

Abstract
The p19 protein (p19) encoded from Tombusvirus is involved in various activities such as pathogenicity and virus transport. Recent studies have found that p19 is a plant suppressor of RNA silencing, which binds to short interfering RNAs (siRNAs) with high affinity. We use molecular dynamics (MD) simulations of the wild-type and mutant p19 protein (W39 and W42G) binding with a 21-nt siRNA duplex to study the p19-siRNA recognition mechanism and mutation effects. Our simulations with standard MD and steered molecular dynamics have shown that the double mutant structure is indeed much less stable than the wild-type, consistent with the recent experimental findings. Comprehensive structural analysis also shows that the W39/42G mutations first induce the loss of stacking interactions between p19 and siRNA, Trp42-Cyt1 (Cyt1 from the 5′ to 3′ strand) and Trp39-Gua′19 (Gua19 from the 3′ to 5′ strand), and then breaks the hydrophobic core formed by W39-W42 with nucleotide basepairs in the wild-type. The steered molecular dynamics simulations also show that the mutant p19 complex is “decompounded” very fast under a constant separation force, whereas the wild-type remains largely intact under the same steering force. Moreover, we have used the free energy perturbation to predict a binding affinity loss of 6.98 ± 0.95 kcal/mol for the single mutation W39G, and 12.8 ± 1.0 kcal/mol loss for the double mutation W39/42G, with the van der Waals interactions dominating the contribution (∼90%). These results indicate that the W39/42G mutations essentially destroy the important p19-siRNA recognition by breaking the strong stacking interaction between Cyt1 and Gua′19 with end-capping tryptophans. These large scale simulations might provide new insights to the interactions and co-evolution relationship between RNA virus proteins and their hosts.
Introduction
RNA silencing plays an important role in gene expression at the transcriptional or posttranscriptional level in eukaryotes (1–3). Double-stranded RNAs (dsRNA) or self-complementary fold back RNAs are first cleaved into short interfering RNA (siRNA) or microRNA (miRNA) of 21–24 nucleotide (nt) by an RNase III-like enzyme DICER (4). Next, these small RNAs are unwound and loaded into a multi-protein complex known as RNA induced silencing complex (RISC). Finally, the active RISC combines to the target messenger RNA and guides its degradation or stops its translation (5–8).
Many plant viruses of single-stranded RNA genomes can form dsRNA during the replication in host cells (1). These dsRNAs can be joined into RNA silencing system and specifically target cognate viral RNA for degradation with RISC programming (3). Meanwhile, plant viruses evolve a defense mechanism to suppress RNA silencing (9–13). P19 is such a plant suppressor encoded from Tombusvirus as a counter-defense against plants (14–25). It shows a high affinity in binding to siRNA, especially 21 nt siRNA (double-stranded) with 2 nt, 3′ overhangs in cytosolic (16). Recent studies have shown that p19 not only suppresses RNA interference (RNAi) in Tomato Bushy stunt virus (TBSV) hosts but also in other plant, insect (26), and human cells (27,28), such as Arabidopsis thaliana (27,29,30), Drosophila (31), and Caenorhabditis elegans (2,26). Furthermore, p19 can interact with an RNA binding host protein and bind to a short RNA in mouse embryonic stem cells (32). All these evidences indicate that p19 can bind to small RNAs both in plant and mammalian cells and could be a useful tool to dissect the RNAi pathway in vivo or in vitro. It has been suggested previously that p19 first binds to siRNAs and then sequesters them, preventing their incorporation into a RISC (14–16,22). However, the exact molecular mechanism by which this suppression occurs remains somewhat unclear (33,34).
The structure of p19 binding to 21 nt siRNA duplex in Carnation Italian ringspot virus was reported by Hall et al. (34), and the structure in TBSV was reported by Ye et al. (35). These structures show that p19 forms a homodimer with a concave surface made of eight β-strands in the middle of the dimer, where the RNA duplex binds ( Fig. 1). Residues Trp39 and Trp42 from each monomer of the p19 act as caliper measuring and bracketing the siRNA. These two tryptophans, along with Arg43 and Asn46, form a glove to hold each side of the nucleic acids at the terminal of siRNA. The contacts between p19 protein and siRNA seem to be sequence independent because there are no interactions between the protein and the RNA bases, other than stacking interactions with the last basepairs at each end of the duplex region (see below) (34).
Figure 1.
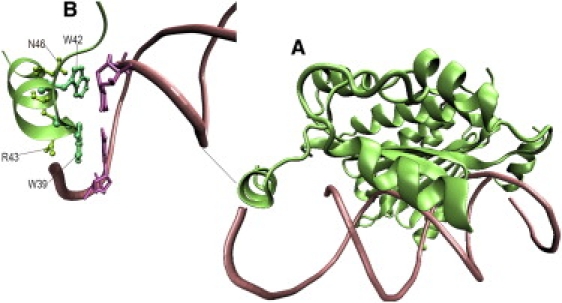
Stereo view of p19-siRNA complex structure. (A) The siRNA is rendered as licorice, and the p19 dimer is represented as green ribbons. (B) Residues Trp39 and Trp42 (turquoise sticks) in the N-terminal subdomain with the interaction of nucleic acids Gua′19 and Cyt1 (pink sticks) in siRNA.
Experiments suggest that mutations on the two tryptophan residues W39 and W42 of p19 strongly reduces the RNA silencing suppression (33,34), as the two hydrophobic residues Trp39 and Trp42 are key amino acids for p19 dimer to contact and recognize the two siRNA terminals (34). The crystal structures of p19 provide details on how Trp39 and Trp42 interact with siRNA statically, however, the molecular details on how exactly the Trp39 and Trp42 residues interact and recognize the 21-nt siRNA dynamically, how much binding affinity each Trp residue contributes, what physical interactions dominate, and what structural changes the mutations introduce are still unclear. To address these questions, we have carried out molecular dynamics (MD) simulations for both the wild-type and mutant p19 protein siRNA complexes. These large scale simulations can complement experiments for better understanding of the p19-siRNA molecular recognition mechanism by providing atomic details that are often inaccessible in experiment due to resolution limits, even with the currently available sophisticated experimental techniques (36–47). Besides normal MD simulations, steered molecular dynamics (SMD), and free energy perturbation (FEP) calculations are also used to investigate the interaction between p19 protein and 21-nt siRNA. In all these simulations (unless otherwise explicitly stated), three types of p19 complex are simulated: the wild-type, the single mutant W39G, and the double mutant W39/42G. The simulation results and discussions are presented in the following sections.
Methods
The starting structure of the p19 protein from Carnation Italian ringspot virus bound to a 21-nt siRNA duplex is taken from the x-ray crystal structure deposited in the Protein Data Bank (PDB, 1RPU.pdb) (34). As shown in Fig. 1, p19 is a homodimer, with each monomer containing five α-helices and four β-strands, and it binds to a 21nt siRNA duplex. The conformation of the 21 nt siRNA duplex is A-Form double helix with 2 nt 3′ overhangs (numbers 1–19 are Watson-Crick basepairs and numbers 20 and 21 are unpaired). For description purpose, we name residues from the first monomer as XXX (e.g., Trp42) and the same residue from the second monomer as XXX′ (e.g., Trp′42). In a similar fashion, nucleotides from the 5′ to 3′ strand of the siRNA duplex is named as YYY (e.g., Cyt1), and the ones from the other strand as YYY′ (e.g., Gua′19). To investigate the importance of end-capping mechanism of p19, which is considered to be the key for siRNA recognition and combination, we simulated the p19-siRNA complex for both the p19 wild-type and mutants. Either one or two tryptophans (Trp39, Trp42) were substituted with glycines for each monomer in the wild-type structure (experiments done on W39G single mutation and W39/42R double mutations (34); and to observe more significant effects in MD simulations within a reasonable time, we also simulated a more drastic double mutation W39/42G, see below). Therefore, for a double mutation, actually four Trp residues in the p19 dimmer are mutated (Trp39, Trp42, and Trp′39, Trp′42). Both the wild-type and mutants were then solvated in water boxes with size ∼70 × 60 × 90 Å3. A total of 39 Na+ counterions were then added to neutralize the solvated systems. We have also simulated the system in 100 mM NaCl salt solution to mimic the biological environment, where 23 more pairs of Na+ and Cl− ions are further added (see below). The total size of the solvated systems was ∼30,000 atoms.
The NAMD2 (48,49), program was used for the MD simulations with the NPT (constant pressure 1 atm and constant temperature 305 K, same as the experiment) ensemble for data collection. The CHARMM (CHARMM32, parameter set c32b1) (50–52) force field was used for the protein and RNA, and the TIP3P (53,54) water model was used for the explicit solvent. We treated the long-range electrostatic interactions with the particle mesh Ewald (PME) (55,56) method and used a typical 10 Å cutoff for the van der Waals interactions. Before data collection, both the wild-type and mutant p19 complex systems were equilibrated. First, a 10,000 steps minimization was run to remove bad contacts due to the solvation process. Then, the minimized configurations were used as the starting point for another 1,000 ps NPT equilibration at 1 atm and 305 K. The time step for equilibration was 0.5 fs. Up to five different configurations from the last 500 ps trajectory were chosen as the starting configurations for production runs. The time step for all production runs was 1.5 fs.
In the normal MD, only the wild-type and double mutant were simulated (for SMD and FEP, the single mutation was also included, see below) to observe meaningful differences in regular MD within a reasonable simulation time. Two long trajectories, one for the wild-type and one for the double mutant, were simulated up to 100 ns to monitor the differences in complex stability. An additional of eight shorter trajectories, four for the wild-type and four for the double mutant starting from different initial configurations, were also simulated up to 10 ns. On the other hand, all three systems, the wild-type, the single mutant, and the double mutant were simulated in the SMD. SMD simulations also were carried out to study the interaction between p19 protein and the siRNA under steering force or “perturbation” starting from the equilibrated structures. Both the wild-type and mutants were given a constant force along the z-axis (the direction in alignment with the siRNA strand). The residues of Ala81, Asn96, His132, and Leu145 from each p19 monomer, which are far away from the active region, were fixed in space to hold the p19 dimer in a proper position during the SMD simulations. Six different constant forces, 0.001, 0.005, 0.01, 0.02, 0.03, and 0.05 kcal/mol/Å, were applied to the siRNA in the wild-type, single mutant and double mutant p19 protein siRNA complexes, resulting a total of 18 SMD simulations with each up to 10 ns. For the constant force of 0.01 kcal/mol/Å case, additional trajectories were also run starting from different initial configurations (chosen from the first 1 ns normal MD trajectory; see Results).
A rigorous FEP method (57,58) is further used to calculate the binding affinity changes for both the single and double mutations. The Helmholtz free energy of a system can be expressed as
| (1) |
where Z is the partition function and H (p,q) is the Hamiltonian of the system. With a thermal integration approach, the binding free energy change ΔG due to a mutation such as W39G in p19 can then be calculated as
| (2) |
| (3) |
where V(λ) = (1 − λ) V 1 + λ V 2, and V 1 represents the potential energy of the wild-type, and V 2 represents the potential energy of the mutant. The FEP parameter λ changes from 0 (V 1) to 1 (V 2) when the system changes from the wild-type to the mutant, and 〈…〉λ represents the ensemble average at potential V(λ). In typical FEP calculations, for a single mutation from residue A (e.g., W39) to residue B (e.g., G39), many perturbation windows have to be used to have a “smooth” transition from state A to B, with typically more windows near the two ends to enhance the sampling statistics.
In general, it is difficult to directly calculate the binding affinity change ΔG A for the binding process between a protein and a siRNA due to the complicated binding process. However, we can avoid this problem by designing a thermodynamical cycle to calculate the relative binding free energy change, i.e., ΔΔG AB. Instead of calculating the difficult direct binding energies ΔG A and ΔG B, we calculate the free energy changes for the same mutation in both the bound state (i.e., p19 and siRNA already bounded, ΔG 1) and the free state (i.e., p19 and siRNA not bounded, ΔG 2) (57–59). Within a complete thermodynamical cycle, the total free energy change should be zero, which gives the relative binding affinity due to the mutation from A → B as
| (4) |
In the current setup, a 22-window scheme has been adopted. For each mutation, single (W39G) or double (W39G and 42G), a total of 10 independent runs starting from different initial configurations (some taken from the 100 ns long normal MD trajectory) are carried out for better convergence. It is widely recognized that for effective FEP calculations, a sufficient conformational space sampling is often critical (57,59,60,62,63). Many groups have developed efficient sampling methods to tackle this problem (57,59,62,63). Warshel et al. (65–67) also pointed out that a satisfactory convergence in FEP is not only related to the extensive sampling, but also to the proper electrostatic boundary condition and long range treatment (such as local reaction field). Our current total simulation length is 264 ns (22 windows × 0.6 ns × 10 runs × 2 states) for each mutation, which is much longer than most FEP calculations reported currently in the literature (57–59).
Results and Discussion
Normal MD not sufficient to show the difference within the simulation length
To examine the effect of mutation on p19 structure, we plot the trajectories of the backbone RMSD from the crystal structure for both the wild-type and the double mutant p19 in Fig. 2. Somewhat larger fluctuations in the double mutant were observed during the normal MD simulations. For both the wild-type and double mutant p19 protein, the backbone RMSDs plateaued at ∼2.5 Å from the crystal structure for all trajectories simulated for 10 ns, indicating that the p19 protein is fairly stable in normal MD (that is why the single mutation W39G was not simulated in normal MD and not shown). In the longer 100 ns simulations, both increased to ∼3 Å before 80 ns, and then the double mutant showed a larger RMSD than the wild-type after ∼80 ns, rising to ∼4 Å, but the difference was not remarkable (Fig. 2 A), indicating that the normal MD is not sufficient in distinguishing the two within the simulation length (that is why we carry out further SMD and FEP simulations; see below). Nevertheless, the normal MD simulations do show some meaningful changes in local regions already.
Figure 2.
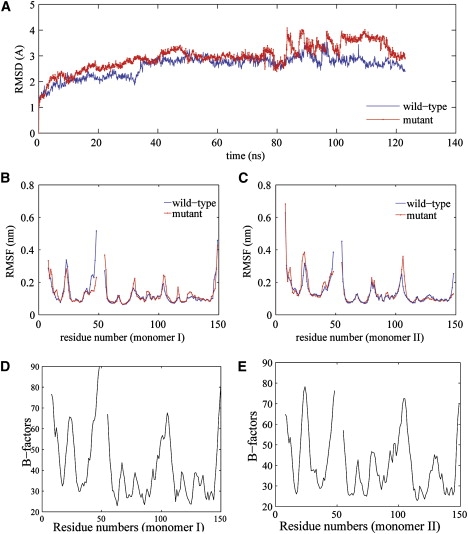
(A) Comparison of backbone RMSDs from the starting crystal structures for both the wild-type (blue) and the double mutant (red) trajectories. The results are obtained from 305 k NPT simulations with the simulation time of 130 ns. Overall, these trajectories show comparable RMSDs from the initial structures. The double mutant p19 dimer experiences a larger increase in RMSD than wild-type during 80–115 ns. (B and C) Comparison of Cα-RMSFs of p19 dimers for the wild-type (blue) and the double mutant (red). (D and E) Measured B-factors from the x-ray structure for Cα atoms in both monomers of wild-type. Our RMSFs agree well with B-factors.
To characterize the local fluctuations of the wild-type and mutant p19 dimmer, we calculated the root mean-square fluctuations (RMSF) of the Cα atoms and also compared with the measured B-factors from x-ray structure (34), as shown in Fig. 2. Overall, our RMSF curves agree with B-factors very well for the wild-type (there is no x-ray structure for the mutant yet), with major peaks near residues Ser23, Leu74, and Ala105 all well captured except that the Ala105 B-factor peak is somewhat broader. The mutant p19 experiences slightly larger fluctuations than the wild-type in local regions near residues Leu74, Ala105, and Phe110 in monomer I, and near residues Ser23 and Ala105 in monomer II. These observations indicate that the W39/42G double mutation indeed causes some of the local contacts to be more flexible.
Furthermore, a detailed examination on the mutation site, Trp39/Trp42, shows a noticeable decrease in local stacking interactions in the double mutant. The van der Waals interactions, or “stacking interactions”, between the indole rings of Trp42 (Trp39) and the bases of Cyt1 (Gua′19) in the wild-type are basically lost in the mutant due to the loss of indole ring in the Gly side chains. In addition, the original stackings between Arg43 and Asn46 with Trp39 and Trp42, respectively, in the wild-type, which prevent the solvent exposure of hydrophobic tryptophan side chains, are also gone in the double mutant. The hydrogen bonds between Trp39, Gua′19, and Trp42, Cyt1 (between the imino nitrogen of Trp39/Trp42 and the phosphate group of Gua′19/Cyt1) are also largely destroyed in the mutant. We further observed an increase of non-native-like contacts among Arg43 and Gua′19; Asn46, Asp47, and Cyt1 in the mutant (see below). This is probably due to the much smaller size of the Gly side chain as compared to Trp, which leaves more space for Cyt1 and Gua′19 to contact closely with neighboring residues in p19. As a result, Cyt1 and Gua′19 cross over Gly39 and Gly42, and make contacts with Arg43, Asn46, and Asp47, instead. To confirm this observation, we then calculated the average distance between each p19 residue and the W/G39, W/G42 residues. The mutant G39/G42 residues indeed have notably shorter distances with neighboring residues 43–48 (R43, L44, Y45, N46, D47, E48) when compared to the wild-type, as shown in Fig. 3. This indicates that the glove formed by residues W39, W42, R43, and N46, etc. was largely crunched in the mutant.
Figure 3.

Change in the average distance between a target residue (W/G39, W/G42) and other residues in the p19 dimer, when mutating from the wild-type to the double mutant (double mutant minus wild-type). The distance difference for residues. (A) W/G39 in monomer I. (B) W/G42 in monomer II. Compared to the wild-type, the residues G39 and G42 in the double mutant have a significantly shorter average distance with other residues 43–48 (R43, L44, Y45, N46, D47, and E48; yellow background).
p19 mutants showing stronger tendency to be decompounded in SMD
To further characterize the interaction between 21-nt siRNA and p19 residue sites W/G39, W/G42, we carried out SMD simulations, by applying an external constant force on the siRNA to “pull apart” the complex. These SMD trajectories of both the wild-type and the mutants show clearly that the siRNA and p19 protein were still bound together during our 10 ns simulation when a small force was applied, indicating the initial complex structure was fairly stable. Only when the external force increased to a certain value (≥0.01 kcal/mol/Å), the complex started to be decompounded, with the mutants, particularly the double mutant, showing a much stronger tendency of falling apart. Fig. 4 A shows the RMSD comparison for the wild-type and the double mutant during the 10ns SMD simulation with an external constant force of 0.01kcal/mol/Å (for better statistics, total 6 trajectories were run, with 3 each, for this case starting from different initial configurations). The double mutant undergoes a much faster increase in the backbone RMSD, indicating that the double mutant structure was less stable compared to the wild-type. Similar results were also obtained from other trajectories with large external forces. However, at too large forces (e.g. 0.05 kcal/mol/Å and above), the complex was decompounded too fast for both the wild-type and the mutant, thus overwhelmed the intrinsic difference due to the mutation. The RMSD changes of the single mutant (W39G) displayed similar effect as well, even though not as dramatic as of the double mutant.
Figure 4.

(A) Comparison of the backbone RMSDs for the wild-type (blue) and double mutant (red) during the SMD simulation with a constant force of 0.01 kcal/mol/Å. In the absence of the stacking interactions, the double mutant shows a much faster increase in the RMSD from the crystal structure. (B and C) The separation between p19 and siRNA for both the wild-type (blue) and double mutant (red) during the SMD simulation with a constant force of 0.01 kcal/mol/Å. (B) The distance between nucleic acid Gua′19 of siRNA and p19 residue W/G39. (C) The distance between nucleic acid Cyt1 of siRNA and p19 residue W/G42. The exponential increase at the curve end indicates that p19 dimer and siRNA are separated. The separation time is ∼4 ns for the mutant and ∼8 ns for the wild-type.
Fig. 4, B and C, show the distance between siRNA nucleic acids (Cyt1 or Gua′19) and p19 residues (W/G39 or W/G42) as a measure of the separation between p19 and siRNA during the SMD simulation (force constant of 0.01 kcal/mol/Å). A sharp increase (>20 Å/ns) in the separation distance for the double mutant is seen around 4.0 ns, indicating the complex was decompounded already after 4 ns. In contrast, the wild-type shows a much milder increase (<0.2 Å/ns) in the separation distance up to 8 ns; and the complex starts to be decompounded only after ∼8 ns with the same external force (Fig. 4, B and C). Overall, at the 0.01 kcal/mol/Å constant force a relatively smooth separation process can be observed in a reasonable timescale; therefore, in the following discussions we use this case for further analysis, unless otherwise stated explicitly.
Loss of stacking interactions responsible for less stable mutant structures
To provide insight into the decompounding mechanism of p19-siRNA complex, we examine the snapshots of the SMD trajectories in detail. Fig. 5 shows some representative ones at 0, 1, 2, 3, and 4 ns for both the wild-type and the double mutant. Because the majority of the contacts between p19 dimer and siRNA are hydrogen bonds between p19 basic and polar residues with siRNA phosphate/sugar groups, we followed these hydrogen bonds (many broken during the SMD simulation) as a measure of complex stability. Here, we define a hydrogen bond with a distance cutoff of 3.5 Å (measured form the donor to the acceptor) and the X–H …Y angle cutoff of 150°. Obviously, the exact time and location of hydrogen bond losses depend on the particular trajectories, but the overall conclusion described below is qualitatively consistent.
Figure 5.
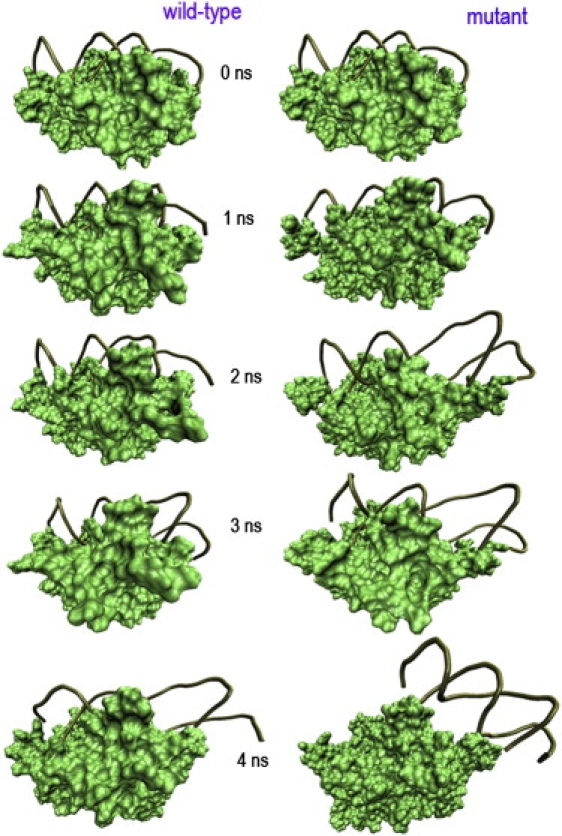
Snapshots of p19 complex at 0, 1, 2, 3, and 4 ns of SMD simulations with a constant force 0.01 kcal/mol/Å. The p19 dimer is rendered as a green solid surface. The siRNA is represented as a brown tube. See text for detailed descriptions.
Let us consider the wild-type first. The first hydrogen bond loss was between Gua2 phosphate and Lys60 imino nitrogen (Lys60-Gua2), which happened around ∼500 ps, whereas the similar one Arg18-Gua2 was broken at ∼1 ns, and subsequently Lys′67-Ura9 and Arg′11-Cyt10 ones were lost around 1.5 ns. A series of hydrogen bonds, such as Gln107-Gua13, Gln107-Cyt14 and Thr′40-Ura21 were lost at ∼2.0 ns. The loss of these hydrogen bonds resulted in the distortion of the siRNA structure (Fig. 5 A), which subsequently introduced more hydrogen bond losses during 2–5 ns simulation time. Surprisingly, the stacking interactions between Trp42-Cyt1 and Trp39-Gua′19 remained intact during this time period, with the two siRNA ends well capped. This supports the experimental findings of the strong staking interactions between Trp42/Cyt1 and Trp39/Gua′19 by Hall et al. (34). This stacking interaction provides a capping as well as a recognition signature on the exposed RNA basepairs at each end of the siRNA duplex. Our results also indicate that the major cleavage events actually start at the middle of siRNA instead of the two ends on external pulling forces. Only after ∼7.3 ns, the distortion force caused the end-cap stacking interactions to be broken in the first strand (5′ to 3′). Similarly, the second strand (3′ to 5′) also went through a serious of hydrogen bond losses before the end-cap was broken. In conclusion, the wild-type p19-siRNA complex undergoes a “middle cleavage before two capping ends” mechanism on an external pulling force.
On the other hand, the double mutant W39/42G displayed a very different behavior from the wild-type. Without the stacking interactions between G42-Cyt1 and G39-Gua′19, the separation of siRNA became much easier and much faster. The first hydrogen bond loss between Cyt14 phosphate group and Gln107 imino nitrogen (Gln107-Cyt14) happened extremely fast, with only 57 ps in the 3′ to 5′ strand. Other similar hydrogen bonds were also lost fast, with Lys60-Gua2 and Lys67-Ura9 ones lost around 250 ps. At ∼800 ps hydrogen bond Arg18-Gua2 was broken, which resulted in a slight shift of the 5′ to 3′ strand with respect to the p19 dimmer. Interestingly, a number of new hydrogen bonds were formed between G39/G42 with neighboring nucleotides Cyt1, Gua2, etc.; however, they were not as stable as the original ones because they form and break frequently. This observation is consistent with above normal MD results, where the formation of non-native-like on-and-off local contacts was also found near Gua′19 and Cyt1, but they were not as stable as the native-contacts in the wild-type. These newly formed hydrogen bonds in the mutant are not strong enough to offset the loss of the stacking interactions, as indicated by the much faster decompounding of the mutant p19-siRNA complex. The sliding of the 3′ to 5′ strand was observed after 1.3 ns, followed by the 5′ to 3′ strand. All of the native hydrogen bonds were broken after 1.8 ns simulation time. Therefore, different from the wide-type's middle cleavage before two capping ends mechanism, the mutant displays a “relatively uniform sliding” mechanism due to the lack of stacking interactions. This relative importance of stacking interactions over electrostatic interactions (hydrogen bonds) between Trp39/Trp42 residues and Gua′19/Cty1 nucleotides can also be seen from another mutation experiment with W39/42R (34). The mutation effect of W39R on the virus silencing suppression is not as dramatic as that of W39G. In other words, the W39G mutation results in a more dramatic plant recovery from the mutant virus infection than the W39R mutation (34), indicating that even the enhanced electrostatic interactions between the basic residue Arg39 with phosphate group of Gua′19 is not strong enough to offset the loss of the stacking interaction (van der Waals interaction) between Trp39 and Gua′19.
FEP showing lower binding affinity for p19 mutants
A total of 10 independent FEP runs were carried out for both the single and double mutations starting from different initial configurations (some taken from the 100 ns long normal MD trajectory), with a total simulation time of 264 ns for each mutation. These extensive FEP calculations show a binding affinity decrease of 6.98 ± 0.95 kcal/mol for the single mutation W39G, and a 12.8 ± 1.0 kcal/mol decrease for the double mutation W39/42G. The direct measurement of the wild-type p19-siRNA dissociation constant (K d) shows a K d value of 0.17 ± 0.02 nM at room temperature (34), which is equivalent to a binding affinity of 13.3 ± 1.5 kcal/mol. Even though no direct experimental K d values are available for the mutants, our FEP binding affinities seem to be in line with the experimental value for the wild-type, given that the double mutant results in a complete dissociation of the p19 from siRNA (i.e., a binding affinity near zero). It will be interesting to see more experimental results on these binding affinity changes due to mutations to validate our current predictions.
It is of interest to see a decomposition of the total binding free energy into its van der Waals and electrostatic components because it might offer useful information about the energetic interactions involved in the p19-siRNA binding. However, the free energy decomposition might be path-dependent (i.e., turning on van der Waals interactions first or electrostatic interactions first). Moreover, there are controversies in literature about the meaningfulness of breaking the total free energy into components (68–71). Nevertheless, such a decomposition can offer some clues in the contributions of various physical interactions in determining the binding mechanism. In this study, we use a straightforward decomposition in FEP by collecting van der Waals and electrostatic interaction contributions separately, i.e., V(λ) = V(λ)elec + V(λ)vdW, in the same ensemble with full interactions in Eq. 2 (see System and Methods). Due to the nonlinearity of the FEP formulation, there might be a small coupling term in this approach (71). Using W39G as an example, we found the following detailed numbers for free energy changes, ΔG 1, ΔG 2, and ΔΔG, in Eq. 4 and their components. For the bound state, out of −6.73 kcal/mol total free energy change ΔG 1, 6.49 kcal/mol is from the electrostatic interactions, −13.52 kcal/mol from van der Waals, and ∼0.34 kcal/mol from the coupling term. Therefore, the net electrostatic interactions unfavor the binding, whereas the van der Waals interactions dominate the binding or recognition in this process. Similarly, for the free state, out of the total −13.71 kcal/mol total free energy change ΔG 2, 5.75 kcal/mol is from the electrostatic interactions, −19.76 kcal/mol from van der Waals, and again ∼0.30 kcal/mol from the coupling term. In the final binding affinity change ΔΔG (ΔΔG = ΔG 1 − ΔG 2 = 6.98 kcal/mol), the van der Waals interactions (6.24 kcal/mol) again dominate the contribution, with ∼90% contribution, whereas the electrostatic interactions (0.74 kcal/mol) only contribute ∼10%. Similar conclusions can be obtained from the double mutation FEP data. A visual inspection of the FEP mutation trajectories also shows the gradual loss of the packing interactions during the Trp → Gly mutation process.
For further validation, we carried out FEP simulations in 100 mM NaCl salt solution to mimic the biological environment. Twenty-three pairs of Na+ and Cl− ions were added to the equilibrated simulation box to obtain an ∼100 mM NaCl solution. The resulting salted systems were then further equilibrated for another 3 ns. Five FEP simulations starting from different initial configurations (picked from the last 1 ns of the equilibration trajectory) were simulated with the same simulation length (22 windows × 0.6 ns) for both the single and double mutations. These new FEP calculations show a binding affinity decrease of 7.13 ± 0.89 kcal/mol for the single mutation W39G, and a 13.3 ± 1.2 kcal/mol decrease for the double mutation W39/42G. Overall, these simulations from the 100 mM NaCl solution show a slightly larger binding affinity decrease, but these changes are still within the SD, therefore, we conclude there is no significant change in the binding affinity for our current p19-siRNA complex with or without the 100 mM NaCl salt. Again, the van der Waals interactions are found to be the dominant force with ∼90% contribution, whereas the electrostatic interactions contribute the remaining 10%.
Therefore, these extensive FEP calculations support the above observations on the strong stacking interaction mechanism for the recognition of siRNA by p19 in the WT. Even though a direct quantitative comparison with experiment is not available for these mutants, these binding affinity results seem to be consistent with the wild-type dissociation constant (K d) measurement mentioned above (K d = 0.17 ± 0.02 nM at room temperature (34), which is equivalent to a binding affinity of 13.3 ± 1.5 kcal/mol), and also consistent with the plants symptom development experiment using single or double mutations, with double mutation showing more dramatic plant recovery (34). In summary, our FEP calculations, as well as MD and SMD simulations, all found that the stacking interactions of Trp42-Cyt1 and Trp39-Gua′19 are the most important interactions to hold the p19-siRNA complex together. This stacking interaction might also explain why p19 is an effective 21-nt siRNA combiner. It prevents siRNA within the complex being deprived by other small RNA receiver, like Argonaute (Ago) proteins in plants or mammals.
Conclusion
In this study, we have carried out a series of molecular dynamics simulations, including SMD and FEP, to investigate the interaction and recognition mechanism between the RNA silencing suppression virus protein p19 and a 21-nt siRNA, as well as the effects of mutations to two important hydrophobic capping residues.
The double mutation W39/42G was found to cause significantly higher fluctuations in the local structure of p19 near the mutation sites (Gly39 and Gly42) due to the smaller size of Gly side chain (as compared to Trp). The SMD simulations show that the mutant p19 complex was decompounded much faster than the wild-type under an external steering force applied on siRNA. Detailed structural analysis indicates that the stacking interactions of Trp42-Cyt1 (Cyt1 from the 5′ to 3′ strand) and Trp39-Gua′19 (Gua19 from the 3′ to 5′ strand) are the most important interactions to hold the p19-siRNA complex together. In other words, Trp39 and Trp42 from each monomer of p19 form strong van der Waals interactions with bases of Gua′19 and Cyt1, respectively, thus capping the exposed RNA basepairs at each end of the siRNA duplex. Under the pulling of an external force, the wild-type displayed a “middle cleavage before two capping ends” mechanism, whereas the mutant displayed a “relatively uniform sliding” mechanism due to the loss of stacking interactions at the two end-caps. The FEP calculations also show a binding affinity decrease of 6.98 ± 0.95 kcal/mol for the single mutation W39G, and 12.8 ± 1.0 kcal/mol decrease for the double mutation W39/42G, with the van der Waals interactions dominating the contributions (90%). The additional FEP calculations in 100 mM NaCl solution (to mimic the biological environment) show similar binding affinity changes. Another change in W39/42G double mutant is the deformation of the local hydrophobic core, which includes the end-capping tryptophan residues and the basepairs on each end of the siRNA (34).
The recent discovery of p19 suppression on RNA silencing has led to its widespread use as an RNAi-probing tool in various plant and animal models. However, our insufficient understanding of the biochemical mechanism of p19/RNA complex formation limits its further applications (72). The current MD simulations provided a rich molecular picture of the critical roles of stacking interactions and the hydrophobic core effect between the end-capping tryptophan residues and siRNA nucleotides, which explains the p19-siRNA size selectivity and recognition mechanism (34). These large scale simulations might also complement experiment to better understand the evolutional correlation between viral pathogenicity and RNA silencing or other transcriptional and posttranscriptional gene silencing in host species.
Acknowledgments
We thank Jingyuan Li, Bruce Berne, David Silverman, Isidore Rigoutsos, and Payel Das for helpful discussions. We also thank Sameer Kumar for numerous help with porting NAMD2, particularly the free energy perturbation (FEP) module, onto IBM BlueGene/L.
Contributor Information
Jun Zhu, Email: jzhu@zju.edu.cn.
Ruhong Zhou, Email: ruhongz@us.ibm.com.
References
- 1.Ahlquist P. RNA-dependent RNA polymerases, viruses, and RNA silencing. Science. 2002;296:1270–1273. doi: 10.1126/science.1069132. [DOI] [PubMed] [Google Scholar]
- 2.Fire A., Xu S.Q., Montgomery M.K., Kostas S.A., Driver S.E. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 3.Novina C.D., Sharp P.A. The RNAi revolution. Nature. 2004;430:161–164. doi: 10.1038/430161a. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein E., Caudy A.A., Hammond S.M., Hannon G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 5.Tomari Y., Zamore P.D. Perspective: machines for RNAi. Genes Dev. 2005;19:517–529. doi: 10.1101/gad.1284105. [DOI] [PubMed] [Google Scholar]
- 6.Schwarz D.S., Hutvagner G., Du T., Xu Z.S., Aronin N. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 7.Pham J.W., Pellino J.L., Lee Y.S., Carthew R.W., Sontheimer E.J. A Dicer-2-dependent 80S complex cleaves targeted mRNAs during RNAi in Drosophila. Cell. 2004;117:83–94. doi: 10.1016/s0092-8674(04)00258-2. [DOI] [PubMed] [Google Scholar]
- 8.Matranga C., Tomari Y., Shin C., Bartel D.P., Zamore P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 9.Voinnet O. RNA silencing as a plant immune system against viruses. Trends Genet. 2001;17:449–459. doi: 10.1016/s0168-9525(01)02367-8. [DOI] [PubMed] [Google Scholar]
- 10.Hamilton A.J., Baulcombe D.C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 11.Li W.X., Ding S.W. Viral suppressors of RNA silencing. Curr. Opin. Biotechnol. 2001;12:150–154. doi: 10.1016/s0958-1669(00)00190-7. [DOI] [PubMed] [Google Scholar]
- 12.Baulcombe D. Viral suppression of systemic silencing. Trends Microbiol. 2002;10:306–308. doi: 10.1016/s0966-842x(02)02387-9. [DOI] [PubMed] [Google Scholar]
- 13.Lakatos L., Csorba T., Pantaleo V., Chapman E.J., Carrington J.C. Small RNA binding is a common strategy to suppress RNA silencing by several viral suppressors. EMBO J. 2006;25:2768–2780. doi: 10.1038/sj.emboj.7601164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu W.P., Park J.W., Scholthof H.B. Tombusvirus p19-mediated suppression of virus-induced gene silencing is controlled by genetic and dosage features that influence pathogenicity . Mol. Plant Microbe Interact. 2002;15:269–280. doi: 10.1094/MPMI.2002.15.3.269. [DOI] [PubMed] [Google Scholar]
- 15.Qu F., Morris T.J. Efficient infection of Nicotiana benthamiana by Tomato bushy stunt virus is facilitated by the coat protein and maintained by p19 through suppression of gene silencing. Mol. Plant Microbe Interact. 2002;15:193–202. doi: 10.1094/MPMI.2002.15.3.193. [DOI] [PubMed] [Google Scholar]
- 16.Silhavy D., Molnar A., Lucioli A., Szittya G., Hornyik C. A viral protein suppresses RNA silencing and binds silencing-generated, 21- to 25-nucleotide double-stranded RNAs. EMBO J. 2002;21:3070–3080. doi: 10.1093/emboj/cdf312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voinnet O. Induction and suppression of RNA silencing: insights from viral infections. Nat. Rev. Genet. 2005;6:206–220. doi: 10.1038/nrg1555. [DOI] [PubMed] [Google Scholar]
- 18.Kasschau K.D., Carrington J.C. A counter defensive strategy of plant viruses: suppression of posttranscriptional gene silencing. Cell. 1998;95:461–470. doi: 10.1016/s0092-8674(00)81614-1. [DOI] [PubMed] [Google Scholar]
- 19.Qu F., Morris T.J. Suppressors of RNA silencing encoded by plant viruses and their role in virus infection. FEBS Lett. 2005;579:5958–5964. doi: 10.1016/j.febslet.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 20.Baulcombe D. RNA silencing in plants. Nature. 2004;431:356–363. doi: 10.1038/nature02874. [DOI] [PubMed] [Google Scholar]
- 21.Omarov R.T., Ciomperlik J.J., Scholthof H.B. RNAi-associated ssRNA-specific ribonucleases in Tombusvirus P19 mutant-infected plants and evidence for a discrete siRNA-containing effector complex. Proc. Natl. Acad. Sci. USA. 2007;104:1714–1719. doi: 10.1073/pnas.0608117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vionnet O., Pinto Y.M., Baulcombe D.C. Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses of plants. Proc. Natl. Acad. Sci. USA. 1999;96:14147–14152. doi: 10.1073/pnas.96.24.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roth B.M., Pruss G.J., Vance V.B. Plant viral suppressors of RNA silencing. Virus Res. 2004;102:97–108. doi: 10.1016/j.virusres.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 24.Anandalakshmi R., Pruss G.J., Ge X., Marathe R., Mallory A.C. A viral suppressor of gene silencing in plants. Proc. Natl. Acad. Sci. USA. 1998;95:13079–13084. doi: 10.1073/pnas.95.22.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szittya G., Molnar A., Silhavy D., Hornyik C., Burgyán J. Short defective interfering RNAs of tombusviruses are not targeted but trigger post-transcriptional gene silencing against their helper virus. Plant Cell. 2002;14:1–15. doi: 10.1105/tpc.010366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W.X., Li H.W., Lu R., Li F., Dus M. Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc. Natl. Acad. Sci. USA. 2004;101:1350–1355. doi: 10.1073/pnas.0308308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunoyer P., Lecellier C.H., Parizotto E.A., Himber C., Voinnet O. Probing the microRNA and small interfering RNA pathways with virus-encoded suppressors of RNA silencing. Plant Cell. 2004;16:1235–1250. doi: 10.1105/tpc.020719. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Lecellier C.H., Dunoyer P., Arar K., Lehmann-Che J., Eyquem S. A cellular MicroRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- 29.Chapman E.J., Prokhnevsky A.I., Gopinath K., Dolja V.V., Carrington J.C. Viral RNA silencing suppressors inhibit the microRNA pathway at an intermediate step. Genes Dev. 2004;18:1179–1186. doi: 10.1101/gad.1201204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papp I., Mette M.F., Aufsatz W., Daxinger L., Schauer S.E., Ray A., van der Winden J., Matzke M., Matzke A.J. Evidence for nuclear processing of plant microRNA and short-interfering RNA precursors. Plant Physiol. 2003;132:1382–1390. doi: 10.1104/pp.103.021980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lakatos L., Szittya G., Silhavy D., Burgyan J. Molecular mechanism of RNA silencing suppression mediated by p19 protein of tombusviruses. EMBO J. 2004;23:876–884. doi: 10.1038/sj.emboj.7600096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calabrese J.M., Sharp P.A. Characterization of the short RNAs bound by the P19 suppressor of RNA silencing in mouse embryonic stem cells. RNA. 2006;12:2092–2102. doi: 10.1261/rna.224606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu M., Desvoyes B., Turina M., Noad R., Scholthof H.B. Genetic dissection of tomato bushy stunt virus p19-protein-mediated host-dependent symptom induction and systemic invasion. Virology. 2000;266:79–87. doi: 10.1006/viro.1999.0071. [DOI] [PubMed] [Google Scholar]
- 34.Vargason J.M., Szittya G., Burgyan J., Hall T.M.T. Size selective recognition of siRNA by an RNA silencing suppressor. Cell. 2003;115:799–811. doi: 10.1016/s0092-8674(03)00984-x. [DOI] [PubMed] [Google Scholar]
- 35.Ye K.Q., Malinina L., Patel D.J. Recognition of small interfering RNA by a viral suppressor of RNA silencing. Nature. 2003;426:874–878. doi: 10.1038/nature02213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fersht A.R., Daggett V. Protein folding and unfolding at atomic resolution. Cell. 2002;108:573–582. doi: 10.1016/s0092-8674(02)00620-7. [DOI] [PubMed] [Google Scholar]
- 37.Brooks C.L., Onuchic J.N., Wales D.J. Statistical thermodynamics—taking a walk on a landscape. Science. 2001;293:612–613. doi: 10.1126/science.1062559. [DOI] [PubMed] [Google Scholar]
- 38.Brooks C.L., Gruebele M., Onuchic J.N., Wolynes P.G. Chemical physics of protein folding. Proc. Natl. Acad. Sci. USA. 1998;95:11037–11038. doi: 10.1073/pnas.95.19.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou R.H., Huang X.H., Margulis C.J., Berne B.J. Hydrophobic collapse in multidomain protein folding. Science. 2004;305:1605–1609. doi: 10.1126/science.1101176. [DOI] [PubMed] [Google Scholar]
- 40.Liu P., Huang X.H., Zhou R.H., Berne B.J. Observation of a dewetting transition in the collapse of the melittin tetramer. Nature. 2005;437:159–162. doi: 10.1038/nature03926. [DOI] [PubMed] [Google Scholar]
- 41.Snow C.D., Nguyen N., Pande V.S., Gruebele M. Absolute comparison of simulated and experimental protein-folding dynamics. Nature. 2002;420:102–106. doi: 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- 42.Garcia A.E., Onuchic J.N. Folding a protein in a computer: an atomic description of the folding/unfolding of protein A. Proc. Natl. Acad. Sci. USA. 2003;100:13898–13903. doi: 10.1073/pnas.2335541100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kokubo H., Okamoto Y. Prediction of transmembrane helix configurations by replica-exchange simulations. Chem. Phys. Lett. 2004;383:397–402. [Google Scholar]
- 44.Straub J.E., Thirumalai D. Theoretical probes of conformational fluctuations in S-peptide and RNase A/3′-UMP enzyme product complex. Proteins. 1993;15:360–373. doi: 10.1002/prot.340150404. [DOI] [PubMed] [Google Scholar]
- 45.Eleftheriou M., Germain R., Royyuru A., Zhou R. Thermal denaturing of mutant lysozyme with both OPLSAA and CHARMM force fields. J. Am. Chem. Soc. 2006;128:13388–13395. doi: 10.1021/ja060972s. [DOI] [PubMed] [Google Scholar]
- 46.Li X., Li J., Eleftheriou M., Zhou R. Hydration and dewetting near fluorinated superhydrophobic plates. J. Am. Chem. Soc. 2006;128:12439–12447. doi: 10.1021/ja057944e. [DOI] [PubMed] [Google Scholar]
- 47.Tucker M.J., Oyola R., Gai F. A novel fluorescent probe for protein binding and folding studies: p-cyano-phenylalanine. Biopolymers. 2006;83:571–576. doi: 10.1002/bip.20587. [DOI] [PubMed] [Google Scholar]
- 48.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar S., Huang C., Zheng G., Bohm E., Bhatele A., Phillips J.C., Yu H., Kale L.V. Scalable molecular dynamics with NAMD on Blue Gene/L. IBM.J. Res. Dev. 2008;52:177–188. [Google Scholar]
- 50.Brooks B.R., Bruccoeri R.E., Olafson B.D., States D.J., Swaminathan S. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 51.MacKerell A.D., Jr., Banavali N., Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 52.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 53.Jorgensen W.L., Chandrasekhar J., Madura J., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 54.Neria E., Karplus M. A position dependent friction model for solution reactions in the high friction regime: proton transfer in triosephosphate isomerase (TIM) J. Chem. Phys. 1996;105:10812–10818. [Google Scholar]
- 55.Deserno M., Holm C. How to mesh up Ewald sums. I. A theoretical and numerical comparison of various particle mesh routines. J. Chem. Phys. 1998;109:7678–7693. [Google Scholar]
- 56.Deserno M., Holm C. How to mesh up Ewald sums. II. An accurate error estimate for the particle-particle-particle-mesh algorithm. J. Chem. Phys. 1998;109:7694–7701. [Google Scholar]
- 57.Deng Y.Q., Roux B. Calculation of standard binding free energies: aromatic molecules in the T4 lysozyme L99A mutant. J. Chem. Theory Comput. 2006;2:1255–1273. doi: 10.1021/ct060037v. [DOI] [PubMed] [Google Scholar]
- 58.Tirado-Rives J., Jorgensen W.L. Contribution of conformer focusing to the uncertainty in predicting free energies for protein-ligand binding. J. Med. Chem. 2006;49:5880–5884. doi: 10.1021/jm060763i. [DOI] [PubMed] [Google Scholar]
- 59.Thorpe I.F., Brooks C.L., 3rd Molecular evolution of affinity and flexibility in the immune system. Proc. Natl. Acad. Sci. USA. 2007;104:8821–8826. doi: 10.1073/pnas.0610064104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jayachandran G., Shirts M.R., Park S., Pande V.S. Parallelized-over-parts computation of absolute binding free energy with docking and molecular dynamics. J. Chem. Phys. 2006;125:084901. doi: 10.1063/1.2221680. [DOI] [PubMed] [Google Scholar]
- 61.Reference deleted in proof.
- 62.Pan Y., Gao D., Yang W., Cho H., Zhan C.G. Free energy perturbation (FEP) simulation on the transition states of cocaine hydrolysis catalyzed by human butyrylcholinesterase and its mutants. J. Am. Chem. Soc. 2007;129:13537–13543. doi: 10.1021/ja073724k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang W., Bitetti-Putzer R., Karplus M. Free energy simulations: use of reverse cumulative averaging to determine the equilibrated region and the time required for convergence. J. Chem. Phys. 2004;120:2618–2628. doi: 10.1063/1.1638996. [DOI] [PubMed] [Google Scholar]
- 64.Reference deleted in proof.
- 65.Warshel A. Simulating the energetics and dynamics of enzymatic reactions in specificity in biological interactions. Pontificiae Academiae Scientiarum Scripta Varia. 1984;55:60–81. [Google Scholar]
- 66.Aqvist J., Warshel A. Energetics of ion permeation through membrane channels. Solvation of Na+ by gramicidin A. Biophys. J. 1989;56:171–182. doi: 10.1016/S0006-3495(89)82662-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Warshel A., Sharma P.K., Kato M., Parson W.W. Modeling electrostatic effects in proteins. Biochim. Biophys. Acta. 2006;1764:1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 68.Mark A.E., van Gunsteren W.F. Decomposition of the free energy of a system in terms of specific interactions. Implications for theoretical and experimental studies. J. Mol. Biol. 1994;240:167–176. doi: 10.1006/jmbi.1994.1430. [DOI] [PubMed] [Google Scholar]
- 69.Boresch S., Karplus M. The meaning of component analysis: decomposition of the free energy in terms of specific interactions. J. Mol. Biol. 1995;254:801–807. doi: 10.1006/jmbi.1995.0656. [DOI] [PubMed] [Google Scholar]
- 70.Brady G.P., Sharp K.A. Decomposition of interaction free energies in proteins and other complex systems. J. Mol. Biol. 1995;254:77–85. doi: 10.1006/jmbi.1995.0600. [DOI] [PubMed] [Google Scholar]
- 71.Bren M., Florian J., Mavri J., Bren U. Do all pieces make a whole? Thiele cumulants and the free energy decomposition. Theor. Chem. Acc. 2007;117:535–540. [Google Scholar]
- 72.Scholthof H.B. Timeline—the Tombusvirus-encoded P19: from irrelevance to elegance. Nat. Rev. Microbiol. 2006;4:405–411. doi: 10.1038/nrmicro1395. [DOI] [PubMed] [Google Scholar]