

Abstract
DD K, a peptide first isolated from the skin secretion of the Phyllomedusa distincta frog, has been prepared by solid-phase chemical peptide synthesis and its conformation was studied in trifluoroethanol/water as well as in the presence of sodium dodecyl sulfate and dodecylphosphocholine micelles or small unilamellar vesicles. Multidimensional solution NMR spectroscopy indicates an α-helical conformation in membrane environments starting at residue 7 and extending to the C-terminal carboxyamide. Furthermore, DD K has been labeled with 15N at a single alanine position that is located within the helical core region of the sequence. When reconstituted into oriented phosphatidylcholine membranes the resulting 15N solid-state NMR spectrum shows a well-defined helix alignment parallel to the membrane surface in excellent agreement with the amphipathic character of DD K. Proton-decoupled 31P solid-state NMR spectroscopy indicates that the peptide creates a high level of disorder at the level of the phospholipid headgroup suggesting that DD K partitions into the bilayer where it severely disrupts membrane packing.
Abbreviations used: DD, dermadistinctin (dermaseptins from P. distincta); CD, circular dichroism; FID, free induction decay; HSQC, heteronuclear single quantum correlation; MALDI, matrix-assisted laser desorption ionization; NOE, nuclear Overhauser effect; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPG, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol; RP-HPLC, reversed phase high performance liquid chromatography; SUV, small unilamellar vesicles; TFA, 2,2,2-trifluoro acetic acid; TFE, 2,2,2-trifluoroethanol
Introduction
The increasing resistance of pathogens against many commonly used antibiotics creates an urgent need to search for new bactericidal and fungicidal compounds. In the past a variety of antibiotic peptides have been isolated from natural sources including plants and animals, which produce, store and secrete antibiotic peptides in exposed tissues, or synthesize such compounds on induction. The availability of these molecules establishes a defense system that can be set into action immediately when infections occur (1,2). Nearly 1000 antimicrobial peptides have been identified and listed in the corresponding databases (http://www.bbcm.univ.trieste.it/∼tossi/amsdb.html or http://aps.unmc.edu/AP/main.php). They have been intensely investigated with the goal to develop alternative agents against pathogenic bacteria and fungi (2) and, indeed, derivatives with improved activities have been designed. Antimicrobial peptides often also exhibit virucidal, tumorcidal, and anti-parasite activities (3,4).
Linear amphipathic peptides are present in many species including fungi, plants, amphibians, insects, or humans (1,2,5). Although a wide variety of sequences have been identified, which differ in amino acid composition, length, and structure, many of these molecules share common physico-chemical properties in carrying an overall cationic charge and by adopting an amphipathic structure when interacting with membranes. There is good evidence that the antimicrobial peptides either exert their antibiotic activities by permeabilizing bacterial membranes or that they interact and cross those membranes to reach internal targets (6,7).
The generally accepted models agree that cationic peptides are attracted by the anionic surface of bacterial membranes (8), where they undergo conformational changes to adopt an amphipathic structure at the level of the bilayer interface and thereafter intercalate into the membrane. However, the mechanism of how the peptides permeabilize the bacterial membranes, why they are selective for bacteria and/or fungi without killing healthy vertebrate cells and how they exert their antimicrobial activity remains a matter of debate and several mechanisms have been proposed to describe the interactions between peptides and lipid bilayers (9–11). Some of the best studied linear peptide antibiotics are those that were found early on in amphibian skins (reviewed in Bechinger (9)) and more recently new compounds have been identified from species living in natural environments and have been characterized (12–14). The frog skin is a valuable source of biologically active peptides, including antimicrobials.
Dermaseptins are a family of cationic linear peptides of 28–34 residues produced by Phyllomedusa secretions, which are very active against Gram-positive and Gram-negative bacterial strains (15). The DD K peptide belongs to the dermaseptin family and has been isolated from the Phyllomedusa distincta frog, which is found in Brazilian Southeast Atlantic forests. DD K is a linear peptide composed of 33 amino acid residues and shows a broad spectrum of activities against several pathogens, including strains from Enteroccus faecalis, Escherichia coli, Staphylococcus aureus, and Pseudomonas aeruginosa, some of which are resistant against commonly used antibiotics (16). More recently, anti-Trypanosoma cruzi activity has also been shown (12). These properties together with the low hemolytic activity of the peptide make DD K a good prototype as an antimicrobial molecule with low activity on mammalian cells (16).
Recent investigations by isothermal titration calorimetry and by fluorescence spectroscopy showed that addition of cholesterol to phosphatidylcholine mimetic membranes led to a diminution of DD K membrane interactions and the concomitant disruption of the lipid bilayers (17). Other investigations by atomic force microscopy indicated that DD K is able to disrupt anionic membranes typical of bacterial membranes at L/P ratios of 100 (18). To obtain detailed information on the peptide conformations in aqueous buffer or in bilayer environments, respectively, and to understand the mechanism of action, more data on the structure and interactions of these peptides with membranes is required.
Solid-state NMR spectroscopy has proven to be a valuable tool for the structural analysis of biomolecules when immobilized within macromolecular aggregates in particular for the investigation of polypeptides associated with extended lipid bilayers (19–24). Whereas all NMR interactions strongly depend on the alignment of the molecules relative to the magnetic field direction, fast rotational diffusion in solution results in averaging and, therefore, only the isotropic chemical shift values as well as scalar couplings are observed in such environments. However, in static solid or semisolid samples averaging is anisotropic or absent and as a consequence the most pronounced features of the spectra can be attributed to this orientational dependence of NMR interactions. This property can be used to deduce valuable information about the relative alignment of bonds and molecules either from uniaxially oriented samples or in nonoriented preparations when fast motional averaging around the membrane normal occurs (25–27). In many studies the orientation-dependent 15N or 13C chemical shifts have been used to derive structural information from membrane-associated polypeptides, but dipolar interactions or quadrupolar splittings have also been investigated (19,23,28).
In particular the 15N chemical shift interaction has proven to provide information on the alignment of α-helical polypeptides relative to the membrane in a straightforward manner (25). Whereas transmembrane helical peptides exhibit 15N chemical shifts in the 200 ppm range those that align parallel to the surface resonate at < 100 ppm. Such measurements have thus been used as an analytical tool during membrane polypeptide structural (19,24,29,30) or biophysical investigations (31,32).
Similar principles apply to the investigation of phospholipid membranes by proton-decoupled 31P solid-state NMR spectroscopy. The anisotropy of the chemical shift interactions results in a orientation-dependent variation of the 31P NMR peak position within a range of 45 ppm for liquid crystalline phosphatidylcholine membranes (33). Furthermore, the corresponding 31P NMR line shape is an indicator of rotational diffusion within the bilayer and as a consequence the macroscopic phase properties of the membrane (34). Therefore the combination of 15N and 31P solid-state NMR spectroscopy provides a more complete view on the peptide-lipid interactions and the resulting peptide topologies.
Materials and Methods
Phosphatidylcholine was purchased from Avanti Polar Lipids (Birmingham, AL). The DD K peptide with the sequence GLWSK IKAAG KEAAK AAAKA AGKAA LNAVS EAV was prepared by solid-phase peptide synthesis on a Millipore 9050 automatic peptide synthesizer using Fmoc (9-fluorenylmethyloxycarbonyl) chemistry and the standard synthesis cycles of the automate. At the underlined position the 15N-labeled alanine was incorporated. The synthetic product was purified using semi-preparative RP-HPLC carried out on a ProtonSIL 300 C4 column (5 μm, 300 Å, 150 × 20 cm). A linear gradient and the following conditions were applied: Solvent A, 10% acetonitrile in 0.1% TFA/water; solvent B, 100% acetonitrile in 0.1% TFA; flow rate, 8 mL/min and detection at a wave length of 210 nm; the ratio of solvent B changed from 17% to 29% in 20 min. The identity of the product was confirmed by MALDI mass spectrometry. For additional CD and liquid-phase NMR experiments DD K was prepared manually by conventional methods of Fmoc solid phase peptide synthesis and purified and characterized as described above.
Vesicle preparation
The appropriate amounts of lipid (POPC) and peptide were dissolved in TFE (2,2,2-trifluoroethanol) and in TFE/H2O 100/5 v/v, respectively. The two solutions were mixed, the bulk of the solvent removed under a stream of nitrogen gas and the remaining traces of solvent by exposure to high vacuum over night. Large multilamellar vesicles were formed from the dry lipid-peptide film by the addition of buffer (10 mM NaH2PO4/Na2HPO4, pH = 7.0) and extensive vortexing. Thereafter the samples were equilibrated by three cycles of vortexing, tip sonication for at least 15 min (Bandelin, Sonorex super RK 514 BH; Berlin, Germany), freezing and thawing. Due to the final freeze/thaw cycle large vesicle are obtained by this procedure. Small unilamellar vesicles were prepared by extruding the POPC preparation twenty times through polycarbonate filters of 100 nm pore size (Avestin, Ottawa, Canada). Alternatively, SUVs of 10 mg/mL POPG were obtained by 2 min of tip sonication until the suspension became transparent.
CD measurements
CD spectra were recorded using Jasco spectropolarimeter J-810 equipped with a Jasco PFD-425S Peltier system for temperature control. The spectra were analyzed using the CDPro software (35,36) and/or by evaluating the circular dichroism at 222 nm (37). The peptide was dissolved in water or water/TFE at 0.2 mg/mL concentrations and transferred into a quartz cuvette of 1 mm path length. Spectra were recorded between 190 and 250 nm using the following parameters: 10 nm min−1 scan speed, 0.2 nm data pitch, 1 s response time, 1 nm band width, and 2 accumulations at 25°C. Alternatively, 10 mg/mL stock solutions of micelles or SUVs were titrated in a stepwise manner to aqueous DD K solutions and the resulting spectra measured at 25°C at scan speeds of 50 nm/min, 0.5 nm data pitch, and 2 s response time. For the CD spectra 8 (micellar samples) to 50 scans (vesicles) were accumulated and the background was subtracted.
Multidimensional solution NMR spectroscopy
Two different samples were prepared for NMR structure determination. First, peptide was dissolved in a mixture of TFE-d2/H2O (50:50, v/v) at 4 mM concentrations. The pH was adjusted to 7.0 with 20 mM aqueous phosphate buffer. Second, a sample containing 1 mM of DDK, 400 mM DPCd38, 5% (v/v) D2O, and 10 μM 2,2-dimethyl-2-silapentane sulfonate (DSS as internal reference) was prepared. The pH of this sample was 6.0.
The solution NMR experiments of the TFE sample were carried out at 20°C on a Bruker Avance DRX spectrometer (Brazilian National NMR Center, Rio de Janeiro, Brazil) operating at 600.043 MHz for the 1H frequency. The DPC sample was investigated at 45°C on a Bruker Avance-III instrument operating at a 1H frequency of 800 MHz at the same center. Triple-resonance (1H/13C/15N) gradient probes (5 mm sample diameter) were used in both cases for all the experiments. Water suppression was achieved by using presaturation techniques (38). All NMR spectra were processed using NMRPIPE (39).
Total correlation spectroscopy spectra were acquired using the MLEV-17 pulse sequence (39). For the TFE sample at 600 MHz the spectral width was chosen to be 6900 Hz, 512 t1 increments were collected with eight transients of 4096 points. At 800 MHz the parameters were: spectral width 9615 Hz, 512 t1 increments, and 32 transients of 4096 points. NOESY spectra (38,40) were acquired using mixing times of 100, 150, 200, 300, and 400 ms (TFE sample) and 80, 100, 120, 140, 160 ms (micellar sample), respectively. At 600 MHz the spectral width was 6900 Hz, 512 t1 increments were collected with 16 transients of 4096 points for each FID, at 800 MHz these parameters were: spectral width 9615 Hz, 512 t1 increments, and 32 transients of 4096 points. Of the TFE sample 1H-13C HSQC spectra were acquired at 600 MHz with F1 and F2 spectral widths of 27,160 Hz and 8993 Hz respectively. Four hundred t1 increments were collected with 56 transients of 1024 points. The experiment was acquired in an edited mode in such a way that CH and CH3 correlations show positive phase and CH2 correlations show negative phase (40,41). 1H-15C HSQC spectra were acquired with F1 and F2 spectral widths of 27,160 Hz and 8993 Hz respectively. Eighty t1 increments were collected with 400 transients of 1024 points for each free induction decay (40,41).
NOE data analysis and structure calculations
The NMR spectra were analyzed using the NMRVIEW software, version 5.0.3 (42). NOE intensities obtained at mixing times of 120 ms (micellar solution) or 200 ms (TFE solution), respectively were converted into semi-quantitative distances by using the calibration by Hybert et al. (42). The upper limits of the distances thus obtained were 2.8, 3.4, and 5.0 Å (for strong, medium, and weak NOEs, respectively). Structure calculations were carried out using the Xplor-NIH software, version 2.14 (43). Starting with an extended conformation 500 structures were generated using a simulated annealing protocol. This was followed by 20,000 steps of simulated annealing at 1000 K and a subsequent decrease in temperature in 15,000 steps in the first slow-cool annealing stage. The stereochemical quality of the lowest energy structures was analyzed by PROCHECK-NMR (44). The display, analysis, and manipulation of the three-dimensional structures were carried out with the program MOLMOL (45). The atomic coordinates of the most stable structures have been deposited in the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) carrying the accession codes PDB 2JX6 and RCSB 100402 (structure in TFE) as well as PDB 2K9B and RCSB 100838 (structure in DPC).
Solid state NMR spectroscopy
Sample preparation
A clear solution of DD K peptide in H2O or TFE/H2O was prepared and mixed with naphthalene in CHCl3 (46) and lipid in TFE. Thereafter the solvent volume was reduced by exposure to a nitrogen stream and applied onto ultra thin coverglass (9 × 22 mm2; Marienfeld, Lauda-Königshofen, Germany), first dried in air and thereafter in high vacuum over night. After the samples have been equilibrated at 93% relative humidity, the glass plates were stacked on top of each other, and the samples sealed with Teflon tape and plastic wrappings.
Solid-state NMR measurements
Solid-state NMR spectra were recorded on a Bruker AMX400 wide-bore NMR spectrometer operating at 9.4 Tesla. Proton-decoupled 15N solid-state NMR spectra were acquired using a commercial double-resonance solid-state NMR probe modified with flattened coils (47) of inner dimensions 15 × 4 × 9 mm3. The sample was introduced into the magnetic field of the NMR spectrometer with the membrane normal parallel to the magnetic field direction. An adiabatic cross polarization sequence (48) was applied with the following typical acquisition parameters: 90° pulse width, 8 μs; spin lock time, 700 μs; recycle delay, 3 s; 512 data points; 70,000 acquisitions; and spectral width, 33 kHz. Before Fourier transformation an exponential apodization function corresponding to a line broadening of 100 Hz was applied. NH4Cl was used as a reference (41.5 ppm). During the solid-state NMR measurements the samples were cooled with a stream of humidified air.
Proton-decoupled 31P solid-state NMR spectra were recorded using a commercial static double-resonance solid-state NMR probe (Bruker, Rheinstetten, Germany). A Hahn echo pulse sequence with high power proton decoupling was used (49). The following spectral parameters were used: spectral width, 75 kHz; acquisition time, 6.4 ms; 2k time domain data points; 90° pulse width, 2.5 μs; interpulse delay, 40 μs; recycle delay, 5 s; number of scans, 100. Before Fourier transformation zero filling to 2048 points and an exponential multiplication corresponding to a line broadening of 150 Hz (100 Hz in case of Fig. 6 A) were applied. The mosaic spread of the oriented solid-state NMR spectra was calculated as described previously (50,51).
Figure 6.
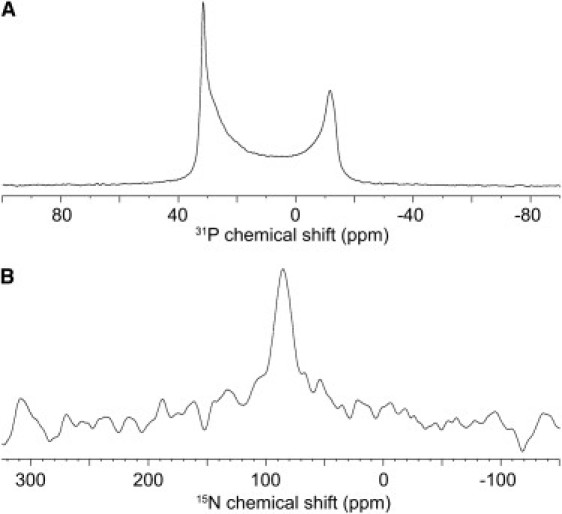
DD K labeled with 15N at its Ala-17 position and reconstituted at 1 mol % into oriented POPC bilayers. (A) Proton-decoupled 31P NMR and (B) proton-decoupled 15N solid-state NMR spectrum of the same sample.
Results and Discussion
To better understand the biological activities and the membrane interactions of DD K the structural alterations of the peptide were first investigated in the presence of membranes, micelles or in membrane-mimetic environments using CD spectroscopy. Whereas the CD spectrum of DD K in aqueous solution is indicative of a predominantly random coil conformation (Fig. 1 A), the stepwise addition of detergent micelles to 42 μM DD K in 20 mM phosphate buffer, 100 mM NaCl, pH 7 results in the appearance of pronounced minima around 208 and 222 nm indicating that the peptides adopt α-helical structures (Fig. 1, A and B). At the highest detergent concentrations the helix content is ≥80%. When investigated in isotropic TFE/water solutions representing the hydrophobic environment of the membrane, the helix content increases with the TFE concentration (Fig. 1 C). Line shape analysis reveals a helix content of ∼80% at TFE concentrations ≥30% v/v (Fig. 1 C). This value is larger than the value calculated for DD K in other alcohols such as propanol, where the peptide helicity reaches 50% (18). In the presence of ≥50 μM POPG in the form of SUV the helical content is ∼70% (Fig. 1 D) and an apparent partitioning constant in the 105 M−1 range can be estimated from the titration experiment (Fig. 1 D). At cationic peptide/POPG ratios close to charge neutrality of the complexes the CD spectra exhibit distorted line shapes (Fig. 1, D and A. Marquette and B. Bechinger, unpublished). In the presence of 1 mM POPC (SUV) ∼50% of the peptides appear in helical conformations (not shown), however, it should be noted that the partition constants of cationic linear peptides to zwitterionic vesicles are typically 2–3 orders of magnitude decreased (52,53) and therefore this value represents the average of membrane-associated and free DD K.
Figure 1.
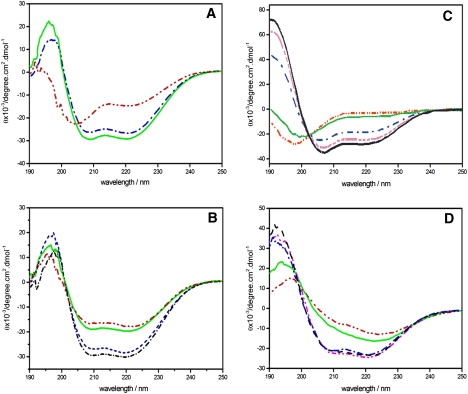
(A) CD spectra of 42 μM DD K in 100 mM NaCl, 20 mM phosphate buffer, pH 7 (dash, dot-dot), and after addition of 200 μM SDS (dash, dot) or 1 mM SDS (solid line). (B) CD spectra of 42 μM DD K in the presence of 200 μM DPC (dash, dot-dot), 400 μM DPC (solid line), 1 mM DPC (dash-dash), or 3 mM DPC (dash, dot). (C) CD spectra of 55 μM DD K in water (solid light line), 10% TFE (dash, dot-dot), 20% TFE (dash, dot), 30% TFE (dash, dot-dot-dot), and 60% TFE (solid black line). (D) CD spectra of 40 μM DD K in from (top to bottom) 10 μM POPG in the form of small unilamellar vesicles (dash, dot-dot), 20 μM POPG (solid line), 50 μM POPG (dash, dot), 100 μM POPG (dash-dash), and 200 μM POPG (dash, dot-dot).
Clearly the DD K peptide associates with and adopts a high degree of helix conformations in the presence of both detergent micelle preparations. As the micellar media match the apolar environment of the membrane as well as its interfacial properties, and in addition they are known to be suitable for high-resolution NMR structure determination of small polypeptides additional measurements were carried out using one-dimensional solution NMR spectroscopy (see the Supporting Material). Solution NMR spectra were recorded of DD K in the presence of 400 mM SDS-d25 or 400 mM DPC-d38, respectively. When compared to the spectra in the aqueous solution or in the presence of 50% TFE the line shapes are considerably broadened in the presence of detergents (Fig. S1) indicating that the peptides associate with the micelles concomitant with a reduction of rotational correlation times. Interestingly, the line shapes ameliorate when the concentration of SDS-d25 is increased from 30 mM to 400 mM (Fig. S2). Furthermore, in the presence of low concentrations of SDS (<20 mM) precipitates are observed that is probably due to the neutralization of cationic charges by the anionic detergent.
The structure of DD K was therefore investigated in the presence of 400 mM DPC-d38, a detergent that forms micelles whose interface is thought to be related closely to that found in phosphatidylcholine bilayers. The determination of the three-dimensional structures of small polypeptides by multidimensional solution NMR spectroscopy has become routine and many high-resolution structures have been obtained using this approach. However, the high redundancy of thirteen alanine and six lysine residues makes the assignment of resonances more challenging than would be expected from a peptide of this size. The proton resonances of DD K in the presence of DPC micelles were attributed using sequential assignments. In the presence of DPC micelles many NOE correlations are observed suggesting a significant structural arrangement of DD K (Fig. 2). The graphical summary of the sequential and medium range NOEs of DD K in the presence of DPC micelles is presented in Fig. 3 B.
Figure 2.
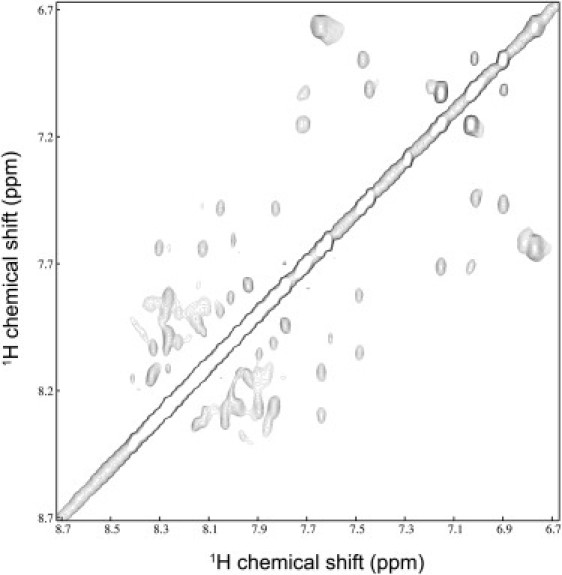
NH-HN region of the NOESY spectrum of 1 mM DD K in a micellar solution of 400 mM DPCd38/H2O 5% D2O.
Figure 3.

Graphical presentation of the NOEs observed for (A) 4 mM DD K in TFE/water 50:50 v/v, and (B) 1 mM DD K in 400 mM DPCd38/H2O and 5% D2O, pH 6. The dNN, dαN, and dβN are sequential NOEs, where strong and weak crosspeaks are indicated by thick and thin lines, respectively. The dαN (i, i + 3), dαβ(i, i + 3), and dαN (i, i + 4) medium-size connectivities link atoms of residues spaced as indicated by the horizontal lines. The outlines of the helical conformation are also indicated.
Medium range NOE crosspeaks involving the amidic protons are observed from the seventh residue up to C-terminus, indicating that the molecule presents a well defined helical arrangement (Fig. 3 B). Notably, the NOESY spectra exhibit many correlations involving protons of residues close to the C-terminus. This is an important observation, because C-terminal amidated cationic peptides often exhibit a higher degree of structuration (54) and are more active in biological assays when compared to the nonamidated analogs (55,56). Some interresidue correlations involving Trp-3 and Ser-4 indicate that this portion of the peptides exhibits some structural order albeit to a lesser extent than the C-terminus.
A total of 270 distance restrains derived from NOE correlations were available for the calculations of the DD K structures in the presence of DPC micelles and the lowest energy structures are shown in Fig. 4, B–D. A well defined helical segment is observed from Lys-7 to the C-terminus in addition to a structured region involving Lys-5 and Ile-6. The statistics of the structural analysis of DD K in the presence of DPC micelles are summarized in Table 1. The RMSD values obtained for the ensemble of all residues are suggestive of considerable conformational flexibility, however, these values significantly drop when only the helical segment is considered. Most of the residues are found in the most favored or in the additionally allowed regions of the Ramachandran plot indicating the good quality of the structures. The few residues observed in the disallowed regions are from residues close to the N-terminus, i.e., the nonstructured part of the peptide.
Figure 4.
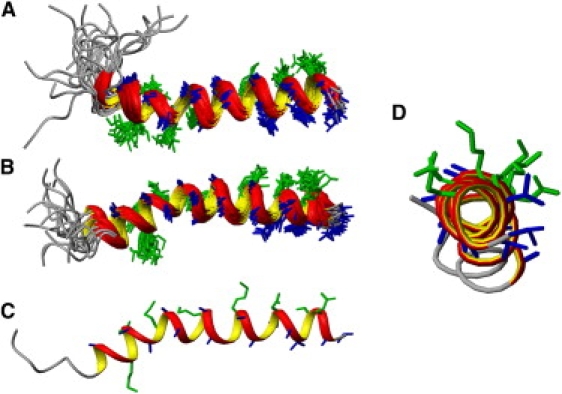
Solution NMR structures of DD K in (A) TFE/water 50:50 v/v, and in (B–D) a micellar solution of DPCd38 in water. (A and B) The 20 lowest energy structures. The hydrophobic residues are represented in blue, the hydrophilic residues in green. (C and D) The lowest energy structure in the presence of detergent viewed along the helix axis and from the side, respectively.
Table 1.
Summary of the structural restraints and statistical analysis of the calculated structures of 1 mM DD K in 400 mM DPCd38/H2O and 5% D2O and of 4 mM DD K in TFE/water 50:50 v/v
| NOE restrains | DPC sample | TFE sample |
|---|---|---|
| Total number of distance restrains | 270 | 294 |
| Number of intraresidue restrains | 169 | 178 |
| Number of sequential restrains (i, i + 1) | 70 | 78 |
| Number of medium range restrains (i, i + j)j=2,3,4 | 31 | 38 |
| RMSD (Å)—all residues∗ | ||
| Backbone | 2.32 | 3.45 |
| Backbone and heavy atoms | 3.02 | 4.53 |
| RMSD (Å)—helical segment∗† | ||
| Backbone | 1.31 | 1.37 |
| Backbone and heavy atoms | 1.83 | 1.88 |
| Ramachandran plot analysis‡ | ||
| Residues in most favored regions | 75.2 | 72.4 |
| Residues in additional allowed regions | 22.1 | 23.3 |
| Residues in generously allowed regions | 2.1 | 4.3 |
| Residues in disallowed regions | 0.7 | 0.0 |
Data from MOLMOL using the 20 lowest energy structures.
From K-7 to V-33.
Data form PROCHECK_NMR.
This structural analysis was extended to an isotropic solvent mixture that mimics the polarity of membranes (TFE/water 50:50 v/v). NOE crosspeaks involving the NH protons ranging from the fifth residue to the C-terminus are observed, indicating that most of the molecule presents a well defined helical structure (Fig. 3 A). Again, the NOESY spectra exhibit many correlations involving protons of residues close to the C-terminus and the C-terminal amide residues. Interresidue NOEs involving protons of Trp-3 and Ser-4 side chains indicate that the N-terminal portion of the peptides is also structured, although to a lesser degree than the C-terminus. The dNN(i,i+1) NOE correlations in TFE-d2/H2O (50:50, v/v) are very similar to those of DD K in the presence of DPC micelles, suggesting the existence of related folds in both media.
Using the NOE restraints 500 structures of DD K were calculated (Table 1). The 20 lowest energy structures in TFE environments are shown in Fig. 4 A. The data are indicative of a predominantly helical conformation starting at residue 5 and extending up to the C-terminus. A small distortion results in a slight bend of the helix involving positions 10 to 16. The high degree of helical structures is in excellent agreement with the CD spectroscopic analysis under similar conditions (Figs. 1 and 4).
Although the observed degree of helicity of DD K is very similar in both environments, an interesting difference becomes obvious when the two structures are compared to each other. Whereas a subtle helix bend is observed at Lys-15 residue for DD K in TFE-d2/H2O (50:50, v/v) (Fig. 4 A), a more pronounced bent is observed at Lys-19 for DD K in the presence of DPC micelles (Fig. 4 B). Furthermore, in the representations shown in Fig. 4, A and B, where the hydrophobic face of the helix is presented in front, the N-terminus points upward for DDK in TFE-d2/H2O (50:50, v/v) but downward in the presence of DPC micelles. One can speculate that this different long-range structure might be due to the curvature imposed by the micellar system. The helix content obtained by NMR structural analysis in TFE/water or in the presence of detergent micelles agrees well with the CD data obtained in the presence of POPG SUVs suggesting that the deposited PDB structures represent reasonably well the conformation of the peptide in phospholipids bilayers.
To investigate the interactions of DD K with biological membranes this peptide antibiotic was reconstituted into oriented phospholipid bilayers. In a first step the effect of the peptide on the organization of the lipid bilayer was investigated and increasing amounts of peptide were mixed with 10 mg of POPC, applied to six glass plates (9 × 22 mm) and the proton-decoupled 31P solid-state NMR spectra of the phospholipid headgroups recorded (Fig. 5). Due to the anisotropic properties of the chemical shift interaction the 31P resonance frequency represents the alignment of the phospholipids relative to the magnetic field direction. For POPC phospholipid bilayers a signal at 30 ppm is observed when the long axis of the phospholipid molecules are aligned parallel to the magnetic field direction, a result that is confirmed by our control experiment (Fig. 5 A). This signal moves to −15 ppm when the sample is tilted to perpendicular orientations (33). Up to peptide/lipid ratios of 1:50 the membranes maintain a good alignment (Fig. 5, B–E). At peptide/lipid ratios of 1:33 much of the order of the lipid headgroup is lost with strong intensities covering the whole chemical shift range of liquid crystalline phosphatidylcholine membranes (Fig. 5 E).
Figure 5.
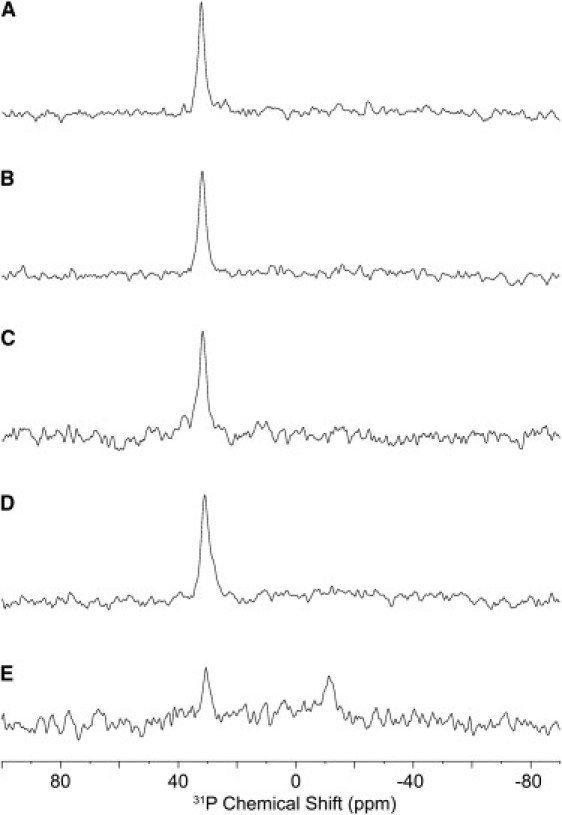
Proton-decoupled 31P solid-state NMR spectra of POPC bilayers in the presence of DD K as a function of peptide concentration: (A) in the absence of peptide; and in the presence of (B) 0.5 mol %; (C) 1 mol %; (D) 2 mol %; and (E) 3 mol % DD K. The bilayers are oriented with the membrane normal parallel to the magnetic field direction.
To increase the size of the samples to permit investigations of the less sensitive 15N nucleus, oriented membranes encompassing 75 mg of lipid and 3.5 mg of peptide were prepared on 28 glass slides (Fig. 6). In this arrangement, the number of membranes stacked in between each pair of glass plates is ∼2–3-fold elevated when compared to the sample shown in Fig. 5 C. Although the peptide/lipid ratio remains the same the phospholipid headgroup order is much reduced in the thick stacks when compared to the smaller samples (Figs. 5 C and 6 A). The 31P NMR spectrum shown in Fig. 6 A can be simulated by considering that ∼50% the phospholipid headgroups are randomly aligned and the other 50% are oriented with a set of populations exhibiting mosaic spreads between 2° and 30°. Related 31P solid-state NMR line shapes have been observed in the presence of other amphipathic peptides (50,57–59) and recent simulations show that related line shapes are obtained from toroidal pore geometries (60).
In a next step the alignment of the peptide within a lipid membrane was investigated by 15N solid state NMR. The sample of Fig. 6 A, encompassing micromole amounts of DD K labeled with 15N at the alanine-17 position, was also investigated by proton-decoupled 15N solid-state NMR spectroscopy (Fig. 6 B). The spectrum shows a well oriented peak at a chemical shift position of (86 ± 3) ppm indicative of helix orientations approximately parallel to the membrane surface (25). It remains possible that a powder pattern line shape of about equal intensity is hidden within the baseline noise of the 15N spectrum that would be in agreement with models where pores are formed by high local curvature and the concomitant rearrangement of the supramolecular peptide-lipid assemblies. This data is in line with results from a variety of linear peptide antibiotics where the formation of amphipathic conformations in membrane environments, helical or other, is a key feature and essential for antibiotic action (1,2,11,61). When several helical sequences, including magainins, dermaseptins, maculatins, or model sequences have been investigated, alignments close to parallel to the membrane surface has been observed also for these peptides using different biophysical approaches including solid-state NMR spectroscopy (8,9,62,63). Furthermore, the antibiotic properties of designed histidine-containing amphipathic model peptides have been correlated to experimental conditions where the peptides adopt in-plane alignments (31,53).
This partitioning of amphipathic structures into the membrane matches the separation of polar and apolar properties at the level of the bilayer interface (Fig. 7). However, a comparison of the molecular dimensions shows that the diameter of amphipathic α-helices is insufficient to completely fill the space liberated by the lipids for intercalation of the peptide thereby causing the disruption of the packing arrangements. Whereas the typical hydrophobic monolayer thickness of a biological membrane is ∼15 Å the hydrophobic radius of an in-plane oriented amphipathic α-helix is only ∼5–6 Å (64). As a consequence, the lipid fatty acyl chains exhibit increased disorder (65) and membrane thinning (66) as well as membrane curvature strain are augmented (67,68). Furthermore, transient openings have been observed at low peptide concentrations and the membranes become unstable at high peptide/lipid ratios (9,11).
Figure 7.

Amphipathic helical wheel of DD K, residues 5–33. The inner circle shows amino acids 5–23, the outer residues 24–33. The charged side chains are circled. It remains possible that the polar C-terminus, including residue E31 is unstructured or distorted (hatched circle) in lipid bilayers thereby providing a more amphipathic structure to the polypeptide.
Whereas transmembrane peptides, such as alamethicin, zervamicin, or gramicidin A have been reconstituted into phospholipid bilayers without much effect on the alignment of the lipid headgroups even at peptide/lipid ratios up to 1:8 (69,70), the preparation of uniformly oriented membranes has often proven difficult when amphipathic peptides are present. In the case of DD K even relatively small concentrations cause considerable misalignment of the phospholipids (Figs. 5 E and 6 A) when at the same time the peptide inserted into the membrane exhibits a well-oriented line shape (Fig. 6 B). This observation suggests that although the sample follows the order imposed by the glass surfaces the lipids adapt to compensate for the geometrical disturbances imposed by the in-plane oriented peptide. This effect is more pronounced when the sample thickness increases (Figs. 5 and 6 A) suggesting that the disorder induced by the peptide in one bilayer propagates from one membrane to the next and is thereby enhanced within the stack. The loss of lipid order as a function of membrane thickness has also been observed previously with pure lipid bilayers as well as in the presence of model peptides. Interestingly, this effect is most pronounced with peptides that mismatch the bilayer thickness or that are oriented parallel to the membrane surface (71). Membrane disordering or membrane disruptive properties have also been observed in the presence of other amphipathic helical peptides and proteins such as magainins (58,64), apolipoproteins (72), myelin basic protein (73), glucagon (74), melittin (75,76), pardaxin (57), signal sequences (77,78), basic amphiphilic model peptides (59,79), as well as of lysolipids (80) or detergents (81). Furthermore, electrostatic interactions can lead to local conformational changes of the lipid headgroup region that might also be reflected in the 31P NMR line shape (82).
Given the parallels in the distribution of hydrophobic and polar/charged domains in both membrane-associated cationic peptides and detergents, as well as of many other physico-chemical properties, a model has been proposed where the interactions of these peptides are best described by phase diagrams (9,11). Such a model describes the biophysical and biological activities of the peptides as a function of peptide concentration, membrane lipid composition, temperature, hydration and other environmental parameters whereby certain areas of the phase diagrams represent more detailed views of the supramolecular aggregates such as the carpet (62) or the wormhole model (60,83). The model is supported by many experiments as it has been possible, for example, to provide evidence of the detergent-like action of magainin peptide antibiotics by monitoring the macroscopic phase properties of such peptide-lipid mixtures using 31P solid-state NMR spectroscopy (64). This comparison with detergents explains observations made with peptides such as increased membrane permeability, pore formation (84), lipid flip-flop (85), and membrane lysis (64). A recent detailed study of the kinetics of pore formation by cecropin A also agrees with such a model (86). Importantly, the investigation also shows that dye release from vesicles involves the formation of large pores without any specific peptide arrangements and where the vesicle contents are released in an all-or-none fashion. An all-or-none mechanism would also be expected when macroscopic phase transitions of the peptide-lipid mixtures are involved in membrane permeabilization. Indeed our 31P NMR data indicate that on addition of DD K the order of the bilayer is maintained until a threshold concentration is reached where “nonoriented” contributions appear (Fig. 5). These line shapes, among other possibilities, may indicate major re-arrangements in the membrane supramolecular architecture. Furthermore, biophysical investigations indicate that the membrane negative surface charge helps membrane association of cationic peptides (8,86) but does not affect the channel structures themselves (86). This is in agreement with our CD data where association with POPG lipid bilayers is considerably increased when compared to POPC (Fig. 1).
We have shown that the helical structure of DD K when associated with phospholipid bilayers results in an amphipathic distribution of side chains and an alignment of the peptide parallel to the membrane surface. As a consequence the regular packing of the membrane is disrupted even at small membrane concentrations of DD K suggesting that this peptide exhibits membrane modifying properties.
Acknowledgments
We are grateful for the financial support of C.M.M. by a CAPES postdoctoral grant, and to CNPq for master and PhD student fellowship to M.V., a PhD fellowship to J.M.R. and a senior fellowship to D.P.V. We thank the Brazilian-French programme CAPES-COFECUP/EGIDE (487/05) that permitted several exchange visits to promote this study. The Bechinger team acknowledges the Institute of Supramolecular Chemistry of the University of Strasbourg for hosting the laboratory.
This work was supported by Fundação da Amparo à Pesquisa do Estado de Minas Gerais and by Vaincre la Mucovicidose (TG0501).
Footnotes
Marcelo Porto Bemquerer's present address is Laboratório de Espectrometria de Massa, Empresa Brasileira de Pesquisa Agropecuária (EMBRAPA)-Recursos Genéticos e Biotecnologia, Estação Parque Biológico, Final W5, Asa Norte, Brasília, DF, 70770-900, Brazil.
Supporting Material
References
- 1.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 2.Boman H.G. Antibacterial peptides: basic facts and emerging concepts. J. Intern. Med. 2003;254:197–215. doi: 10.1046/j.1365-2796.2003.01228.x. [DOI] [PubMed] [Google Scholar]
- 3.Powers J.P., Hancock R.E. The relationship between peptide structure and antibacterial activity. Peptides. 2003;24:1681–1691. doi: 10.1016/j.peptides.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 4.Lehrer R.I. Primate defensins. Nat. Rev. Microbiol. 2004;2:727–738. doi: 10.1038/nrmicro976. [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Olmedo F., Molina A., Alamillo J.M., Rodriguez-Palenzuela P. Plant defense peptides. Biopolymers. 1999;47:479–491. doi: 10.1002/(SICI)1097-0282(1998)47:6<479::AID-BIP6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 6.Hancock R.E., Scott M.G. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. USA. 2000;97:8856–8861. doi: 10.1073/pnas.97.16.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 8.Bechinger B. Membrane-lytic peptides. Crit. Rev. Plant Sci. 2004;23:271–292. [Google Scholar]
- 9.Bechinger B. The structure, dynamics and orientation of antimicrobial peptides in membranes by solid-state NMR spectroscopy. Biochim. Biophys. Acta. 1999;1462:157–183. doi: 10.1016/s0005-2736(99)00205-9. [DOI] [PubMed] [Google Scholar]
- 10.Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 11.Bechinger B., Lohner K. Detergent-like action of linear cationic membrane-active antibiotic peptides. Biochim. Biophys. Acta. 2006;1758:1529–1539. doi: 10.1016/j.bbamem.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Brand G.D., Leite J.R., Silva L.P., Albuquerque S., Prates M.V. Dermaseptins from Phyllomedusa oreades and Phyllomedusa distincta. Anti-Trypanosoma cruzi activity without cytotoxicity to mammalian cells. J. Biol. Chem. 2002;277:49332–49340. doi: 10.1074/jbc.M209289200. [DOI] [PubMed] [Google Scholar]
- 13.Apponyi M.A., Pukala T.L., Brinkworth C.S., Maselli V.M., Bowie J.H. Host-defense peptides of Australian anurans: structure, mechanism of action and evolutionary significance. Peptides. 2004;25:1035–1054. doi: 10.1016/j.peptides.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Brand G.D., Leite J.R., de Sa Mandel S.M., Mesquita D.A., Silva L.P. Novel dermaseptins from Phyllomedusa hypochondrialis (Amphibia) Biochem. Biophys. Res. Commun. 2006;347:739–746. doi: 10.1016/j.bbrc.2006.06.168. [DOI] [PubMed] [Google Scholar]
- 15.Mor A., Nicolas P. Isolation and structure of novel defensive peptides from frog skin. Eur. J. Biochem. 1994;219:145–154. doi: 10.1111/j.1432-1033.1994.tb19924.x. [DOI] [PubMed] [Google Scholar]
- 16.Batista C.V., da Silva L.R., Sebben A., Scaloni A., Ferrara L. Antimicrobial peptides from the Brazilian frog Phyllomedusa distincta. Peptides. 1999;20:679–686. doi: 10.1016/s0196-9781(99)00050-9. [DOI] [PubMed] [Google Scholar]
- 17.Verly R.M., Rodrigues M.A., Daghastanli K.R., Denadai A.M., Cuccovia I.M. Effect of cholesterol on the interaction of the amphibian antimicrobial peptide DD K with liposomes. Peptides. 2008;29:15–24. doi: 10.1016/j.peptides.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 18.Silva L.P., Leite J.R., Brand G.D., Regis W.B., Tedesco A.C. Dermaseptins from Phyllomedusa oreades and Phyllomedusa distincta: liposomes fusion and/or lysis investigated by fluorescence and atomic force microscopy. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2008;151:329–335. doi: 10.1016/j.cbpa.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 19.Cross T.A. Solid-state nuclear magnetic resonance characterization of gramicidin channel structure. Methods Enzymol. 1997;289:672–696. doi: 10.1016/s0076-6879(97)89070-2. [DOI] [PubMed] [Google Scholar]
- 20.Griffin R.G. Dipolar recoupling in MAS spectra of biological solids. Nature. 1998;5:508–512. doi: 10.1038/749. [DOI] [PubMed] [Google Scholar]
- 21.Davis J.H., Auger M. Static and magic angle spinning NMR of membrane peptides and proteins. Prog. Nucl. Magn. Reson. Spectrosc. 1999;35:1–84. [Google Scholar]
- 22.Watts A., Burnett I.J., Glaubitz C., Grobner G., Middleton D.A. Membrane protein structure determination by solid state NMR. Nat. Prod. Rep. 1999;16:419–423. doi: 10.1039/a801189c. [DOI] [PubMed] [Google Scholar]
- 23.Bechinger B., Aisenbrey C., Bertani P. Topology, structure and dynamics of membrane-associated peptides by solid-state NMR spectroscopy. Biochim. Biophys. Acta. 2004;1666:190–204. doi: 10.1016/j.bbamem.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Hong M. Solid-state NMR studies of the structure, dynamics, and assembly of beta-sheet membrane peptides and alpha-helical membrane proteins with antibiotic activities. Acc. Chem. Res. 2006;39:176–183. doi: 10.1021/ar040037e. [DOI] [PubMed] [Google Scholar]
- 25.Bechinger B., Sizun C. Alignment and structural analysis of membrane polypeptides by 15N and 31P solid-state NMR spectroscopy. Concepts Magn. Reson. 2003;18A:130–145. [Google Scholar]
- 26.Prongidi-Fix L., Bertani P., Bechinger B. The membrane alignment of helical peptides from non-oriented 15N chemical shift solid-state NMR spectroscopy. J. Am. Chem. Soc. 2007;129:8430–8431. doi: 10.1021/ja072668k. [DOI] [PubMed] [Google Scholar]
- 27.Cady S.D., Goodman C., Tatko C.D., DeGrado W.F., Hong M. Determining the orientation of uniaxially rotating membrane proteins using unoriented samples: a 2H, 13C, and 15N solid-state NMR investigation of the dynamics and orientation of a transmembrane helical bundle. J. Am. Chem. Soc. 2007;129:5719–5729. doi: 10.1021/ja070305e. [DOI] [PubMed] [Google Scholar]
- 28.Smith R., Separovic F., Milne T.J., Whittaker A., Bennett F.M. Structure and orientation of the pore-forming peptide, melittin, in lipid bilayers. J. Mol. Biol. 1994;241:456–466. doi: 10.1006/jmbi.1994.1520. [DOI] [PubMed] [Google Scholar]
- 29.Bechinger B., Zasloff M., Opella S.J. Structure and orientation of the antibiotic peptide magainin in membranes by solid-state NMR spectroscopy. Protein Sci. 1993;2:2077–2084. doi: 10.1002/pro.5560021208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Epand R.F., Ramamoorthy A., Epand R.M. Membrane lipid composition and the interaction of pardaxin: the role of cholesterol. Protein Pept. Lett. 2006;13:1–5. [PubMed] [Google Scholar]
- 31.Bechinger B. Towards membrane protein design: pH dependent topology of histidine-containing polypeptides. J. Mol. Biol. 1996;263:768–775. doi: 10.1006/jmbi.1996.0614. [DOI] [PubMed] [Google Scholar]
- 32.Aisenbrey C., Kinder R., Goormaghtigh E., Ruysschaert J.M., Bechinger B. Interactions involved in the realignment of membrane-associated helices: An investigation using oriented solid-state NMR and ATR-FTIR spectroscopies topologies. J. Biol. Chem. 2006;281:7708–7716. doi: 10.1074/jbc.M513151200. [DOI] [PubMed] [Google Scholar]
- 33.Seelig J. 31P NMR and the head group structure of phospholipids in membranes. Biochim. Biophys. Acta. 1978;515:105–140. doi: 10.1016/0304-4157(78)90001-1. [DOI] [PubMed] [Google Scholar]
- 34.Cullis P.R., De Kruijff B. Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim. Biophys. Acta. 1979;559:399–420. doi: 10.1016/0304-4157(79)90012-1. [DOI] [PubMed] [Google Scholar]
- 35.Sreerama N., Woody R.W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 36.Sreerama N., Woody R.W. On the analysis of membrane protein circular dichroism spectra. Protein Sci. 2004;13:100–112. doi: 10.1110/ps.03258404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruch M.D., Dhingra M.M., Gierasch L.M. Side chain-backbone hydrogen bonding contributes to helix stability in peptides derived from an α-helical region of carboxypeptidase A. Proteins Struct. Funct. Genet. 1991;10:130–139. doi: 10.1002/prot.340100206. [DOI] [PubMed] [Google Scholar]
- 38.Clarigde T.D.W. Pergamon Press; Amsterdam: 1999. High-Resolution NMR Techniques in Organic Chemistry. Tetrahedron Organic Chemistry Series. [Google Scholar]
- 39.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 40.Braun S., Kalinowski H.O., Berger S. Wiley-VCH, Weinheim; Germany: 1998. One Hundred and Fifty and More Basic NMR Experiments—A Practical Curse. [Google Scholar]
- 41.Wilker W., Leibfritz D., Kerssebaum R., Bernel W. Gradient selection in inverse heteronuclear spectroscopy. Magn. Reson. Chem. 1993;31:287–292. [Google Scholar]
- 42.Hybert S.G., Goldberg M.S., Havel T.F., Wagner G. The solution structure of Eglin C based on measurements of many NOEs and coupling constants and its comparison with x-ray structures. Protein Sci. 1992;1:736–751. doi: 10.1002/pro.5560010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwieters C.D., Kuszewski J.J., Tjandra N., Glore G.M. The Xplor-NIH NMR molecular structure determination package 1996. J. Magn. Reson. 2003;160:66–74. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 44.Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 45.Koradi R., Billeter M., Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 46.Hallock K.J., Henzler W.K., Lee D.K., Ramamoorthy A. An innovative procedure using a sublimable solid to align lipid bilayers for solid-state NMR studies. Biophys. J. 2002;82:2499–2503. doi: 10.1016/S0006-3495(02)75592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bechinger B., Opella S.J. Flat-coil probe for NMR spectroscopy of oriented membrane samples. J. Magn. Reson. 1991;95:585–588. [Google Scholar]
- 48.Pines A., Gibby M.G., Waugh J.S. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973;59:569–590. [Google Scholar]
- 49.Rance M., Byrd R.A. Obtaining high-fidelity spin-1/2 powder spectra in anisotropic media: phase-cycled Hahn echo spectroscopy. J. Magn. Reson. 1983;52:221–240. [Google Scholar]
- 50.Aisenbrey C., Bechinger B. Tilt and rotational pitch angles of membrane-inserted polypeptides from combined 15N and 2H solid-state NMR spectroscopy. Biochemistry. 2004;43:10502–10512. doi: 10.1021/bi049409h. [DOI] [PubMed] [Google Scholar]
- 51.Aisenbrey C., Bertani P., Henklein P., Bechinger B. Structure, dynamics and topology of membrane polypeptides by oriented 2H solid-state NMR spectroscopy. Eur. Biophys. J. 2007;36:451–460. doi: 10.1007/s00249-006-0122-2. [DOI] [PubMed] [Google Scholar]
- 52.Wieprecht T., Beyermann M., Seelig J. Binding of antibacterial magainin peptides to electrically neutral membranes: thermodynamics and structure. Biochemistry. 1999;38:10377–10378. doi: 10.1021/bi990913+. [DOI] [PubMed] [Google Scholar]
- 53.Vogt T.C.B., Bechinger B. The interactions of histidine-containing amphipathic helical peptide antibiotics with lipid bilayers: the effects of charges and pH. J. Biol. Chem. 1999;274:29115–29121. doi: 10.1074/jbc.274.41.29115. [DOI] [PubMed] [Google Scholar]
- 54.Resende J.M., Moraes C.M., Prates M.V., Cesar A., Almeida F.C.L. Solution NMR structures of the antimicrobial peptides phylloseptin-1, -2, and -3 and biological activity: the role of charges and hydrogen bonding interactions in stabilizing helix conformations. Peptides. 2008;29:1633–1644. doi: 10.1016/j.peptides.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 55.Ali M.F., Soto A., Knoop F.C., Conlon J.M. Antimicrobial peptides isolated from skin secretions of the diploid frog, Xenopus tropicalis (Pipidae) Biochim. Biophys. Acta. 2001;1550:81–89. doi: 10.1016/s0167-4838(01)00272-2. [DOI] [PubMed] [Google Scholar]
- 56.Katayama H., Ohira T., Aida K., Nagasawa H. Significance of a carboxyl-terminal amide moiety in the folding and biological activity of crustacean hyperglycemic hormone. Peptides. 2002;23:1537–1546. doi: 10.1016/s0196-9781(02)00094-3. [DOI] [PubMed] [Google Scholar]
- 57.Hallock K.J., Lee D.K., Omnaas J., Mosberg H.I., Ramamoorthy A. Membrane composition determines pardaxin's mechanism of lipid bilayer disruption. Biophys. J. 2002;83:1004–1013. doi: 10.1016/S0006-3495(02)75226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hallock K.J., Lee D.K., Ramamoorthy A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys. J. 2003;84:3052–3060. doi: 10.1016/S0006-3495(03)70031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ouellet M., Doucet J.D., Voyer N., Auger M. Membrane topology of a 14-mer model amphipathic peptide: a solid-state NMR spectroscopy study. Biochemistry. 2007;46:6597–6606. doi: 10.1021/bi0620151. [DOI] [PubMed] [Google Scholar]
- 60.Wi S., Kim C. Pore structure, thinning effect, and lateral diffusive dynamics of oriented lipid membranes interacting with antimicrobial peptide protegrin-1: P-31 and H-2 solid-state NMR study. J. Phys. Chem. B. 2008;112:11402–11414. doi: 10.1021/jp801825k. [DOI] [PubMed] [Google Scholar]
- 61.Epand R.M., Vogel H.J. Diversity of antimicrobial peptides and their mechanism of action. Biochim. Biophys. Acta. 1999;1462:11–28. doi: 10.1016/s0005-2736(99)00198-4. [DOI] [PubMed] [Google Scholar]
- 62.Shai Y. Mechanism of the binding, insertion, and destabilization of phospholipid bilayer membranes by a-helical antimicrobial and cell non-selective lytic peptides. Biochim. Biophys. Acta. 1999;1462:55–70. doi: 10.1016/s0005-2736(99)00200-x. [DOI] [PubMed] [Google Scholar]
- 63.Balla M.S., Bowie J.H., Separovic F. Solid-state NMR study of antimicrobial peptides from Australian frogs in phospholipid membranes. Eur. Biophys. J. 2004;33:109–116. doi: 10.1007/s00249-003-0342-7. [DOI] [PubMed] [Google Scholar]
- 64.Bechinger B. Detergent-like properties of magainin antibiotic peptides: a 31P solid-state NMR study. Biochim. Biophys. Acta. 2005;1712:101–108. doi: 10.1016/j.bbamem.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 65.Mason A.J., Bechinger B. Zwitterionic phospholipids and sterols modulate antimicrobial peptide-induced membrane destabilization. Biophys. J. 2007;93:4289–4299. doi: 10.1529/biophysj.107.116681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ludtke S., He K., Huang H. Membrane thinning caused by magainin 2. Biochemistry. 1995;34:16764–16769. doi: 10.1021/bi00051a026. [DOI] [PubMed] [Google Scholar]
- 67.Matsuzaki K., Sugishita K., Ishibe N., Ueha M., Nakata S. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry. 1998;37:11856–11863. doi: 10.1021/bi980539y. [DOI] [PubMed] [Google Scholar]
- 68.Ramamoorthy A., Thennarasu S., Lee D.K., Tan A., Maloy L. Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 2006;91:206–216. doi: 10.1529/biophysj.105.073890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moll F., 3rd, Cross T.A. Optimizing and characterizing alignment of oriented lipid bilayers containing gramicidin D. Biophys. J. 1990;57:351–362. doi: 10.1016/S0006-3495(90)82536-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bechinger B., Skladnev D.A., Ogrel A., Li X., Swischewa N.V. 15N and 31P solid-state NMR investigations on the orientation of zervamicin II and alamethicin in phosphatidylcholine membranes. Biochemistry. 2001;40:9428–9437. doi: 10.1021/bi010162n. [DOI] [PubMed] [Google Scholar]
- 71.Harzer U., Bechinger B. The alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: a CD, 15N and 31P solid-state NMR spectroscopy investigation. Biochemistry. 2000;39:13106–13114. doi: 10.1021/bi000770n. [DOI] [PubMed] [Google Scholar]
- 72.Segrest J.P., Garber D.W., Brouillette C.G., Harvey S.C., Anantharamaiah G.M. The amphipathic alpha-helix: a multifunctional structural motif in plasma apolipoproteins. Adv. Protein Chem. 1994;45:303–369. doi: 10.1016/s0065-3233(08)60643-9. [DOI] [PubMed] [Google Scholar]
- 73.Roux M., Nezil F.A., Monck M., Bloom M. Fragmentation of phospholipid bilayers by myelin basic protein. Biochemistry. 1994;33:307–311. doi: 10.1021/bi00167a040. [DOI] [PubMed] [Google Scholar]
- 74.Jones A.J.S., Epand R.M., Lin K.F., Walton D., Vail W.J. Size and shape of the model lipoprotein complex formed between glucagon and dimyristolglycerophosphocholine. Biochemistry. 1978;17:2301–2307. doi: 10.1021/bi00605a007. [DOI] [PubMed] [Google Scholar]
- 75.Dufourcq J., Faucon J.-F., Fourche G., Dasseux J.-L., Le Maire M. Morphological changes of phosphatidylcholine bilayers induced by melittin: vesicularization, fusion, discoidal particles. Biochim. Biophys. Acta. 1986;859:22–48. doi: 10.1016/0005-2736(86)90315-9. [DOI] [PubMed] [Google Scholar]
- 76.Dempsey C.E., Sternberg B. Reversible disc-micellization of dimyristoylphosphatidylcholine bilayers induced by melittin and [Ala-14]melittin. Biochim. Biophys. Acta. 1991;1061:175–184. doi: 10.1016/0005-2736(91)90283-e. [DOI] [PubMed] [Google Scholar]
- 77.Killian J.A., de Jong A.M., Bijvelt J., Verkleij A.J., De Kruijff B. Induction of non-bilayer lipid structures by functional signal peptides. EMBO J. 1990;9:815–819. doi: 10.1002/j.1460-2075.1990.tb08178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Batenburg A.M., Demel R.A., Verkleij A.J., De Kruijff B. Penetration of the signal sequence of Escherichia coli PhoE protein into phospholipid model membranes leads to lipid-specific changes in signal peptide structure and alterations of lipid organization. Biochemistry. 1988;27:5678–5685. doi: 10.1021/bi00415a043. [DOI] [PubMed] [Google Scholar]
- 79.Reynaud J.A., Grivet J.P., Sy D., Trudelle Y. Interactions of basic amphiphilic peptides with dimyristoylphosphatidylcholine small unilamellar vesicles: optical, NMR and electron microscopy studies and conformational calculations. Biochemistry. 1993;32:4997–5008. doi: 10.1021/bi00070a005. [DOI] [PubMed] [Google Scholar]
- 80.Inoue K., Suzuki K., Nojima S. Morphology of lipid micelles containing lysolecithin. J. Biol. Chem. 1977;81:1097–1106. doi: 10.1093/oxfordjournals.jbchem.a131534. [DOI] [PubMed] [Google Scholar]
- 81.Sanders C.R., 2nd, Prestegard J.H. Magnetically orientable phospholipid bilayer containing small amounts of a bile salt analogue, CHAPSO. Biophys. J. 1990;58:447–460. doi: 10.1016/S0006-3495(90)82390-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Scherer P.G., Seelig J. Electric charge effects on phospholipid headgroups. phosphatidylcholine in mixtures with cationic and anionic amphiles. Biochemistry. 1989;28:7720–7727. doi: 10.1021/bi00445a030. [DOI] [PubMed] [Google Scholar]
- 83.Ludtke S.J., He K., Heller W.T., Harroun T.A., Yang L. Membrane pores induced by magainin. Biochemistry. 1996;35:13723–13728. doi: 10.1021/bi9620621. [DOI] [PubMed] [Google Scholar]
- 84.Duclohier H., Molle G., Spach G. Antimicrobial peptide magainin I from Xenopus skin forms anion-permeable channels in planar lipid bilayers. Biophys. J. 1989;56:1017–1021. doi: 10.1016/S0006-3495(89)82746-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsuzaki K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta. 1998;1376:391–400. doi: 10.1016/s0304-4157(98)00014-8. [DOI] [PubMed] [Google Scholar]
- 86.Gregory S.M., Cavenaugh A., Journigan V., Pokorny A., Almeida P.F.F. A quantitative model for the all-or-none permeabilization of phospholipid vesicles by the antimicrobial peptide cecropin A. Biophys. J. 2008;94:1667–1680. doi: 10.1529/biophysj.107.118760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.