expansion. The results showed that individual fusion steps were all stimulated by cholesterol but to different extents. The maximal effect was seen at the hemifusion step. Remarkably, hemifusion accelerated as much as 30-fold in the presence of 40 mol % cholesterol. However, the stimulatory effect trickled down to only a three-fold increase for pore expansion. There was also a six-fold increase of the SNARE complex formation.
The fact that stimulation by cholesterol was successively reduced along the fusion pathway after hemifusion was quite intriguing. It suggests that cholesterol negatively impact the rate constants for fusion pore formation, as well as for its expansion. Such fusion step-specific effects of cholesterol, i.e., stimulatory for hemifusion and the lesser stimulation for fusion pore formation, seem to be consistent with the prediction based on the cholesterol's intrinsic negative spontaneous curvature. Hemifusion involves creation of negatively curved surfaces. Therefore, the inverted cone-shaped cholesterol would stabilize the transition state for the hemifusion step. In contrast, fusion pore formation requires formation of positively curved surfaces, for which cholesterol is expected to be unfavorable.
Meanwhile, dilation of the fusion pore accompanies the relaxation of the acute positive curvature at the small pore stage. For this reason, cholesterol is expected to serve as a promoter for pore dilation. In fact, such a positive effect of cholesterol on pore expansion has been seen for membrane fusion induced by influenza hemagglutinin (42), as well as for protein-free membrane fusion (43). For SNARE-mediated fusion, however, it seems that cholesterol stimulated pore dilation only very mildly, which is contradictory to the prediction based on the cholesterol's spontaneous negative curvature. What would then be the molecular basis for such a minimal effect of cholesterol on pore expansion? It is highly likely that pore expansion is structurally and energetically coupled to heteromeric association of v- and t-SNARE TMDs. One possible scenario is that cholesterol does not favor the association of v- and t-SNARE TMDS, buttressing the expansion of the pore.
Our results show cholesterol stimulates SNARE complex formation. However, this effect cannot fully account for the dramatic stimulation of hemifusion. It was shown previously that cholesterol has the ability to affect the lateral distribution of SNAREs (23,25) that could influence the kinetics of SNARE assembly. There are also other reasons that might have contributed to the acceleration of SNARE assembly by cholesterol. First, the strong stimulation of hemifusion could reduce the rate of the dissociation of the docked vesicles (Fig. 1 A), which would result in a faster overall reaction rate for complex formation. Second, there are unknown fractions of t- and v-SNAREs that do not participate in membrane fusion but assemble into the complex after the completion of fusion, specifically after pore expansion. Therefore, the rate of SNARE complex formation partly reflects the rate of pore expansion that exhibits net stimulation by cholesterol. It seems paradoxical that the rate of outer leaflet mixing is faster than SNARE complex formation at 40 mol % (Fig. 5 B). This seemingly counterintuitive result can be also attributed to the same belated SNARE complex formation. The newly developed single-fusion assay (17), which can dissect the individual fusion steps for a single fusion event, might be an adequate tool to address such complex issues inherent for the ensemble assays. In addition, C-C FRET reflects not only association of the TMDs of v- and t-SNAREs participating in fusion but also the colocalization of SNAREs not participating in the fusion reaction. Therefore, C-C FRET has its shortcomings as a general method to detect the expansion of the fusion pore.
Recently, it has been found the hemifusion state is an important intermediate that serves as a substrate for the fusion activators (44,45). For example, sea urchin cortical granules are hemifused with the egg plasma membrane before fusion (45). In neurons, the majority of synaptic vesicles are found to be at the hemifused state on the presynaptic plasma membrane (18,46). In this work, cholesterol is shown to be highly effective in promoting hemifusion. Therefore, cholesterol may play a role in easing formation of the primed hemifused state waiting for the fusion trigger. On the other hand, cholesterol may also work as a negative regulator for fusion pore formation, rendering the improved opportunity for the regulator proteins to be able to control membrane fusion tightly.
Very recently, it was discovered that SNAREs are involved in homotypic fusion of lipid droplets, which has implications in obesity as well as in diabetes (47). Fusion between two lipid droplets involved just hemifusion and no pore formation. Therefore, it is interesting to speculate what the effects of cholesterol would be on this medically important fusion event.
In summary, the fluorescence fusion assays show that cholesterol dramatically stimulates membrane hemifusion but mildly stimulates fusion pore opening and widening. The results show that the cholesterol effect cannot be fully explained with the intrinsic negative curvature and that the effect on the protein conformation must be considered, too.
Footnotes
Jinyoung Chang and Sun-Ae Kim contributed equally to the work.
References
- 1.White J.M. Membrane fusion. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- 2.Jahn R., Lang T., Sudhof T.C. Membrane fusion. Cell. 2003;112:519–533. doi: 10.1016/s0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 3.Rothman J.E. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 4.Sollner T., Whiteheart S.W., Brunner M., Erdjument-Bromage H., Geromanos S. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 5.Weber T., Zemelman B.V., McNew J.A., Westermann B., Gmachl M. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 6.Jahn R., Scheller R.H. SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 7.Hanson P.I., Roth R., Morisaki H., Jahn R., Heuser J.E. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90:523–535. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- 8.Lin R.C., Scheller R.H. Structural organization of the synaptic exocytosis core complex. Neuron. 1997;19:1087–1094. doi: 10.1016/s0896-6273(00)80399-2. [DOI] [PubMed] [Google Scholar]
- 9.Poirier M.A., Xiao W., Macosko J.C., Chan C., Shin Y.K. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat. Struct. Biol. 1998;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- 10.Katz L., Hanson P.I., Heuser J.E., Brennwald P. Genetic and morphological analyses reveal a critical interaction between the C-termini of two SNARE proteins and a parallel four helical arrangement for the exocytic SNARE complex. EMBO J. 1998;17:6200–6209. doi: 10.1093/emboj/17.21.6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutton R.B., Fasshauer D., Jahn R., Brunger A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 12.Lu X., Zhang Y., Shin Y.K. Supramolecular SNARE assembly precedes hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 2008;15:700–706. doi: 10.1038/nsmb.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson M.B., Chapman E.R. Fusion pores and fusion machines in Ca2+-triggered exocytosis. Annu. Rev. Biophys. Biomol. Struct. 2006;35:135–160. doi: 10.1146/annurev.biophys.35.040405.101958. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y., Zhang F., Su Z., McNew J.A., Shin Y.K. Hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 2005;12:417–422. doi: 10.1038/nsmb921. [DOI] [PubMed] [Google Scholar]
- 15.Lu X., Zhang F., McNew J.A., Shin Y.K. Membrane fusion induced by neuronal SNAREs transits through hemifusion. J. Biol. Chem. 2005;280:30538–30541. doi: 10.1074/jbc.M506862200. [DOI] [PubMed] [Google Scholar]
- 16.Giraudo C.G., Hu C., You D., Slovic A.M., Mosharov E.V. SNAREs can promote complete fusion and hemifusion as alternative outcomes. J. Cell Biol. 2005;170:249–260. doi: 10.1083/jcb.200501093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon T.Y., Okumus B., Zhang F., Shin Y.K., Ha T. Multiple intermediates in SNARE-induced membrane fusion. Proc. Natl. Acad. Sci. USA. 2006;103:19731–19736. doi: 10.1073/pnas.0606032103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu T., Wang T., Chapman E.R., Weisshaar J.C. Productive hemifusion intermediates in fast vesicle fusion driven by neuronal SNAREs. Biophys. J. 2008;94:1303–1314. doi: 10.1529/biophysj.107.107896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdulreda M.H., Bhalla A., Chapman E.R., Moy V.T. Atomic force microscope spectroscopy reveals a hemifusion intermediate during soluble N-ethylmaleimide-sensitive factor-attachment protein receptors-mediated membrane fusion. Biophys. J. 2008;94:648–655. doi: 10.1529/biophysj.107.114298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jun Y., Wickner W. Assays of vacuole fusion resolve the stages of docking, lipid mixing, and content mixing. Proc. Natl. Acad. Sci. USA. 2007;104:13010–13015. doi: 10.1073/pnas.0700970104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takamori S., Holt M., Stenius K., Lemke E.A., Gronborg M. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 22.Churchward M.A., Rogasevskaia T., Hofgen J., Bau J., Coorssen J.R. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J. Cell Sci. 2005;118:4833–4848. doi: 10.1242/jcs.02601. [DOI] [PubMed] [Google Scholar]
- 23.Lang T., Bruns D., Wenzel D., Riedel D., Holroyd P. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001;20:2202–2213. doi: 10.1093/emboj/20.9.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin H., McCaffery J.M., Grote E. Ergosterol promotes pheromone signaling and plasma membrane fusion in mating yeast. J. Cell Biol. 2008;180:813–826. doi: 10.1083/jcb.200705076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sieber J.J., Willig K.I., Kutzner C., Gerding-Reimers C., Harke B. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 2007;317:1072–1076. doi: 10.1126/science.1141727. [DOI] [PubMed] [Google Scholar]
- 26.Tsui-Pierchala B.A., Encinas M., Milbrandt J., Johnson E.M., Jr. Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002;25:412–417. doi: 10.1016/s0166-2236(02)02215-4. [DOI] [PubMed] [Google Scholar]
- 27.Edidin M. The state of lipid rafts: from model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 28.Chamberlain L.H., Burgoyne R.D., Gould G.W. SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proc. Natl. Acad. Sci. USA. 2001;98:5619–5624. doi: 10.1073/pnas.091502398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lafont F., Verkade P., Galli T., Wimmer C., Louvard D. Raft association of SNAP receptors acting in apical trafficking in Madin-Darby canine kidney cells. Proc. Natl. Acad. Sci. USA. 1999;96:3734–3738. doi: 10.1073/pnas.96.7.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bacia K., Schuette C.G., Kahya N., Jahn R., Schwille P. SNAREs prefer liquid-disordered over “raft” (liquid-ordered) domains when reconstituted into giant unilamellar vesicles. J. Biol. Chem. 2004;279:37951–37955. doi: 10.1074/jbc.M407020200. [DOI] [PubMed] [Google Scholar]
- 31.Churchward M.A., Rogasevskaia T., Brandman D.M., Khosravani H., Nava P. Specific lipids supply critical negative spontaneous curvature–an essential component of native Ca2+-triggered membrane fusion. Biophys. J. 2008;94:3976–3986. doi: 10.1529/biophysj.107.123984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chernomordik L.V., Kozlov M.M. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 2003;72:175–207. doi: 10.1146/annurev.biochem.72.121801.161504. [DOI] [PubMed] [Google Scholar]
- 33.Ohtani Y., Irie T., Uekama K., Fukunaga K., Pitha J. Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur. J. Biochem. 1989;186:17–22. doi: 10.1111/j.1432-1033.1989.tb15171.x. [DOI] [PubMed] [Google Scholar]
- 34.Wasser C.R., Ertunc M., Liu X., Kavalali E.T. Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. J. Physiol. 2007;579:413–429. doi: 10.1113/jphysiol.2006.123133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheiffele P., Roth M.G., Simons K. Interaction of influenza virus hemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 1997;16:5501–5508. doi: 10.1093/emboj/16.18.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meers P., Ali S., Erukulla R., Janoff A.S. Novel inner monolayer fusion assays reveal differential monolayer mixing associated with cation-dependent membrane fusion. Biochim. Biophys. Acta. 2000;1467:227–243. doi: 10.1016/s0005-2736(00)00224-8. [DOI] [PubMed] [Google Scholar]
- 37.Zhang F., Chen Y., Su Z., Shin Y.K. SNARE assembly and membrane fusion, a kinetic analysis. J. Biol. Chem. 2004;279:38668–38672. doi: 10.1074/jbc.M404904200. [DOI] [PubMed] [Google Scholar]
- 38.Burri L., Lithgow T. A complete set of SNAREs in yeast. Traffic. 2004;5:45–52. doi: 10.1046/j.1600-0854.2003.00151.x. [DOI] [PubMed] [Google Scholar]
- 39.Chen Y., Xu Y., Zhang F., Shin Y.K. Constitutive versus regulated SNARE assembly: a structural basis. EMBO J. 2004;23:681–689. doi: 10.1038/sj.emboj.7600083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arora A., Raghuraman H., Chattopadhyay A. Influence of cholesterol and ergosterol on membrane dynamics: a fluorescence approach. Biochem. Biophys. Res. Commun. 2004;318:920–926. doi: 10.1016/j.bbrc.2004.04.118. [DOI] [PubMed] [Google Scholar]
- 41.Kweon D.H., Kim C.S., Shin Y.K. Regulation of neuronal SNARE assembly by the membrane. Nat. Struct. Biol. 2003;10:440–447. doi: 10.1038/nsb928. [DOI] [PubMed] [Google Scholar]
- 42.Biswas S., Yin S.R., Blank P.S., Zimmerberg J. Cholesterol promotes hemifusion and pore widening in membrane fusion induced by influenza hemagglutinin. J. Gen. Physiol. 2008;131:503–513. doi: 10.1085/jgp.200709932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haque M.E., Lentz B.R. Roles of curvature and hydrophobic interstice energy in fusion: studies of lipid perturbant effects. Biochemistry. 2004;43:3507–3517. doi: 10.1021/bi035794j. [DOI] [PubMed] [Google Scholar]
- 44.Yoon T.Y., Lu X., Diao J., Lee S.M., Ha T. Complexin and Ca2+ stimulate SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 2008;15:707–713. doi: 10.1038/nsmb.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong J.L., Koppel D.E., Cowan A.E., Wessel G.M. Membrane hemifusion is a stable intermediate of exocytosis. Dev. Cell. 2007;12:653–659. doi: 10.1016/j.devcel.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zampighi G.A., Zampighi L.M., Fain N., Lanzavecchia S., Simon S.A. Conical electron tomography of a chemical synapse: vesicles docked to the active zone are hemi-fused. Biophys. J. 2006;91:2910–2918. doi: 10.1529/biophysj.106.084814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bostrom P., Andersson L., Rutberg M., Perman J., Lidberg U. SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nat. Cell Biol. 2007;9:1286–1293. doi: 10.1038/ncb1648. [DOI] [PubMed] [Google Scholar]