

Abstract
Athymic mice, injected with A375 human melanoma cells, were treated daily with intraperitoneal injections of adenosine 5′-triphosphate (ATP). The tumour volume and animal weight were measured over the course of the experiment and the final tumour nodule weight was measured at the end of the experiment. Tumour volume decreased by nearly 50% by 7 weeks in treated mice. Weight loss in untreated animals was prevented by ATP. Histological examination of the excised tumour nodules showed necrosis in the ATP-treated tumours only. The presence of P2Y1 and P2X7 receptors, previously proposed as extracellular targets for melanoma treatment with ATP, were demonstrated in the excised specimens by immunohistochemistry. This paper provides further support for the use of ATP as a treatment for melanoma.
Keywords: Melanoma, Cancer, Purinergic signalling, ATP
Introduction
Malignant melanoma is a very aggressive cancer that originates from melanocytes, the pigment-producing cells of the skin. The incidence of malignant melanoma has doubled every 10 years since the 1950s and it is predicted that by the year 2010, the lifetime risk of the disease will approach 1 in 50 of the United Kingdom population [1]. Around 1,500 patients die each year from malignant melanoma in the UK. Unlike other cancers, metastatic (cancer which has spread around the body) melanoma is virtually untreatable by methods of cancer treatment such as surgery, radiotherapy and chemotherapy. Despite significant advances in the treatment of other cancers, the mortality from melanoma has remained unchanged: 66% of patients with metastatic melanoma will die within 5 years. Alternative forms of treatment for this cancer are, therefore, urgently needed.
Interactions between the nervous system and epidermal melanocytes have been suspected on the basis of their common embryological origin from the neural crest but little published work exists to support this. It has been suggested that melanocytes are innervated by autonomic nerves [2] with both acetylcholine and noradrenaline acting as transmitter molecules [3]. Melanomas, which are tumours of melanocyte origin, have also been reported to have an autonomic innervation which is related to their growth and differentiation [4].
In addition to its key role in cellular metabolism, where it acts as a ubiquitous enzyme co-factor and as the key source of the cellular energy unique to phosphate bond formation, the purine nucleotide adenosine 5′-triphosphate (ATP) also functions as a potent extracellular messenger producing its effects via a distinct family of cell surface receptors [5]. ATP was shown to be a co-transmitter with noradrenaline in sympathetic nerves in smooth muscle cells of the vas deferens [6]. Sympathetic co-transmission has also been clearly demonstrated in a variety of blood vessels [7]. Co-transmission with acetylcholine in parasympathetic nerves has also been proposed in organs such as the urinary bladder [8, 9].
ATP acts on extracellular receptors which have been cloned and characterised to consist of two families: P2X ion channel receptors with seven subtypes and P2Y G protein-coupled receptors with eight subtypes [5]. ATP acting on these receptors is involved with both rapid signalling in neurotransmission and also long-term signalling in cell proliferation, differentiation and apoptosis [10].
It has been shown recently that melanoma cells express P1 receptors [11] and P2X7 receptors [12]. Our work confirms this [13, 14] and also identifies the presence of other P2 receptors; in particular, the P2Y1 and P2Y2 subtypes. In vitro work already performed with purinergic receptor agonists, such as ATP and 2′-3′-O-(4-benzoyl-benzoyl) ATP (BzATP), has shown that they produce a reduction in melanoma cell number [13, 14]. Inoculation of MF-1 immunocompromised mice, with A375 melanoma cells is a well-established model of cancer research [15, 16]. Athymic mice are deficient in matured T cell maturity and make excellent models of cancer growth with a reported take of cancer cells of 100% after subcutaneous injection. Therapeutic plasma levels of ATP or other adenosine nucleotides have been shown to be easily achieved by administration through the intraperitoneal route [17].
The objectives of this study are to establish whether purinergic receptor agonists can prevent or reduce melanoma growth and metastasis in vivo by producing a model of melanoma by subcutaneous injection of A375 melanoma cells into immunocompromised mice. These mice are treated by intraperitoneal injection of ATP and the outcome assessed by measurement of tumour volume, tumour weight and animal weight; in addition to histological analysis of the tumour specimens.
Materials and methods
Cell culture
The melanoma cell line A375 [18] was obtained from the Wellcome Trust Functional Genomics Cell Bank (St Georges Hospital Medical School, London, UK). Cell culture medium and reagents were purchased from Sigma (Sigma Chemical Co., Poole, UK). Melanoma cells were grown in 90% Dulbecco’s Modified Eagle’s Medium (DMEM) and 10% heat-inactivated foetal calf serum supplemented with penicillin (100 U per ml), streptomycin (100 µg per ml) and l-glutamine (2 mM) in 75 cm2 tissue culture flasks (Corning, New York, USA). Cells were incubated at 37°C in 5% CO2/95% air and were subcultured at 70% confluence. Cell number and viability was determined using the trypan blue exclusion method.
In vivo model
Twenty 6- to 10-week-old male nude mice, strain MF-1, weighing 25–35 g were used in this study. They were kept under barrier conditions in a pathogen-free environment and had access to food and water ad libitum. On day 1 of the experiment, 5 × 106 A375 melanoma cells were subcutaneously injected, under light general anaesthesia, to induce localised tumour nodules. The injection site was high on the right rear flank. The cells were injected in a 200-µl suspension consisting of 100 μl of sterile Dulbecco’s Phosphate Buffered Saline (DPBS; Sigma, Poole, UK) and 100 μl of matrigel (BD Biosciences, Erembodegem, Belgium). The animals were then randomly allocated to two groups of ten which were designated as either the treatment or control group. They were allowed to recover from the procedure. At all times, during the full course of the experiment, the mice were monitored for adverse effects. These included: (a) loss of >15% body weight; (b) tumour weight >10% body weight; (c) ulceration over the tumour; and (d) abnormal behaviour or appearance.
This experiment was designed in accordance with the United Kingdom guidelines for the production of solid tumours [19].
Intraperitoneal injection of ATP
Ten days post inoculation with A375 melanoma cells, the treatment group of ten mice commenced daily intraperitoneal injections of ATP. The mice had a mean weight of 30 g. Each mouse was injected with 1 ml of 50 mM ATP in a solution of DPBS which was corrected to a pH 6.2 with 1 M NaOH. This dose was equivalent to injecting a 30-g mouse with 1 mg of ATP per gram of body weight. The mice underwent daily injections for 39 days after which they were killed according to Home Office (UK) regulations covering Schedule 1 procedures and approved by the institutional ethics committee.
Measurement of outcome and statistical analysis
Three-dimensional tumour measurements were performed using callipers and the tumour volume was calculated using the formula: 4/3Π(L/2 × W/2)3 where L is long axis and W is mean midaxis width [20]. The animals were weighed at regular intervals. After the animals were killed the tumours were removed and weighed. The results were analysed using an analysis of variance (ANOVA; Excel, Office XP Professional).
Immunohistochemistry
Excised specimens of melanoma were embedded in paraffin for haematoxylin and eosin staining and P2Y1 receptor immunostaining or frozen for P2X7/TdT-mediated dUTP nick end labelling (TUNEL) staining. Paraffin blocks were sectioned at 4 µm on a Reichert-Jung Microtome, and sections were taken on Snowcoat Extra slides (Surgipath, Cambridgeshire, UK), then dried in an oven for 2 h at 60°C. Sections were de-waxed and rehydrated using xylene and graded concentrations of ethanol. Antigen retrieval was performed by microwaving for 10 min in a solution of 1 mM ethylenediamine tetraacetic acid (Tris–EDTA) at pH 9.0. Endogenous alkaline phosphatase was blocked by 20 min of incubation in 20% acetic acid. Sections were washed and then incubated with avidin D blocking solution, biotin blocking solution and 1:5 normal swine serum (Vector Laboratories).
Polyclonal anti-P2Y1 receptor antibody, corresponding to a 17 peptide sequence of the intracellular portion of the transmembrane receptors was obtained from Alomone Laboratories (Jerusalem, Israel). The antibody was kept frozen at a stock concentration of 0.6 mg/ml and used at a dilution of 1:100. One hundred microlitres of anti-P2Y1 receptor antibody, diluted 1:100, was applied for 12 h at 4°C. One hundred microlitres of biotinylated anti-rabbit antibody (DAKO E0353), diluted 1:200 in DAKO ChemMate was applied for 30 min followed by 100 µl of streptavidin alkaline phosphatase (Vector SA5100) diluted 1:200 in DAKO ChemMate for 30 min. Vector Red substrate (Vector Alkaline phosphatase substrate, SK5100) made up in 200 mM Tris–HCl (pH 8.2) was then applied for 10 min. Positive staining appeared bright pink, nuclei were counterstained with haematoxylin (purple). All sections were subsequently dehydrated, cleared and mounted. Negative controls were performed by either omission of the primary antibody or preabsorption of the primary antibody with the corresponding peptide sequence.
For immunostaining of frozen cryostat sections, slides were fixed in 4% formaldehyde in 0.1 M phosphate buffer for 2 min. Non-specific binding sites were blocked by a 20-min preincubation with 10% normal horse serum (NHS) in 0.1 M phosphate buffer containing 0.05% Merthiolate, followed by incubation with primary P2X7 antibody [13] diluted 1:100, with 0.2% Triton x-100, for 12 h at 4°C. Subsequently, the slides were incubated with donkey anti-rabbit Cy3 (Jackson Immunoresearch, Pennsylvania, USA) diluted 1:300 with 1% NHS in phosphate buffer. Slides were then mounted and examined. Control experiments were carried out with the primary antibody being omitted from the staining procedure or the primary antibody preabsorbed with the corresponding peptide. All other reagents were obtained from Sigma (Poole).
Double-labelling with P2X7 receptor antibodies and TUNEL was performed using a kit (Boehringer Mannheim, Germany). After overnight incubation with P2X7 receptor antibody diluted to 1:100 as above, sections were washed in phosphate buffered saline (PBS) and then incubated with the TUNEL reaction mixture for 1 h at 37°C. As a negative control, sections were incubated with the TUNEL Label solution only. After further washes in PBS, sections were then incubated with Cy3 conjugated secondary antibody for 1 h, were washed in PBS and mounted.
Results
All animals tolerated the subcutaneous injection of cancer cells and the intraperitoneal injections of ATP; they grew and developed normally in other respects and none of the mice had to be killed before the endpoint of the experiment due to any adverse effect.
The time course of the experiment was 49 days. At day 1, the mice were inoculated with cancer cells, at day 10 the injections of ATP commenced and the mice were killed on day 49. After 21 days, there was a clinically obvious development of a palpable tumour nodule in the mice.
Animal weight
The weight of the untreated group of animals decreased over the 6-week course of the experiment from a mean of 30 g to a mean of 28.2 g. However, in the treated group, the weight of the animals increased from a mean of 30 to 31.8 g (Fig. 1a). There was a statistically significant difference in the weight of the animals over the course of the experiment (P = 0.0038).
Fig. 1.

a The weight of the untreated group of animals decreased over the six week course of the experiment from a mean of 30 g to a mean of 28.2 g. However, in the treated group, the weight of the animals increased from a mean of 30 to 31.8 g. There was a statistically significant difference in the weight of the animals over the course of the experiment (P = 0.0038). b There was a statistically significant reduction in tumour volume in the treated group compared to the untreated group (P = 0.0163). Tumours in both the treated and untreated groups continued to grow during the course of the experiment but the rate of growth of the untreated group was much higher than that of the treated group. c At the end of the experiment, the tumour nodules were excised and weighed. There was a statistically significant reduction in final tumour weight in the treated group compared to the untreated group (P = 0.0156). The tumours from the untreated group had a mean weight of 1.92 ± 0.31 g (n = 10) compared to the tumours from the treated group which had a mean weight of 1.15 ± 0.24 g (n = 10)
Tumour volume
There was a statistically significant reduction in tumour volume in the treated group compared to the untreated group (P = 0.0163). Tumours in both the treated and untreated groups continued to grow during the course of the experiment but the rate of growth of the untreated group was much higher than that of the treated group (Fig. 1b). By day 49, there was a nearly 50% reduction in tumour volume in the treated animals.
Tumour weight
At the end of the experiment, the tumour nodules were excised and weighed (Fig. 1c). There was a statistically significant 40% reduction in the final tumour weight in the treated group compared to the untreated group (P = 0.0156). The tumours from the untreated group had a mean weight of 1.92 ± 0.31 g compared to the tumours from the treated group which had a mean weight of 1.15 ± 0.24 g.
Histological examination of excised tumour specimens
The microscopic appearance of the excised tumour nodule from the animal model was similar to the appearance of a normal melanoma when examined with a standard haematoxylin and eosin stain. The tumour nodules excised from the untreated group were solid tumours whereas the specimens from the treated group contained patchy areas of necrosis (Figs. 2a and b).
Fig. 2.

Haematoxylin and eosin staining (a and b) and immunohistochemical staining P2Y1 receptors (c and d) of excised tumour nodules. a Solid tumour from an untreated mouse. b Tumour from a mouse treated with ATP showing patchy necrosis, illustrated by red staining areas with no nuclear counterstain (arrows). c Scant expression of P2Y1 receptors (pink) in a specimen of untreated melanoma with a haematoxylin nuclear counterstain (purple). b Increased P2Y1 receptor staining in the ATP-treated group concentrated around the areas of necrosis. Haematoxylin nuclear counterstain (purple). All calibration bars = 250 µm
In addition, immunohistochemical analysis of the tumour cells for P2Y1 and P2X7 receptors (the two receptor subtypes implicated in causing a decrease in cell number and apoptosis, respectively) showed the presence of these receptors.
P2Y1 receptors were only scarcely distributed in the untreated group of melanomas whereas there was greater P2Y1 receptor staining in the treated group but this was concentrated around the areas of necrosis (Figs. 2c and d). Localisation studies for the P2X7 receptor and TUNEL showed extracellular staining of the P2X7 receptor and cytoplasmic staining for TUNEL (Fig. 3). TUNEL identifies cells undergoing apoptosis by labelling nuclear DNA fragments that have been cleaved during apoptosis [21]. There was some overlap of staining, but that TUNEL largely stained the cytoplasm of melanoma cells undergoing apoptosis in the specimens from the group which was treated with ATP. P2X7 receptor extracellular staining was present in both sets of tissue.
Fig. 3.
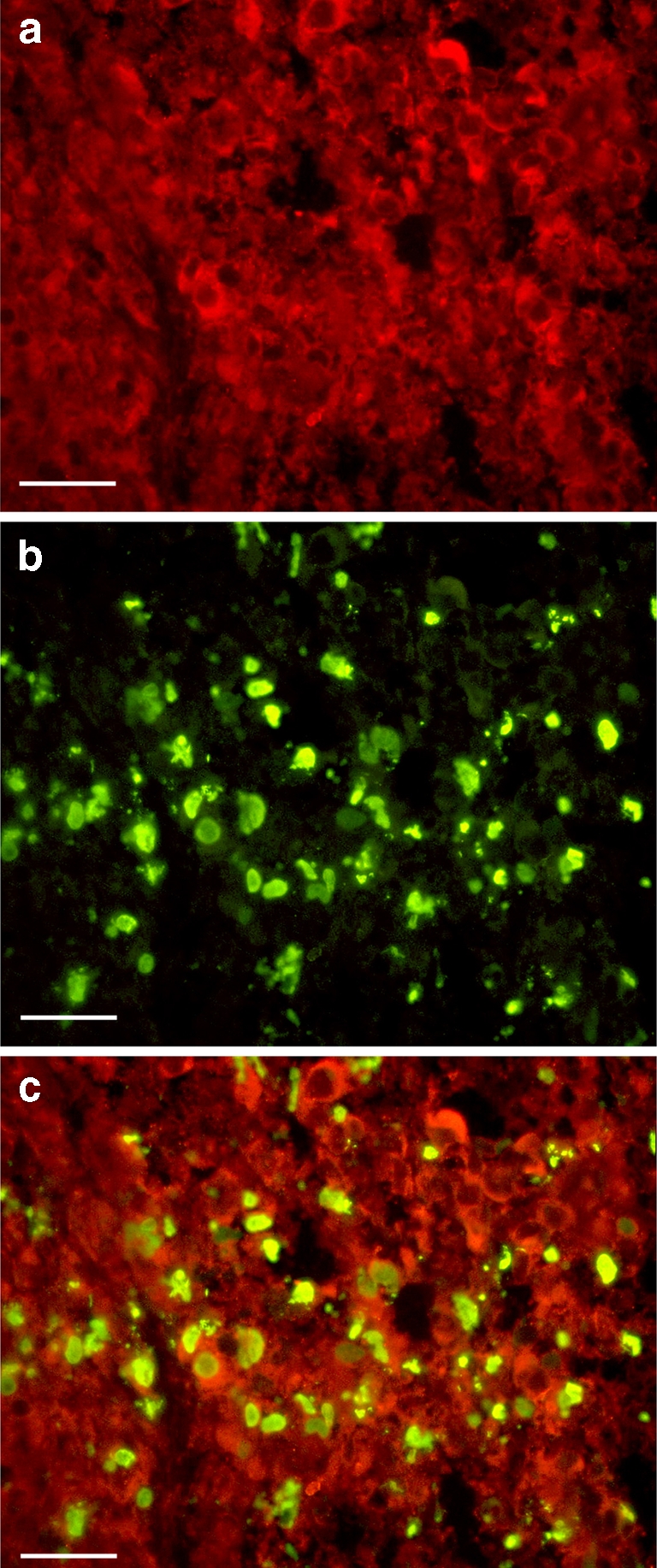
Localisation of P2X7 receptors and TUNEL in an excised melanoma from an ATP-treated mouse. a Red extracellular P2X7 staining. b Green intracellular TUNEL staining. c Localisation of extracellular P2X7 receptors and intracellular TUNEL in ATP-treated melanoma tissue undergoing apoptosis. Calibration bars = 50 μM
Discussion
We have shown that intraperitoneal injections of ATP inhibit the growth of A375 melanoma cells in an in vivo model. The rate of growth of induced tumours was decreased in the group treated with ATP. The final volume and weight, at 49 days, of the induced tumour nodule was less in the treated group than the untreated group. Weight loss was prevented in those tumour-bearing mice which were treated with ATP. It would be desirable to confirm these findings on other melanoma xenograft models, such as 70-W, C 6181, G361, MeWo, SK-MEL-5 and B16-F10 (see [22–24]).
The intraperitoneal route of administration of ATP is the only practical route of access for giving large doses and volumes of ATP in solution to a small animal. Previous experimental work [25] has shown that an intraperitoneal injection of 50 mM of ATP gives a circulating plasma level of 5 μM 5 h post injection, compared to a baseline measurement of 10 nM. The concentrations of ATP needed to produce a decrease in cell number in the previously described in vitro studies [14] are higher than the circulating plasma levels in this in vivo model but still produce an anti-cancer effect. This can be explained by a single dose of ATP being used in the in vitro system compared to multiple daily injections in the in vivo model. Also, the efficacy of the anti-cancer effects of ATP in humans is underestimated in in vivo murine studies. This is due to human blood and tissue having a significantly lower level of enzyme activity to breakdown extracellular ATP than a mouse [26]. Intravenous infusions of relatively low levels of ATP into humans has been shown to produce a rapid elevation of circulating ATP levels in both healthy volunteers [27] and cancer patients [28].
A striking finding in our study is the prevention of cancer-induced weight loss or cachexia. This has been previously described as an effect of ATP treatment of cancer in both murine models of other cancer types [17] and clinical trials [29]. In an in vivo study such as the work presented here it is necessary to ask whether the prevention of weight loss is due to the direct effect of ATP acting on cancer cells or is it mediated by another action of ATP in the animal. Other possible mechanisms of action include the up-regulation of the immune system to combat a growing tumour mediated by increased circulating plasma ATP levels. Purines, such as ATP, have been implicated in priming the immune system and activating immune-response cascades [30]. This may be occurring in a whole animal model of cancer leading to a decreased rate of growth of the cancer cells and less weight loss. Also, the liver plays a major role in cancer cachexia. Cancer-induced weight loss is mediated by increased catabolism in the liver to provide energy for the rapidly growing tumour cells. This causes increased gluconeogenesis which increases ATP consumption and decreased glycolysis which decreases ATP synthesis. Following intraperitoneal injection, ATP will pass through the liver following its absorption into the enteric circulation. Therefore, some of this ATP may be used to reducing any weight loss without being associated with its direct effect on the tumour itself.
In this study, we have strong histological evidence for ATP acting directly on the tumour. Firstly, the tumours treated with ATP show areas of necrosis which is not present in the untreated groups when examined microscopically after routine H and E staining. P2Y1 and P2X7 receptors have been described to have anti-cancer actions in A375 melanoma cells [13, 14] and in HT168-M1 cells [31]. Very few of the melanoma cells in the untreated group expressed the P2Y1 receptor, but there were substantially more cells positive for the P2Y1 receptor in the ATP-treated group and these were concentrated around the areas of necrosis. Finally, we have shown that the P2X7 receptor mediates apoptosis in melanoma cells. In tumour specimens excised from host mice treated with ATP, there was localisation of extracellular P2X7 receptors with intracellular TUNEL, a marker of apoptosis.
Regarding the mechanisms underlying the anti-tumour effects of ATP, it appears that ATP has a dual action. Its action via P2Y1 receptors is to reduce tumour-cell proliferation, while occupation of P2X7 receptors expressed by the tumour cells leads to apoptotic cell death. These anti-tumour effects might be counteracted to some extent by adenosine, produced after breakdown of ATP by ecto-nucleotidase, which is known to promote proliferation of tumour cells, as well as acting as an immunosuppressant [32–36]. However, adenosine has also been shown to have anti-tumour effects by inhibiting tumour growth via A3 receptors [37–39] or by inhibiting apoptosis [40]. Further studies are needed to resolve these contradictory issues.
In summary, ATP has an anti-cancer activity when used to treat athymic MF-1 mice inoculated with A375 melanoma cells and this action appears to be mediated through P2Y1 and P2X7 receptors. This study has provided further evidence for a therapeutic role of ATP in the treatment of cancer (see [41]).
Acknowledgements
This work was supported by a research fellowship from The Royal College of Surgeons of England, a research pump-priming grant from the Royal College of Surgeons of Edinburgh and the Paton/Masser research award from the British Association of Plastic Surgeons.
References
- 1.Grin-Jorgensen CM, Rigel DS, Friedman RJ (1992) The world-wide incidence of malignant melanoma. In: Balch CM (ed) Cutaneous melanoma. Lippincott Williams & Wilkins, Philadelphia, pp 27–39
- 2.Hara M, Toyoda M, Yaar M, Bhawan J, Avila EM, Penner IR, Gilchrest BA (1996) Innervation of melanocytes in human skin. J Exp Med 184:1385–1395. doi:10.1084/jem.184.4.1385 [DOI] [PMC free article] [PubMed]
- 3.Hu DN, Woodward DF, McCormick SA (2000) Influence of autonomic neurotransmitters on human uveal melanocytes in vitro. Exp Eye Res 71:217–224. doi:10.1006/exer.2000.0869 [DOI] [PubMed]
- 4.Brocker EB, Magiera H, Herlyn M (1991) Nerve growth and expression of receptors for nerve growth factor in tumors of melanocyte origin. J Invest Dermatol 96:662–665. doi:10.1111/1523-1747.ep12470585 [DOI] [PubMed]
- 5.Burnstock G (2007) Purine and pyrimidine receptors. Cell Mol Life Sci 64:1471–1483. doi:10.1007/s00018-007-6497-0 [DOI] [PMC free article] [PubMed]
- 6.Sneddon P, Burnstock G (1984) Inhibition of excitatory junction potentials in guinea-pig vas deferens by α, β-methylene-ATP: further evidence for ATP and noradrenaline as cotransmitters. Eur J Pharmacol 100:85–90. doi:10.1016/0014-2999(84) 90318-2 [DOI] [PubMed]
- 7.Burnstock G (1988) Sympathetic purinergic transmission in small blood vessels. Trends Pharmacol Sci 9:116–117. doi:10.1016/0165-6147(88) 90185-X [DOI] [PubMed]
- 8.Hoyle CHV, Burnstock G (1993) Postganglionic efferent transmission to the bladder and urethra. In: Maggi C (ed) The autonomic nervous system, vol. 3. Nervous control of the urogenital system. Harwood, Switzerland, pp 349–383
- 9.Burnstock G (2004) Cotransmission. Curr Opin Pharmacol 4:47–52. doi:10.1016/j.coph.2003.08.001 [DOI] [PubMed]
- 10.Burnstock G, Knight GE (2004) Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 240:31–304. doi:10.1016/S0074-7696(04) 40002-3 [DOI] [PubMed]
- 11.Merighi S, Varani K, Gessi S, Cattabriga E, Iannotta V, Ulouglu C, Leung E, Borea PA (2001) Pharmacological and biochemical characterization of adenosine receptors in the human malignant melanoma A375 cell line. Br J Pharmacol 134:1215–1226. doi:10.1038/sj.bjp. 0704352 [DOI] [PMC free article] [PubMed]
- 12.Slater M, Scolyer RA, Gidley-Baird A, Thompson JF, Barden JA (2003) Increased expression of apoptotic markers in melanoma. Melanoma Res 13:137–145. doi:10.1097/00008390-200304000-00005 [DOI] [PubMed]
- 13.White N, Butler PEM, Burnstock G (2005) Human melanomas express functional P2X7 receptors. Cell Tissue Res 321:411–418. doi:10.1007/s00441-005-1149-x [DOI] [PubMed]
- 14.White N, Ryten M, Clayton E, Butler P, Burnstock G (2005) P2Y purinergic receptors regulate the growth of human melanomas. Cancer Lett 224:81–91 [DOI] [PubMed]
- 15.Allman R, Cowburn P, Mason M (2000) In vitro and in vivo effects of a cyclic peptide with affinity for the ανβ3 integrin in human melanoma cells. Eur J Cancer 36:410–422. doi:10.1016/S0959-8049(99) 00279-8 [DOI] [PubMed]
- 16.Gershwin ME, Ikeda RM, Kawakami RG, Owens RB (1977) Immunobiology of heterotransplanted human tumors in nude mice. J Natl Cancer Inst 58:1455–1461 [DOI] [PubMed]
- 17.Rapaport E, Fontaine J (1989) Generation of extracellular ATP in blood and its mediated inhibition of host weight loss in tumor-bearing mice. Biochem Pharmacol 38:4261–4266. doi:10.1016/0006-2952(89) 90524-8 [DOI] [PubMed]
- 18.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP (1973) In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst 51:1417–1423 [DOI] [PubMed]
- 19.United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) (1998) Guidelines for the welfare of animals in experimental neoplasia (Second Edition). Br J Cancer 77:1–10 [DOI] [PMC free article] [PubMed]
- 20.Amizuka N, Warshawsky H, Henderson JE, Goltzman D, Karaplis AC (1994) Parathyroid hormone-related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol 126:1611–1623. doi:10.1083/jcb.126.6.1611 [DOI] [PMC free article] [PubMed]
- 21.Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119:493–501. doi:10.1083/jcb.119.3.493 [DOI] [PMC free article] [PubMed]
- 22.Fidler IJ (1973) Selection of successive tumour lines for metastasis. Nat New Biol 242:148–149 [DOI] [PubMed]
- 23.Ishikawa M, Fernandez B, Kerbel RS (1988) Highly pigmented human melanoma variant which metastasizes widely in nude mice, including to skin and brain. Cancer Res 48:4897–4903 [PubMed]
- 24.Vad NM, Yount G, Moore D, Weidanz J, Moridani MY (2009) Biochemical mechanism of acetaminophen (APAP) induced toxicity in melanoma cell lines. J Pharm Sci 98:1409–25 [DOI] [PMC free article] [PubMed]
- 25.Rapaport E, Fontaine J (1989) Anticancer activities of adenine nucleotides in mice are mediated through expansion of erythrocyte ATP pools. Proc Natl Acad Sci USA 86:1662–1666. doi:10.1073/pnas.86.5.1662 [DOI] [PMC free article] [PubMed]
- 26.Rapaport E (1988) Experimental cancer therapy in mice by adenine nucleotides. Eur J Cancer Clin Oncol 24:1491–1497. doi:10.1016/0277-5379(88) 90340-9 [DOI] [PubMed]
- 27.Gaba SJ, Bourgouin-Karaouni D, Dujols P, Michel FB, Prefaut C (1986) Effects of adenosine triphosphate on pulmonary circulation in chronic obstructive pulmonary disease. ATP: a pulmonary vasoregulator? Am Rev Respir Dis 134:1140–1144 [DOI] [PubMed]
- 28.Haskell CM, Wong M, Williams A, Lee LY (1996) Phase I trial of extracellular adenosine 5′-triphosphate in patients with advanced cancer. Med Pediatr Oncol 27:165–173. doi:10.1002/(SICI) 1096-911X(199609) 27:3<165::AID-MPO6>3.0.CO;2-C [DOI] [PubMed]
- 29.Agteresch HJ, Dagnelie PV, van der Gaast A, Stijnen T, Wilson JH (2000) Randomized clinical trial of adenosine 5′-triphosphate in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst 92:321–328. doi:10.1093/jnci/92.4.321 [DOI] [PubMed]
- 30.Burnstock G (2001) Overview of P2 receptors: possible functions in immune cells. Drug Dev Res 53:53–59. doi:10.1002/ddr.1170 [DOI]
- 31.Deli T, Varga N, Adam A, Kenessey I, Rásó E, Puskás LG, Tóvári J, Fodor J, Fehér M, Szigeti GP, Csernoch L, Tímár J (2007) Functional genomics of calcium channels in human melanoma cells. Int J Cancer 121:55–65. doi:10.1002/ijc.22621 [DOI] [PubMed]
- 32.Spychala J (2000) Tumor-promoting functions of adenosine. Pharmacol Ther 87:161–173. doi:10.1016/S0163-7258(00) 00053-X [DOI] [PubMed]
- 33.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF, Jackson EK, Apasov S, Abrams S, Sitkovsky M (2006) A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA 103:13132–13137. doi:10.1073/pnas.0605251103 [DOI] [PMC free article] [PubMed]
- 34.Richard CL, Tan EY, Blay J (2006) Adenosine upregulates CXCR4 and enhances the proliferative and migratory responses of human carcinoma cells to CXCL12/SDF-1α. Int J Cancer 119:2044–2053. doi:10.1002/ijc.22084 [DOI] [PubMed]
- 35.Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J (2008) Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells. Int J Oncol 32:527–535 [PubMed]
- 36.Sitkovsky M, Lukashev D, Deaglio S, Dwyer K, Robson SC, Ohta A (2008) Adenosine A2A receptor antagonists: blockade of adenosinergic effects and T regulatory cells. Br J Pharmacol 153(Suppl 1):S457–S464. doi:10.1038/bjp. 2008.23 [DOI] [PMC free article] [PubMed]
- 37.Fishman P, Bar-Yehuda S, Madi L, Cohn I (2002) A3 adenosine receptor as a target for cancer therapy. Anticancer Drugs 13:437–443. doi:10.1097/00001813-200206000-00001 [DOI] [PubMed]
- 38.Madi L, Ochaion A, Rath-Wolfson L, Bar-Yehuda S, Erlanger A, Ohana G, Harish A, Merimski O, Barer F, Fishman P (2004) The A3 adenosine receptor is highly expressed in tumor versus normal cells: potential target for tumor growth inhibition. Clin Cancer Res 10:4472–4479. doi:10.1158/1078-0432.CCR-03-0651 [DOI] [PubMed]
- 39.Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M (2006) Antitumor effect of cordycepin (3′-deoxyadenosine) on mouse melanoma and lung carcinoma cells involves adenosine A3 receptor stimulation. Anticancer Res 26:43–47 [PubMed]
- 40.Saitoh M, Nagai K, Nakagawa K, Yamamura T, Yamamoto S, Nishizaki T (2004) Adenosine induces apoptosis in the human gastric cancer cells via an intrinsic pathway relevant to activation of AMP-activated protein kinase. Biochem Pharmacol 67:2005–2011. doi:10.1016/j.bcp. 2004.01.020 [DOI] [PubMed]
- 41.White N, Burnstock G (2006) P2 receptors and cancer. Trends Pharmacol Sci 27:211–217. doi:10.1016/j.tips.2006.02.004 [DOI] [PubMed]