

Abstract
Putative metal-chelate-type ABC transporter HI1470/1 is homologous with vitamin B12 importer BtuCD but exhibits a distinct inward-facing conformation in contrast to the outward-facing conformation of BtuCD. Normal-mode analysis of HI1470/1 reveals the intrinsic asymmetric conformational flexibility in this transporter and demonstrates that the transition from the inward-facing to the outward-facing conformation is realized through the asymmetric motion of individual subunits of the transporter. This analysis suggests that the asymmetric arrangement of the BtuC dimer in the crystal structure of the BtuCD-F complex represents an intermediate state relating HI1470/1 and BtuCD. Furthermore, a twisting motion between transmembrane domains and nucleotide-binding domains encoded in the lowest-frequency normal mode of this type of importer is found to contribute to the conformational transitions during the whole cycle of substrate transportation. A more complete translocation mechanism of the BtuCD type importer is proposed.
Introduction
ABC transporters belong to a large family of integral membrane proteins that use the energy of ATP binding and hydrolysis to transport substrate across cell membranes. Prokaryotic ABC transporters include importers that are predominantly involved in nutrient uptake, and exporters that export toxins and harmful substances out of the cell. Mammalian ABC transporters are all exporters and many of them, such as the human MDR1, CFTR, and TAP proteins (1–3), are clinically relevant. Most ABC transporters consist of four domains, two transmembrane domains (TMDs), and two nucleotide-binding domains (NBDs), also known as ATP-binding cassettes (ABCs). The TMDs show sequence diversity between different ABC transporters, whereas the NBDs are highly conserved and contain sequence motifs critical for ATP binding and hydrolysis, such as P-loops (Walker A) and signature motifs (LSGGQ) at the ATP binding sites (2,4–6).
Crystal structures of several intact ABC transporters (7–12) and isolated NBD domains (13–21) provide rich information for understanding the working mechanism of transportation. Although the architecture of the TMDs that form the translocation pathway varies among different transporters, the structures can be classified into two major states (5): 1), the outward-facing conformation, in which the substrate translocation pore opens on the periplasmic side; and 2), the inward-facing conformation, in which the pore opens on the cytoplasmic side. The crystal structure of the HI1470/1 transporter from Haemophilus influenza represents one of the inward-facing importer conformations (22). HI1470/1 is a putative metal-chelate-uptake ABC transporter, homologous with the vitamin B12 importer BtuCD. The sequence identities of TMDs and NBDs between HI1470/1 and BtuCD are 24% and 33%, respectively. Crystal structural studies of BtuCD transporter have located two different conformations: an outward-facing conformation of BtuCD, and an asymmetric conformation of BtuCD-F complex. In the outward-facing conformation of BtuCD (11), the translocation pore closes on the cytoplasmic side and opens on the periplasmic side, with the NBD dimer in a more closed conformation than that of HI1470/1. In the other crystal structure of BtuCD associated with its periplasmic binding protein (PBP) BtuF (9), the two TMD subunits exhibit different conformations, with the translocation pore closing on both sides of the membrane.
Because of the homology between HI1470/1 and BtuCD, the outward-facing conformation of HI1470/1 is considered to be one of the intermediate states in the translocation cycle of BtuCD. In this respect, the more open conformation of NBDs in HI1470/1 suggest that, in this type of importer, the outward-facing conformation of the translocation pore corresponds to the ATP-binding state of the NBD dimer, and the inward-facing conformation corresponds to the post-hydrolysis state of the NBDs (22,23), although all three structures are in nucleotide-free states. A previous normal-mode analysis (24) of the outward-facing conformation of BtuCD verified that the opening of the cytoplasmic gate of the translocation pathway couples to the simultaneous opening of the two ATP binding sites of the NBDs. This coupling implies that the hydrolysis of ATP triggers the conformational transition of TMDs and hence substrate translocation. Thus, the transporter must return to the outward-facing conformation to start the next round of transportation. In the putative reverse-conformational transition from inward-facing to outward-facing states, it is straightforward to assume that the pathway of conformational change is also reversible, i.e., NBDs close the two ATP-binding sites to induce the occlusion of the cytoplasmic gate of the translocation pore (11,17,25–27). However, the asymmetry in the crystal structure of the BtuCD-F complex implies an alternative conformational movement during the translocation cycle of the BtuCD transporter. It has been suggested that the asymmetric conformation of BtuCD-F is intermediate between HI1470/1 and BtuCD (9,23). If this is the case, it means that the post-hydrolysis conformational change that resets the transporter to the outward-facing state is quite different from that accompanying the ATP hydrolysis. Therefore, the details of the post-hydrolysis conformational movement must be clarified before we can gain a comprehensive understanding of the molecular mechanism of the translocation cycle of BtuCD-type importers.
In this report, we present a detailed elastic normal-mode analysis of HI1470/1. Recently, the coarse-grained elastic normal-mode analysis method was shown to be efficient and reliable for deriving conformational movement of proteins (28–31). Many effective applications of this method to membrane protein systems have been reported (32–39), and it has been shown that membrane environment does not affect the intrinsic conformational change direction (40). Using this method, we found that the conformational transitions of TMDs to their outward-facing states require the individual motions of the HI1470 and HI1471 subunits, i.e., the intrinsic asymmetric conformational flexibility of the transporter homodimer contributes to the HI1470/1-BtuCD transition. A more detailed picture of the translocation mechanism of BtuCD-type importer is proposed.
Materials and Methods
In the elastic network model, atomistic structures are simplified as one Cα atom per residue, and pairs of Cα atoms that lie within a certain cutoff distance, Rc, of each other are connected by Hookean springs with a harmonic pairwise potential. As an extension of the simplest Gaussian network model, the anisotropic network model (ANM) incorporates the anisotropic effects on fluctuation dynamics (28). A force constant of 1.0 kcal/mol·Å2 with a cutoff distance Rc of 13 Å was employed in the calculation of vibrational modes, a configuration that was shown to work well in a previous study (24).
A component analysis was performed to see how the normal modes of isolated domains contribute to the normal modes of the whole protein. As in the ANM model, the crystal structure is presumed to be at the local minimum on the potential energy surface, and the weights of modes of an isolated domain can be derived from the modes of the whole protein by a direct vector product. The details of the component analysis were described previously (24).
The involvement coefficients (measures of normal-mode contributions in certain conformational transitions) are calculated based on the strongly conserved residue pairs, which are determined according to the structure-based alignment of BtuCDF and HI1470/1 (22). The involvement coefficient was calculated as described previously (24). A few residues are missing in the crystal structure of HI1470/1 (PDBID: 2NQ2), including the residues between the TM4 and TM5 helices in TMD, between the s1 and s2 strands in NBD, and especially in the extracellular loop 1 (ECL1). However, none of the missing residues in HI1470/1 were rebuilt, because the BtuCD and BtuCDF (PDBID: 1L7V and 2QI9) structures show that truncation of the residues in the corresponding positions has a limited effect on the low-frequency modes.
A spin angle is used to describe the relative spin motion between the TMD and NBD dimers. To evaluate the spin angle, the BtuC and BtuD dimers in the BtuCD crystal structure are aligned on the TMD and NBD parts of HI1470/1 or BtuCDF to minimize the root mean-square deviation (RMSD) between conserved residues. As a result, the BtuC and BtuD dimers are in new arrangements that reflect the relative positions of the TMD and NBD domains in HI1470/1 and BtuCD-F. Then the spin angle is evaluated as the angle between the lines connecting the centers-of-mass of the BtuC/BtuD subunits (Fig. 1).
Figure 1.
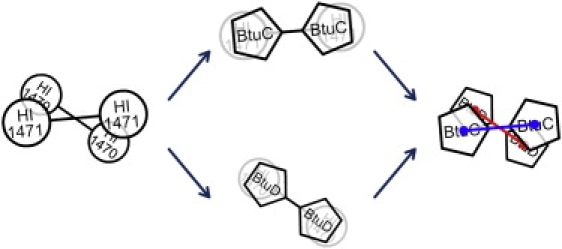
The spin angle definition used to describe the relative positions and torsional motions between the TMD and NBD dimers. To evaluate the spin angle, BtuC and BtuD dimers in the BtuCD crystal structure are overlapped (to minimize RMSD between conserved residues) onto the TMD and NBD parts of HI1470/1 or BtuCDF. As a result, the BtuC and BtuD dimers are in new arrangements that reflect the relative positions of TMD and NBD parts in HI1470/1 or BtuCD-F. The spin angle is then evaluated as the angle between the lines (red and blue) connecting the mass centers of the BtuC/BtuD subunits.
Results
Since the conformational changes accompanying substrate transport are expected to involve communication between the TMD and NBD parts, the normal-mode analysis of HI1470/1 is first performed with isolated TMD and NBD domains. The motions encoded in these isolated domains contribute to the conformational change of the whole transporter. The conformational changes of the whole transporter are examined by analyzing the normal modes of the intact molecular complex HI1470/1 in the context of the motional coupling between TMDs and NBDs.
Normal modes of isolated HI1470 dimer
The three lowest-frequency modes of the HI1470 dimer share significant similarity with the BtuD dimer in the BtuCD transporter. This is expected because of both the sequence and structure similarities between HI1470 and BtuD. A >30% sequence identity results in the same folds for the NBD subunits, and the dimers are also similarly arranged, with RMSD of 3.4Å between the Cα atoms for the conserved residues. Similarly to the normal modes of the BtuD dimer, the low-frequency modes of the HI1470 dimer consist of two symmetric hinge-bending modes, an asymmetric mode, and several modes involving helical subdomain movements.
The two lowest-frequency modes of HI1470 essentially result from rigid-body motions of the two NBD subunits, with the C-terminals acting as the hinge. Along the lowest-frequency mode, each NBD subunit rotates around its respective axis, passing through L5 in β-strand s1, L34 in β-strand s3 preceding the Walker A motif, Q191 in α-helix h7 adjacent to the H-loop, and P246 near the C-terminal of the opposite subunit (Fig. 2 b). Along this mode, the two nucleotide-binding sites, composed of the LSGGQ signature motif and P-loop on opposing HI1470 subunits, open and close simultaneously. A distinct feature of this mode is that the hinge-bending motion changes the distance between the signature motifs of the two opposing HI1470 subunits significantly, thereby changing the distance between the two helical subdomains as well, but the motion hardly affects the relative positions of the two P-loops in the RecA subdomains. This feature also exists in the first mode of the BtuD dimer, but not as obviously as in HI1470. A comparison of the crystal structures of the NBD dimers in intact BtuCD and HI1470/1 shows that although the distance between the P-loops on different NBD subunits changes little between the two transporters, the distance between the two signature motifs changes significantly (22), implying that this mode of the NBD dimer conforms to the conformational change between BtuCD and HI1470.
Figure 2.
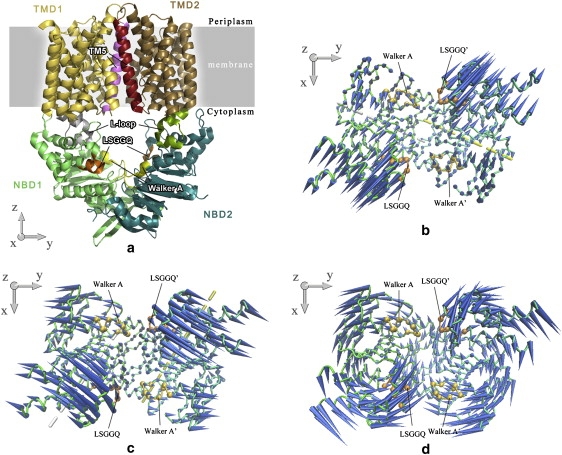
(a) Ribbon diagram of the crystal structure of HI1470/1 transporter. The TM5 helices of each TMD monomer are highlighted in red and magenta, and L-loops are highlighted in green and gray. Walker A or P-loop motifs and LSGGQ signature motifs in NBD dimer are colored yellow and orange, respectively. The first (b), second (c) and third (d) lowest normal modes of the isolated NBD HI1470 dimer are depicted by porcupine plots looking down from the periplasmic side. Cones denote the direction of the movement of each Cα atom, from the base to the tip, with the length of the cone describing the amplitude of the motion. The ATP-binding sites are denoted by the VDW drawing method (yellow for Walker A and orange for LSGGQ signature motif). In the first two lowest-frequency modes, the axes of NBD subunit rotation are indicated by dashed lines.
The hinge bending motion of the second-lowest-frequency mode also results in the simultaneous opening and closing of the two nucleotide-binding sites, and the rigid-body motions of the two NBD subunits resemble those in the BtuD dimer. The rotation axes pass through Y115 of α-helix h4 in the helical subdomain, L171 in α-helix h6, V192 in α-helix h7 beside the H-loop, and E219 near the C-terminals of the respective NBD subunits (Fig. 2 c). The third mode of the HI1470 dimer is very similar to its counterpart mode of the BtuD dimer. The two subunits rotate in an antisymmetric way with respect to the twofold-symmetry axis of the dimer, resulting in the alternative opening and closing of two nucleotide-binding sites (Fig. 2 d).
For several modes with higher frequencies than mode 3, a common feature is the significant intradomain motion of the helical subdomain, such as in modes 4–6 (data not shown). Along these modes, the helical domains show remarkable flexibility and rock with respect to the RecA-like subdomain. In contrast, the counterpart modes of the BtuD dimer do not involve such obvious helical subdomain movements. A crystal structure study of the HI1470/1 transporter revealed that one NBD subunit has a translational shift along the dimer interface in comparison with the BtuD dimer in BtuCD (22). This shift may make the helical subdomain more loosely packed against the opposing RecA-like subdomain with respect to the BtuD dimer, thereby leading to the greater flexibility of the helical subdomain.
Normal modes of isolated HI1471 dimer
The TMD of HI1470/1 adopts an inward-facing conformation, i.e., the substrate translocation pore opens at the cytoplasmic side and closes at the periplasmic side. Therefore, we found in the low-frequency normal modes of the HI1471 dimer that the periplasmic side stays rigid and acts as a hinge while the cytoplasmic side undergoes large-amplitude movements. The two lowest-frequency modes of the HI1471 dimer can be described primarily as reverse rigid-body rotations of the two TMD subunits around their respective axes (Fig. 3, a and b). Along the lowest-frequency mode, each monomer rotates around the axis that passes through L17 of TM1, V72 of TM2, A104 of TM3, and V165 of TM5, tilting an angle of ∼70° with respect to the bilayer normal (Fig. 3 a). The converse rotation of the two HI1471 subunits is symmetric with respect to the twofold axis of the transporter. As the L-loops on the two TMD subunits move apart, the cytoplasmic side of the HI1471 dimer expands. However, the shape or diameter of the translocation pathway characterized by the distance between the two TM5 helices does not show obvious variation (Fig. 3 a).
Figure 3.
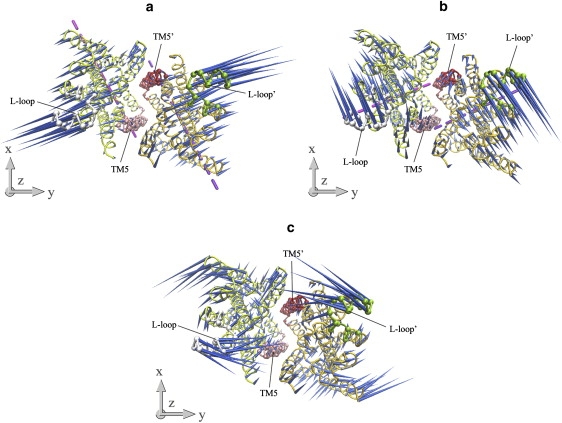
Porcupine plots of the first (a), second (b), and fourth (c) lowest-frequency normal modes of the isolated TMD HI1471 dimer looking up from the cytoplasmic side. TM5 helices are marked with red and pink, and L-loops are green and gray. The dashed lines are axes around which each subunit rotates along the two lowest normal modes.
Monomer rigid-body rotations also dominate the second-lowest mode of the HI1471 dimer. As with the first mode, the converse rotation of the two TMD subunits has twofold symmetry. However, the rotational axes in this mode are nearly perpendicular to those of mode 1 (Fig. 3, a and b). Each rotational axis passes through Q253 of TM7, S312 of TM10, and L164 of TM5 in the opposite monomer, forming an angle of 75° with respect to the bilayer normal (Fig. 3 b). As a result, the L-loops move with the rigid-body motions along a direction nearly perpendicular to that in mode 1 (Fig. 3 b). With the motion of this mode, the tilting angles of the TM5 helices change significantly such that the shape of the translocation pore varies as well.
Mode 4 is another low-frequency mode that significantly changes the shape of the translocation pathway. In contrast to the rigid-body motions in the two lowest modes, mode 4 involves many more local fluctuations and twisting motions. At the cytoplasmic side, TM1, TM3, TM8, and TM9 move outward and make room for the inward motion of TM5 and the L-loops (Fig. 3 c). As a result, when the L-loops move almost directly toward each other, the TM5 helices change their tilting angles and close the cytoplasmic gate. The motional direction of the L-loops resembles that of mode 1, but the TM5 helices undergo similar movements as in mode 2.
Overall, the low-frequency normal modes of HI1471 indicate more flexible behavior on the cytoplasmic side, whereas the periplasmic part of TM5 appears more rigid. The symmetric modes dominate the opening and closing motions of the central pore. Among those modes, modes 2 and 4 can effectively change the shape of the translocation pathway, although the directions of the L-loop movements in these two modes are different.
Normal modes of the intact HI1470/1
The normal modes of intact HI1470/1 transporter are calculated and analyzed by evaluating the local contributions from TMDs and NBDs (Table 1). To select potential functionally relevant normal modes from the low-frequency modes, we examined the pore-shape variations along the vibrational motions of those modes. Fig. 4 illustrates the distance changes between the Cα atoms of the two TM5 helices in different TMD subunits as the nucleotide-binding sites close at the NBD domain in the nine lowest-frequency modes. The modes that change the shape of the translocation pore are considered potential functionally relevant modes. According to this criterion, modes 1, 2, 3, 7, and 8 are excluded from the functionally relevant modes because the shape of the translocation pore is hardly disturbed therein. The remaining modes may be divided into two groups. For one group of modes, which includes modes 4 and 9, the pore opens wider as two NBD subunits close. In contrast, in the other group of modes, which includes modes 5 and 6, the pore closes along with the closure of the NBD dimer. Next, we will discuss the details of these modes along with the lowest mode of the intact transporter.
Table 1.
Weights of rigid-body movements and normal modes of isolated TMD and NBD dimers in the low-frequency modes of intact HI1470/1 complex
| Mode (k) | TMD | NBD | ||||||
|---|---|---|---|---|---|---|---|---|
| P′vib,k(i)∗ | PRT,k† | P′vib,k(i) ∗ | PRT,k† | |||||
| 1 | 50.8(2) | 32.6(1) | 83.3 | 47.5(2) | 35.4(1) | 10.4(5) | 77.7 | |
| 2 | 44.1(3) | 9.3(11) | 96.2 | 34.4(3) | 22.7(4) | 17.5(6) | 93.1 | |
| 3 | 33.3(3) | 24.3(7) | 15.6(9) | 87.1 | 65.6(3) | 25.8(4) | 56.1 | |
| 13.4(5) | ||||||||
| 4 | 85.1(1) | 11.6(2) | 33.2 | 79.2(1) | 26.9 | |||
| 5 | 58.0(2) | 19.9(1) | 39.9 | 51.0(3) | 34.0(2) | 3.1 | ||
| 6 | 62.5(2) | 24.3(1) | 26.6 | 48.1(3) | 31.6(2) | 7.4(5) | 3.1 | |
| 7 | 62.1(3) | 12.1(5) | 44.6 | 30.0(6) | 21.2(3) | 17.9(4) | 64.3 | |
| 7.7(9) | ||||||||
| 8 | 37.8(3) | 7.4(11) | 7.0(7) | 80.8 | 71.3(6) | 9.3(3) | 64.5 | |
| 9 | 43.5(1) | 26.1(4) | 14.7(2) | 27.5 | 42.7(5) | 29.4(8) | 8.2(7) | 30.4 |
| 7.6(9) | ||||||||
Values after renormalization by removing the overall rotation and translation of TMD or NBD dimers. The numbers in parentheses are the indices of the normal modes of isolated TMD or NBD dimers.
The contribution of overall rotation and translation of isolated TMD or NBD dimers.
Figure 4.
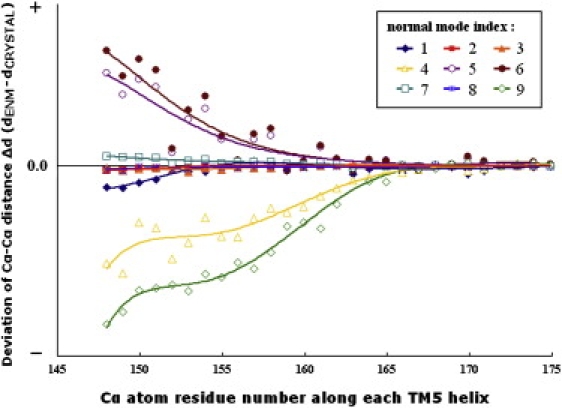
Variations of the central pore in the TMD dimer along the low-frequency normal modes of the HI1470/1 complex. The profile of the pore is evaluated based on the distances between corresponding Cα atoms in the TM5 helices, and the variation values are the deviations of the TM5 distances between the end structure of each mode and that of the crystal structure. The vibrational direction of every mode is chosen so that the NBD subunits move closer to each other.
The lowest mode of HI1470/1 is mainly the combination of the two lowest normal modes of both isolated TMD and NBD dimers, which can be described as the rigid-body spin motions of TMDs and NBDs around the transporter's twofold axis (Fig. 5). Of interest, a similar spin motion is also found in the lowest normal modes of BtuCD (24) and BtuCD-F (data not shown). This spin motion twists the transporter and changes the interface between the TMD and NBD dimers. An increase in the torsion angle between TMDs and NBDs (defined in Materials and Methods) resulting from the rigid-body spin motion is accompanied by the closure of the pore at the cytoplasmic side and the opening of the two ATP-binding sites, although the amplitudes of closure and opening are both very small. It is noticeable that the structural differences among BtuCD, BtuCD-F, and HI1470/1 are consistent with the conformational change direction in this mode; for example, HI1470/1 exhibits a larger torsion angle (35.9°) between TMDs and NBDs than BtuCD-F (31.2°) and BtuCD (29.7°), and its NBD dimer adopts a more open conformation than the other two. We also noticed that in addition to the two lowest normal modes, the fifth mode of the isolated NBDs contributes considerably to the lowest mode of intact transporter (Table 1). In mode 5 of the isolated NBDs of HI1470/1, relative movement of the two NBD subunits shifts them along the direction parallel to the NBD dimer interface (data not shown). This translational shift of NBD subunits is also found in the crystal structure of HI1470/1 with respect to the structure of BtuCD. All of the evidence suggests that this prevailing spin mode with lowest frequency represents a general conformational dynamic property of this type of importer and may play an important role in the conformational transformations during the transportation cycle, although it does not induce significant conformational change at the translocation pore.
Figure 5.
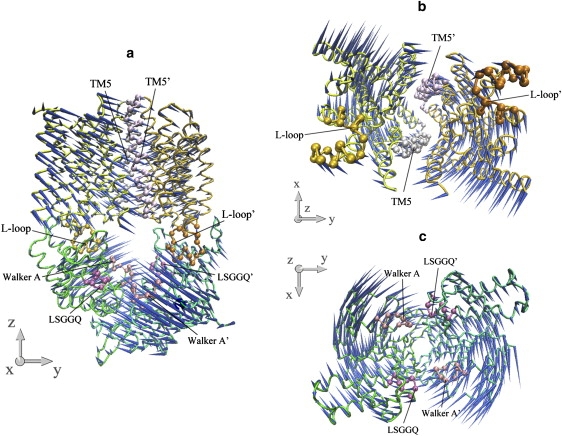
Porcupine plots of the lowest normal mode of the intact HI1470/1 complex. (a) Front view of the motion of intact HI1470/1. (b) Bottom view looking up from the cytoplasmic side of the motion of HI1471 (TMD). (c) Top view looking down from the periplasmic side of the motion of HI1470 (NBD).
Mode 4 is mainly the combination of the lowest modes of both isolated TMD and NBD dimers (Table 1), and the rigid-body movements of the HI1471 or HI1470 monomer dominate this normal mode. Along this mode, the NBD dimer undergoes a tweezer-like motion that opens and closes the NBD dimer in a symmetric fashion. Coupled with the closing of the NBD dimer, the two L-loops move transversely, contracting the cytoplasmic end of the TMD dimer (Fig. 6). The cytoplasmic ends of the TM helices of different HI1471 subunits approach each other, except for those of TM5 and TM10 that define the shape of the translocation pore, i.e., the gate of the translocation pathway opens wider as other TM helices get closer at the cytoplasmic side of the membrane. In addition, there is a stretching movement of the whole transporter along the twofold axis, resulting in remarkable variation of the longitudinal distance between the TMD and NBD dimers along with the conformational change at the TMD-NBD interface (Fig. 6). The main characteristic of this mode is that the closing of the nucleotide-binding sites accompanies the opening of the cytoplasmic gate of the pore.
Figure 6.
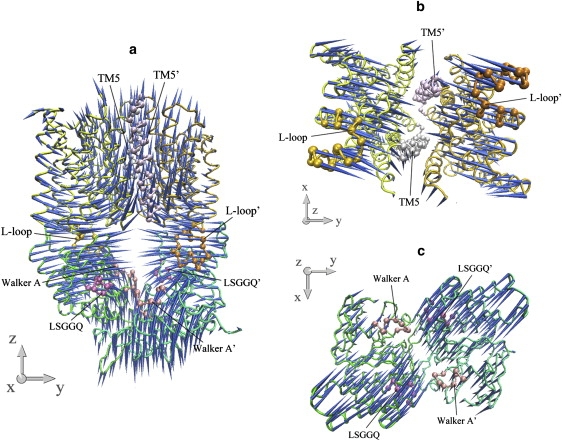
Porcupine plots of the fourth-lowest normal mode of the intact HI1470/1 complex. (a) Front view of the motion of intact HI1470/1. (b) Bottom view looking up from the cytoplasmic side of the motion of HI1471 (TMD). (c) Top view looking down from the periplasmic side of the motion of HI1470 (NBD).
In mode 9, the motion of the NBD dimer involves a large portion of helical subdomain rotation. Projecting this mode onto the normal-mode basis set of isolated TMD and NBD dimers reveals that the fourth mode of the isolated NBD dimer plays an important role in mode 9 (Table 1). The rotation of the helical subdomain is coupled to the cytoplasmic side of the TMD helices through the L-loops, resulting in the large-amplitude conformational change of the TMD dimer at the cytoplasmic end (Fig. 7, b and c). The conformational coupling relationship between TMD and NBD is similar to that of mode 4, i.e., the cytoplasmic gate of the translocation pore opens when the nucleotide-binding sites close at the NBD dimer (Fig. 7 b). However, unlike mode 4, there is no obvious longitudinal shape variation of the whole transporter. Instead, the remarkable conformational change at the NBD-TMD interface results in the whole transporter undergoing a breathing motion with respect to the center of the heterotetramer (Fig. 7 a).
Figure 7.
Porcupine plots of the ninth-lowest normal mode of the intact HI1470/1 complex. (a) Front view of the motion of intact HI1470/1. (b) Bottom view looking up from the cytoplasmic side of the motion of HI1471 (TMD). (c) Top view looking down from the periplasmic side of the motion of HI1470 (NBD).
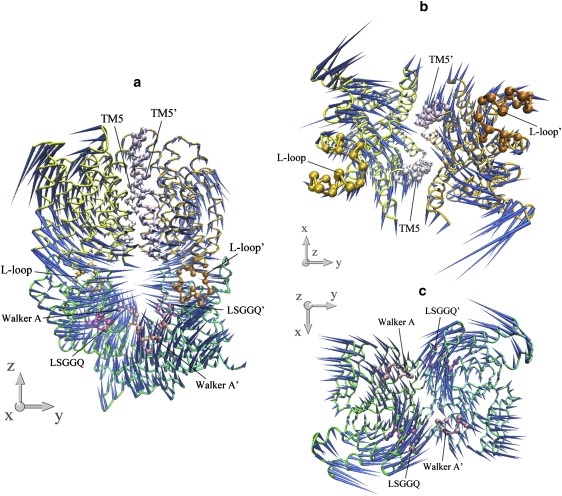
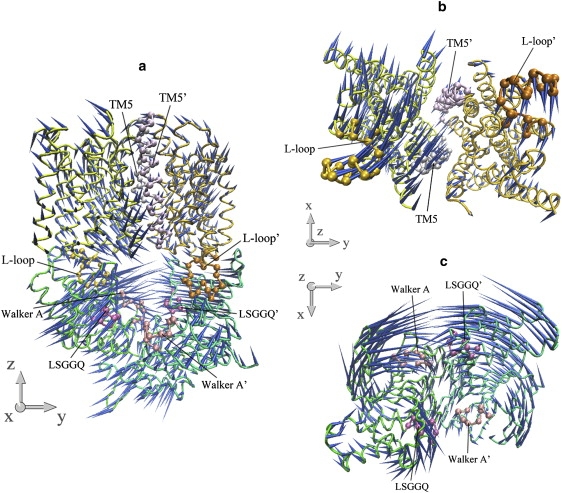
The common feature of modes 5 and 6 is the asymmetric motion of both TMD and NBD dimers. In both modes, the motion of the NBD part is mainly the combination of modes 3 and 2 of the isolated NBD dimer, whereas the two lowest modes of the isolated TMD dimer dominate the motion of the TMD part (Table 1). In each mode, one monomer of the transporter, consisting of one HI1471 and one HI1470 subunit, undergoes a much larger conformational change than the other (Figs. 8 and 9). In a sense, modes 5 and 6 are degenerate modes if the structure of the transporter is strictly twofold symmetric. Because of the antisymmetry of mode 3 of the isolated NBDs (Fig. 2 d), the two nucleotide-binding sites open and close alternatively. Along both modes 5 and 6 of HI1470/1, one binding site opens and closes with much larger amplitude than the other. This asymmetric movement of NBD is coupled to the TMD part, leading to one HI1471 subunit undergoing a much larger conformational change in which one TM5 changes its tilting angle dramatically. As a result, the closing of the nucleotide-binding site that experiences larger amplitude movement is coupled with the closing of the cytoplasmic gate of the translocation pore.
Figure 8.
Porcupine plots of the fifth-lowest normal mode of the intact HI1470/1 complex. (a) Front view of the motion of intact HI1470/1. (b) Bottom view looking up from the cytoplasmic side of the motion of HI1471 (TMD). (c) Top view looking down from the periplasmic side of the motion of HI1470 (NBD).
Figure 9.
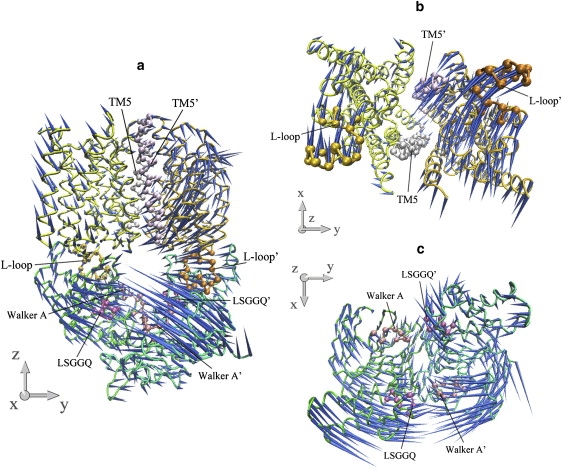
Porcupine plots of the sixth-lowest normal mode of the intact HI1470/1 complex. (a) Front view of the motion of intact HI1470/1. (b) Bottom view looking up from the cytoplasmic side of the motion of HI1471 (TMD). (c) Top view looking down from the periplasmic side of the motion of HI1470 (NBD).
The inward-facing conformation of HI1470/1 is considered to correlate with the post-hydrolysis state of the importer because the NBD dimer of HI1470/1 adopts a more open conformation than that of BtuCD (22). Therefore, in a complete cycle of transportation, the recovery of the inward-facing conformation to the outward-facing conformation corresponds to the closing of the NBD dimer. Among the normal modes discussed above, modes 5 and 6 conform to the conformational change of the post-hydrolysis recovery process, whereas modes 4 and 9 have an opposite coupling relation to the movements of TMD and NBD. The asymmetric characteristic of modes 5 and 6 are reminiscent of the recently reported crystal structure of the BtuCD-F complex, in which the substrate translocation pore of the TMD dimer shows an asymmetric conformation with the two TM5 helices bearing different tilting angles with respect to the membrane normal (9). Fig. 10 shows the conformations of the TM5 helices in the crystal structures of HI1470/1 (22) and the BtuCD-F complex (9) in comparison with the vibrational end structures of mode 6. The conformational changes along the normal mode conform perfectly to the experimental structures (Fig. 10).
Figure 10.
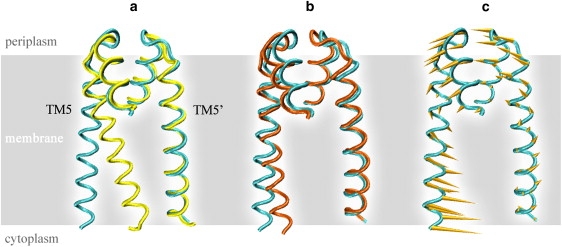
Asymmetric motions of the TM5 helices manifested in the crystal structures and the low-frequency normal modes of HI1470/1. (a) Superposition of TM5 helices in the crystal structures of HI1470/1 (cyan) and the BtuCD-F (yellow) complex. (b) Superposition of TM5 helices in the crystal structures of HI1470/1 (cyan) and in one vibrational end structure of mode 6 (orange). (c) Conformational movement of TM5 helices along mode 6 of HI1470/1.
It is worth noting that the PBP BtuF is associated with BtuCD transporter in the asymmetric conformation, which is presumably the intermediate state between HI1470/1 and BtuCD. This implies that the inward-facing conformation that corresponds to the HI1470/1 structure during the translocation cycle may have the PBP bound after substrate transportation. To examine the effect of the PBP association, we constructed a complex by docking BtuF onto the HI1470/1 transporter at the periplasmic side, using a method similar to that described previously (24,41). The BtuF structure used in the constructed complex is taken from the BtuCD-F complex by considering that the PBP is expected to be substrate-free in the inward-facing conformation of HI1470/1. The positions of the arginine and glutamate residues that form the salt bridges at the docking interface match very well. The results of the normal-mode analysis of this constructed complex show that the association of BtuF changes the order of some of the low-frequency modes with respect to HI1470/1, but the main characteristics of the low-frequency modes discussed above are conserved (data not shown). The degeneracy of modes 5 and 6 (we use the mode indices of HI1470/1 for consistency) disappear, and two low-frequency modes appear bearing characteristics of mode 6. Mode 4 also splits into two modes with similar conformational coupling relationships, whereas the counterparts of modes 1, 5, and 9 are well conserved. Overall, the association of PBP causes some changes in the low-frequency spectrum of the transporter, but it does not change the main characteristic of the conformational movement. This is consistent with the results of the normal-mode analysis of the outward-facing state BtuCD associated with BtuF (24).
Involvement coefficients relating experimental structures
The involvement coefficient is a measure of normal-mode contributions in certain conformational transitions. The involvement coefficients of the HI1470/1 modes in the conformational transformation from HI1470/1 to BtuCDF are calculated and these coefficients are further decomposed into local contributions from the isolated TMD and NBD parts (Table 2).
Table 2.
Some large global and local involvement coefficients associated with the transformation from HI1470/1 to BtuCDF based on the normal modes of HI1470/1
| Mode (k) | Ck∗ | CTMD† | CNBD† | TTMD‡ | TNBD‡ | RTMD§ | RNBD§ |
|---|---|---|---|---|---|---|---|
| 6 | −0.228 | −0.170 | −0.058 | −0.003 | −0.002 | −0.020 | 0.002 |
| 22 | 0.214 | 0.000 | 0.214 | −0.001 | −0.003 | −0.004 | −0.002 |
| 12 | −0.185 | 0.017 | −0.202 | −0.003 | −0.002 | 0.002 | 0.013 |
| 14 | −0.171 | −0.062 | −0.109 | −0.002 | 0.000 | 0.005 | 0.016 |
| 9 | 0.165 | −0.002 | 0.167 | −0.008 | −0.010 | 0.009 | −0.001 |
| 19 | −0.157 | −0.090 | −0.067 | 0.001 | 0.002 | 0.006 | −0.002 |
| 1 | −0.155 | −0.114 | −0.041 | −0.003 | −0.003 | −0.047 | −0.058 |
| 79 | −0.123 | 0.021 | −0.144 | −0.001 | −0.002 | −0.005 | 0.009 |
| 57 | −0.112 | −0.050 | −0.062 | 0.000 | 0.000 | 0.002 | −0.005 |
| 5 | 0.104 | 0.058 | 0.046 | −0.001 | 0.000 | −0.013 | 0.001 |
| 38 | 0.104 | 0.074 | 0.030 | 0.000 | 0.000 | 0.000 | 0.000 |
| 16 | −0.100 | −0.120 | 0.020 | 0.002 | 0.002 | 0.003 | 0.002 |
Global involvement coefficients.
Local involvement coefficients of TMD or NBD parts.
Isolated translational components of overall TMD or NBD in the involvement coefficients.
Isolated rotational components of overall TMD or NBD in the involvement coefficients.
As expected, the asymmetric mode 6 has the largest involvement coefficient, in which the contribution from the TMD dimer (CTMD) is larger than that from the NBD part (CNBD). Furthermore, the value of CTMD of mode 6 is the largest among all HI1470/1 modes, showing that the TMD motion in the conformational change between HI1470/1 and BtuCD-F is well reproduced by mode 6. This is consistent with the above analysis of individual modes. However, the smaller value of CNBD demonstrates that an asymmetric NBD movement of HI1470/1 does not match the symmetric NBD arrangement in the crystal structure of BtuCD-F well.
Some of the modes with large involvement coefficients listed in Table 2 do not have a balanced contribution from TMD and NBD motions; for example, for mode 22 the contribution from TMD motion is nearly zero. In yet other modes, the coefficients CTMD and CNBD have opposite signs, which indicates that the conformational couplings between TMD and NBD in these modes do not conform to the structural differences between the two experimental structures. Therefore, these modes are not suitable for describing the conformational transition between the two structures, even though the global involvement coefficients are large.
Among the remaining modes, mode 1 has a quite large CTMD (−0.114). However, the decomposition of the coefficient shows that the contribution comes mainly from the overall rotational motion of isolated TMD and NBD (RTMD = −0.047, RNBD = −0.058). As mentioned above, the distinguishing feature of the lowest mode is the relative spin motions of the TMD and NBD dimers. Similar spin modes can be found in both BtuCD and BtuCD-F (data not shown), and they contribute to the transition from BtuCD to HI1470/1 (24).
Along the lowest mode, the relative spin motions around the twofold axis of the transporter change the torsion angle (defined in Materials and Methods) between the TMD and NBD dimers. The torsion angle varies among the three crystal structures of BtuCD, BtuCDF, and HI1470/1, and is calculated to be 29.7°, 31.2°, and 35.9°, respectively. Therefore, the spin mode contributes to this structural difference in addition to the conformational change of the translocation pore and nucleotide-binding sites. This spin mode may reveal a general dynamic property of BtuCD and HI1470/1; however, its physiological meaning is still obscure.
Mode 5 is also among the modes with large involvement coefficients with opposite sign to mode 6. However, mode 5's role in the conformational transition should not be confused. It contributes when the other monomer changes its conformation dramatically, since mode 5 is a degenerate mode of mode 6.
Discussion
With an inward-facing translocation pore at the TMD and a more open conformation of the NBD dimer than BtuCD, HI1470/1 represents a distinct conformational state compared with the homologous importer BtuCD. This suggests that in this type of importer, the outward-facing conformation corresponds to the closed state of the NBD dimer with bound ATPs, whereas the inward-facing conformation corresponds to the open state of the NBDs with ATPs hydrolyzed. This notion is also supported by the previous normal-mode analysis of BtuCD (24), which revealed that the conformational transition of TMDs from the outward-facing state to the inward-facing state is coupled to the simultaneous opening of the two ATP-binding sites. In this scenario of substrate translocation, the next step to complete the whole transport cycle would be to return the inward-facing conformation to the outward-facing one of the TMDs.
The low-frequency modes of HI1470/1 show that there are two possible ways in which the TMDs can undergo conformational change in response to the closing of the NBDs. One is represented by modes 4 and 9, in which the cytoplasmic gate of the translocation pore opens wider as the nucleotide-binding sites close. In contrast, the conformational movement represented by modes 5 and 6 tends to close the cytoplasmic gate upon occlusion of the nucleotide-binding sites, which conforms to the conformational change in the recovery of the inward-facing state to the outward-facing state of the transporter. It is interesting that both modes 5 and 6 exhibit asymmetric conformational motion, i.e., the motion of one subunit of HI1470 and HI1471 has much larger amplitude than the other. The movement of individual subunits found in the normal modes coincides perfectly with the structure differences between HI1470/1 and the BtuCD-F complex, in which the translocation pore exhibits an asymmetric conformation (Fig. 10). This strongly suggests that BtuCD-F is an intermediate conformation connecting HI1470/1 and BtuCD, and the inward-facing transporter returns to the outward-facing conformation by resetting each subunit, one by one. On the other hand, the symmetric conformational movement encoded in low-frequency modes, such as modes 4 and 9, may result in futile ATP hydrolysis, since the closure of NBDs does not recover the outward-facing conformation of the translocation pore.
In vitro functional characterization of the BtuCD-F system revealed significant basal ATPase activity in the absence of BtuF and vitamin B12 (42). Although the addition of BtuF can double ATPase activity, the estimated apparent stoichiometry between ATP hydrolysis and B12 uptake is ∼100 ATP per B12. Therefore, substrate transport and ATP hydrolysis are weakly coupled in the BtuCD-F transporter, in contrast to bacterial histidine and maltose importers that show tighter coupling relationships (43,44). The characteristics of the conformational movement of the BtuCD-F/HI1470/1 system demonstrated in the normal-mode analysis may explain the weak coupling relationship between substrate transport and ATP hydrolysis. In both BtuCD and HI1470/1, the low-frequency normal modes involved in significant opening and closing movements at the nucleotide-binding sites do not necessarily induce effective conformational change at the translocation pathway. Some of the modes do not contribute to obvious changes in pore shape, and others induce “wrong” conformational changes at the TMD that could not result in effective substrate translocation, such as modes 4 and 9 of HI1470/1. In other words, the intrinsic conformational flexibility of BtuCD-F-type importers does not dictate a tight conformational coupling between TMD and NBD parts, leaving room for other conformational changes that may result in futile ATP hydrolysis.
The involvement coefficients associated with the transformation from HI1470/1 to BtuCD-F also demonstrate the significant contribution of mode 6. However, the local involvement coefficient CNBD that represents the contribution from NBD movement in mode 6 is much smaller than CTMD, since the asymmetric movement of NBDs in mode 5 or 6 does not conform to the symmetric arrangement of NBDs in both crystal structures. It is notable that the conformational differences of the NBD dimer among the nucleotide-free states of the transporters, such as those of BtuCD (11), HI1470/1 (22), ModBC-ModA (8), and several structures of MalK (17), are significant, in contrast to the highly similar conformations between ATP-bound states (7,10,12,19). This indicates that the NBD dimer has considerable conformational flexibility in the absence of bound nucleotide molecules. The asymmetric movement of NBDs encoded in mode 5 or 6 implies that the two nucleotide-binding sites at the NBD dimer interface close individually by binding one molecule of ATP first, resulting in an asymmetric conformation of the NBD dimer. If this is the case, the asymmetric conformation may not be stable in the absence of nucleotide molecules because of the conformational flexibility of the NBD dimer. In fact, the only NBD dimer structure with an unambiguous asymmetric NBD subunit arrangement keeps an ADP molecule at the binding site (14). The absence of nucleotide molecules may induce the loss of asymmetry in the NBD dimer under crystallographic conditions. A comparison between the structures of BtuCD-F and BtuCD reveals a high similarity between the two BtuD dimer conformations (RMSD = 0.56 Å). On the grounds of motional coupling between TMDs and NBDs in the conformational transformation of the transporter, the highly similar conformations of the NBD dimers in BtuCD-F and BtuCD make it difficult to correlate these two structures along the conformational change pathway. It is anticipated that the intermediate state during the transformation from HI1470/1 to BtuCD adopts both asymmetric conformations at TMD and NBD parts, which may be stabilized in the presence of nucleotide molecules. However, trapping the specific asymmetric transition state related to substrate transportation may be difficult due to significant levels of futile ATP hydrolysis, during which the transporter may experience a variety of conformational changes different from that of substrate translocation.
On the basis of the experimental data and theoretical analysis described above, a refined mechanism of substrate translocation of BtuCD-type importer is proposed and depicted in Fig. 11. In the ATP-binding state of the transporter, the membrane-spanning domain adopts an outward-facing conformation that resembles the BtuCD crystal structure (11). PBP BtuF complexed with substrate docks to the periplasmic side of the TMDs of the ATP-binding transporter. Docking of BtuF may trigger the release of the substrate into the outward-facing pore (41,45). ATP hydrolysis or ADP release at the NBD dimer interface that opens the two nucleotide-binding sites simultaneously can induce a substantial conformational transition at the TMDs, as suggested by the previous normal-mode analysis of BtuCD (24). It is worth noting that the simultaneous opening of two ATP-binding sites does not necessarily require the simultaneous hydrolysis of two ATPs, since we do not know precisely whether nucleotide hydrolysis or release triggers the NBD conformational change. When the NBD dimer opens, the TM helices lining the translocation pore tilt to close the pore at the periplasmic side and open at the cytoplasmic side, transporting the substrate into the cell. After translocation of the substrate, the conformation of the transporter resembles the crystal structure of HI1470/1 (22), albeit with the PBP (BtuF) bound. Next, the asymmetric motion at the NBDs results in the closure of one of the ATP-binding sites by trapping/releasing an ATP/ADP molecule between the P-loop and signature motif, inducing one BtuC subunit to transform to the outward-facing conformation. The TMD conformation of this intermediate state resembles that of the crystal structure of BtuCD-F (9), but possibly with an asymmetric arrangement of NBD dimer as well. Finally, the other BtuC subunit rearranges to an outward-facing conformation upon the association/dissociation of another ATP/ADP molecule at the NBD interface. The BtuF part may dissociate from the transporter at this stage or remain in the bound state. To this end, the transporter recovers to the outward-facing conformation of the ATP-binding state, ready for the next cycle of transportation.
Figure 11.
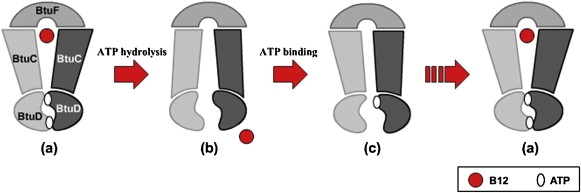
The proposed mechanism of substrate translocation of the BtuCD-type importer. ATP binding induces a closed NBD dimer interface and an outward-facing conformation of the translocation pathway. Then ATP hydrolysis or ADP release opens the two nucleotide-binding sites and results in an inward-facing conformation of the pathway. After translocation of substrate, sequential trapping/releasing of ATPs (or ADPs) induces an asymmetric conformation similar to the BtuCD-F crystal structure. Along with the trapping/releasing of the other ATP/ADP molecule, the transporter recovers to the outward-facing state. See text for a detailed description.
Conclusions
The normal-mode analysis of HI1470/1 reveals that the transporter transforms from the outward-facing conformation to the inward-facing conformation through asymmetric conformational motions of individual subunits of TMD and NBD. In other words, the homodimer of the transporter rearranges the monomers one after another during the conformational transition, implying that the association/dissociation of two molecules of ATP/ADP occurs successively to form the closed NBD dimer. This conformational movement coincides with the structure differences between HI1470/1 and BtuCD-F, which are considered to be intermediate states relating HI1470 and BtuCD. In addition to the dramatic conformational change of the translocation pore coupled with ATP binding and hydrolysis, the transporter also undergoes a twisting motion between TMDs and NBDs all the way along the transportation cycle, changing the torsion angle and interface between NBDs and TMDs. This may represent a general property of ABC transporters of this family.
Acknowledgments
This work was supported by the National Science Foundation of China (20473019), National High Technology Research Program of China (2006AA02A320), National Major Basic Research Program of China (2009CB918600), and Shanghai Leading Academic Discipline Project (B108). J.M. acknowledges support from the Welch Foundation. We are grateful to the Shanghai Supercomputer Center and the Computer Center of Fudan University for their allocation of computer time.
References
- 1.Jones P.M., George A.M. Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol. Lett. 1999;179:187–202. doi: 10.1111/j.1574-6968.1999.tb08727.x. [DOI] [PubMed] [Google Scholar]
- 2.Davidson A.L., Chen J. ATP-binding cassette transporters in bacteria. Annu. Rev. Biochem. 2004;73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- 3.Jones P.M., George A.M. The ABC transporter structure and mechanism: perspectives on recent research. Cell. Mol. Life Sci. 2004;61:682–699. doi: 10.1007/s00018-003-3336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Locher K.P. Structure and mechanism of ABC transporters. Curr. Opin. Struct. Biol. 2004;14:426–431. doi: 10.1016/j.sbi.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Hollenstein K., Dawson R.J.P., Locher K.P. Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Linton K.J. Structure and function of ABC transporters. Physiology (Bethesda) 2007;22:122–130. doi: 10.1152/physiol.00046.2006. [DOI] [PubMed] [Google Scholar]
- 7.Murakami S., Nakashima R., Yamashita E., Matsumoto T., Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- 8.Hollenstein K., Frei D.C., Locher K.P. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213–216. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 9.Hvorup R.N., Goetz B.A., Niederer M., Hollenstein K., Perozo E. Asymmetry in the structure of the ABC transporter-binding protein complex BtuCD-BtuF. Science. 2007;317:1387–1390. doi: 10.1126/science.1145950. [DOI] [PubMed] [Google Scholar]
- 10.Ward A., Reyes C.L., Yu J., Roth C.B., Chang G. Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc. Natl. Acad. Sci. USA. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Locher K.P., Lee A.T., Rees D.C. The E-coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 12.Dawson R.J.P., Locher K.P. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007;581:935–938. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 13.Chen J., Lu G., Lin J., Davidson A.L., Quiocho F.A. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell. 2003;12:651–661. doi: 10.1016/j.molcel.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Lu G., Westbrooks J.M., Davidson A.L., Chen J. ATP hydrolysis is required to reset the ATP-binding cassette dimer into the resting-state conformation. Proc. Natl. Acad. Sci. USA. 2005;102:17969–17974. doi: 10.1073/pnas.0506039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Procko E., Ferrin-O'Connell I., Ng S.L., Gaudet R. Distinct structural and functional properties of the ATPase sites in an asymmetric ABC transporter. Mol. Cell. 2006;24:51–62. doi: 10.1016/j.molcel.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 16.Schmitt L., Benabdelhak H., Blight M.A., Holland B.I., Stubbs M.T. Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains. J. Mol. Biol. 2003;330:333–342. doi: 10.1016/s0022-2836(03)00592-8. [DOI] [PubMed] [Google Scholar]
- 17.Smith P.C. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verdon G., Albers S.V., van Oosterwijk N., Dijkstra B.W., Driessen A.J.M. Formation of the productive ATP-Mg2+-bound dimer of GlcV, an ABC-ATPase from Sulfolobus solfataricus. J. Mol. Biol. 2003;334:255–267. doi: 10.1016/j.jmb.2003.08.065. [DOI] [PubMed] [Google Scholar]
- 19.Yuan Y.R., Blecker S., Martsinkevich O., Millen L., Thomas P.J. The crystal structure of the MJ0796 ATP-binding cassette—implications for the structural consequences of ATP hydrolysis in the active site of an ABC transporter. J. Biol. Chem. 2001;276:32313–32321. doi: 10.1074/jbc.M100758200. [DOI] [PubMed] [Google Scholar]
- 20.Zaitseva J., Jenewein S., Jumpertz T., Holland I.B., Schmitt L. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 2005;24:1901–1910. doi: 10.1038/sj.emboj.7600657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaitseva J., Oswald C., Jumpertz T., Jenewein S., Wiedenmann A. A structural analysis of asymmetry required for catalytic activity of an ABC-ATPase domain dimer. EMBO J. 2006;25:3432–3443. doi: 10.1038/sj.emboj.7601208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinkett H.W., Lee A.T., Lum P., Locher K.P., Rees D.C. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 2007;315:373–377. doi: 10.1126/science.1133488. [DOI] [PubMed] [Google Scholar]
- 23.Parcej D., Tampe R. Caught in the act: an ABC transporter on the move. Structure. 2007;15:1028–1030. doi: 10.1016/j.str.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Weng J.W., Ma J.P., Fan K.N., Wang W.N. The conformational coupling and translocation mechanism of vitamin B-12 ATP-binding cassette transporter BtuCD. Biophys. J. 2008;94:612–621. doi: 10.1529/biophysj.107.110734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janas E., Hofacker M., Chen M., Gompf S., van der Does C. The ATP hydrolysis cycle of the nucleotide-binding domain of the mitochondrial ATP-binding cassette transporter Mdl1p. J. Biol. Chem. 2003;278:26862–26869. doi: 10.1074/jbc.M301227200. [DOI] [PubMed] [Google Scholar]
- 26.Higgins C.F., Linton K.J. The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 27.Rosenberg M.F., Velarde G., Ford R.C., Martin C., Berridge G. Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 2001;20:5615–5625. doi: 10.1093/emboj/20.20.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atilgan A.R., Durell S.R., Jernigan R.L., Demirel M.C., Keskin O. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szarecka A., Xu Y., Tang P. Dynamics of firefly luciferase inhibition by general anesthetics: Gaussian and anisotropic network analyses. Biophys. J. 2007;93:1895–1905. doi: 10.1529/biophysj.106.102780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bahar I., Rader A.J. Coarse-grained normal mode analysis in structural biology. Curr. Opin. Struct. Biol. 2005;15:586–592. doi: 10.1016/j.sbi.2005.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J.P. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure. 2005;13:373–380. doi: 10.1016/j.str.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Sen Taner Z., Kloster M., Jernigan R.L., Kolinski A., Bujnicki J.M. Predicting the complex structure and functional motions of the outer membrane transporter and signal transducer FecA. Biophys. J. 2008;94:2482–2491. doi: 10.1529/biophysj.107.116046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niv M.Y., Filizola M. Influence of oligomerization on the dynamics of G-protein coupled receptors as assessed by normal mode analysis. Proteins. 2008;71:575–586. doi: 10.1002/prot.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng X.L., Lu B., Grant B., Law R.J., McCammon J.A. Channel opening motion of α7 nicotinic acetylcholine receptor as suggested by normal mode analysis. J. Mol. Biol. 2006;355:310–324. doi: 10.1016/j.jmb.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 35.Valadie H., Lacapčre J.J., Sanejouand Y.H., Etchebest C. Dynamical properties of the MscL of Escherichia coli: a normal mode analysis. J. Mol. Biol. 2003;332:657–674. doi: 10.1016/s0022-2836(03)00851-9. [DOI] [PubMed] [Google Scholar]
- 36.Reference deleted in proof.
- 37.Shrivastava I.H., Bahar I. Common mechanism of pore opening shared by five different potassium channels. Biophys. J. 2006;90:3929–3940. doi: 10.1529/biophysj.105.080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taly A., Delarue M., Grutter T., Nilges M., Le Novère N. Normal mode analysis suggests a quaternary twist model for the nicotinic receptor gating mechanism. Biophys. J. 2005;88:3954–3965. doi: 10.1529/biophysj.104.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taly A., Corriginger P.J., Grutter T., de Carvalho L.P., Karplus M. Implications of the quaternary twist allosteric model for the physiology and pathology of nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. USA. 2006;103:16965–16970. doi: 10.1073/pnas.0607477103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Debret G., Valadie H., Stadler A.M., Etchebest C. New insights of membrane environment effects on MscL channel mechanics from theoretical approaches. Proteins. 2008;71:1183–1196. doi: 10.1002/prot.21810. [DOI] [PubMed] [Google Scholar]
- 41.Borths E.L., Locher K.P., Lee A.T., Rees D.C. The structure of Escherichia coli BtuF and binding to its cognate ATP binding cassette transporter. Proc. Natl. Acad. Sci. USA. 2002;99:16642–16647. doi: 10.1073/pnas.262659699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borths E.L., Poolman B., Hvorup R.N., Locher K.P., Rees D.C. In vitro functional characterization of BtuCD-F, the Escherichia coli ABC transporter for vitamin B-12 uptake. Biochemistry. 2005;44:16301–16309. doi: 10.1021/bi0513103. [DOI] [PubMed] [Google Scholar]
- 43.Davidson A.L., Shuman H.A., Nikaido H. Mechanism of maltose transport in Escherichia coli—transmembrane signaling by periplasmic-binding proteins. Proc. Natl. Acad. Sci. USA. 1992;89:2360–2364. doi: 10.1073/pnas.89.6.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu C.E., Liu P.Q., Ames G.F.L. Characterization of the adenosine triphosphatase activity of the periplasmic histidine permease, a traffic ATPase (ABC transporter) J. Biol. Chem. 1997;272:21883–21891. doi: 10.1074/jbc.272.35.21883. [DOI] [PubMed] [Google Scholar]
- 45.Karpowich N.K., Huang H.H., Smith P.C., Hunt J.F. Crystal structures of the BtuF periplasmic-binding protein for vitamin B12 suggest a functionally important reduction in protein mobility upon ligand binding. J. Biol. Chem. 2003;278:8429–8434. doi: 10.1074/jbc.M212239200. [DOI] [PubMed] [Google Scholar]