

Abstract
Two arginine residues, Arg-181 and Arg-268, are conserved throughout the known family of FMN-containing enzymes that catalyze the oxidation of α-hydroxyacids. In the lactate oxidase from Aerococcus viridans, these residues have been changed to lysine in two single mutations and in a double mutant form. In addition, Arg-181 has been replaced by methionine to determine the effect of removing the positive charge on the residue. The effects of these replacements on the kinetic and thermodynamic properties are reported. With all mutant forms, there are only small effects on the reactivity of the reduced flavin with oxygen. On the other hand, the efficiency of reduction of the oxidized flavin by l-lactate is greatly reduced, particularly with the R268K mutant forms. The results demonstrate the importance of the two arginine residues in the binding of substrate and its interaction with the flavin, and are consistent with a previous hypothesis that they also play a role of charge neutralization in the transition state of substrate dehydrogenation. The replacement of Arg-268 by lysine also results in a slow conversion of the 8-CH3- substituent of FMN to yield 8-formyl-FMN, still tightly bound to the enzyme, and with significantly different physical and chemical properties from those of the FMN-enzyme.
The enzyme l-lactate oxidase from Aerococcus viridans (LOX) is a member of the family of flavoproteins that catalyze the oxidation of α-hydroxyacids (1). The kinetic characteristics of the enzyme have been reported by our group, but still the mechanism of the reductive half-reaction by α-hydroxyacids is one of central interest. Our previous study of the reactivity of LOX with a series of para-substituted mandelates indicated little development of charge in the transition state of the reductive half reaction (2). This result is compatible with either a hydride mechanism or with a synchronous mechanism in which the α-CH- of the substrate is formally abstracted as a proton, whereas the resulting charge is simultaneously neutralized by another event. We proposed the participation of two arginine residues, Arg-181 and Arg-268, which are located around the FMN and which could be part of the substrate binding site, based on the x-ray crystal structures of glycolate oxidase (3, 4) and flavocytochrome b2 (5, 6). These two arginine residues are conserved in all family members, as shown in Fig. 1. It should be noted that there is a 20- to 30-aa residue insertion in l-lactate monooxygenase and l-mandelate dehydrogenase between these two arginine residues.
Figure 1.

Conserved two-arginine residues in members of the family of FMN-containing flavoproteins that catalyze the oxidation of α-hydroxyacids. Only parts of the sequences are shown; those on the left containing the recently recognized catalytically important arginine residue, those on the right also containing the active site histidine residue. Abbreviations: LOX, lactate oxidase of Aerococcus viridans, (GenBank accession no. E02499); LMO, lactate monooxygenase from Mycobacterium smegmatis (J05402); GLO, glycolate oxidase from spinach, (J03492); b2′ flavocytochrome b2 from Saccharomyces cerevisiae (X03215); MDH, mandelate dehydrogenase from Pseudomonas putida (J05293); LHO, long chain α-hydroxyacid oxidase from rat (X67156).
In this paper, we have replaced these two residues by either lysine as a relatively mild perturbation or methionine as removing positive charge (R268K, R181K, R181M, and R181K + R268K) and studied the characteristics of the resulting enzymes to try to elucidate the functions of these arginine residues. We found that both residues are involved in the reductive half reaction and in the interaction of substrates and ligands with the FMN prosthetic group.
Materials and Methods
Site-Directed Mutagenesis.
Amino acid substitutions R181K, R268K, and their double substitution were carried out by a site-directed mutagenesis of the lactate oxidase gene in the expression vector, pAOX8, which is composed of a 1.2-kb insert containing the coding region for lactate oxidase in pACYC184. Two fragments were generated by PCR: one contained the sequence from the HindIII site of the pACYC184 plasmid to the site of mutagenesis and the other the sequence from the site of mutagenesis to the BamHI site of the expression plasmid. These two fragments were annealed at the site of mutagenesis and extended enzymatically to yield the coding region with the expected mutation. The resulting sequence was amplified by PCR using the HindIII and BamHI site primers. After treatment with HindIII and BamHI, the DNA product was ligated into these restriction sites in pACYC184 to yield the expression plasmid, pAOX8, with the appropriate mutation. The double mutant was generated by using the primers for both mutations together with the primers that generated the HindIII and BamHI sites, resulting in three DNA fragments, which were annealed, extended, and ligated into the expression vector. The mutation of R181M was carried out by the method of Kunkel et al. (7).
Flavin Extraction from the Enzymes.
Buffer salts in solutions of R268K-LOX and wild-type LOX were replaced by water by repeated ultrafiltration with Centricon 30 filters. Trifluoroacetic acid (TFA) (25 μl) was added to 0.5 ml enzyme solution. After 2 min of incubation, the samples were centrifuged in a microfuge for 2 min. Diethyl ether (10 ml) was added to the supernatant and shaken well in a separating funnel to remove TFA from the water phase. This process was repeated until the pH value of the water phase became neutral.
HPLC Analysis.
RP-HPLC analysis was carried out with a Hitachi instrument, Hitachi model 655 pump and Hitachi model L-6200 intelligent pump with a Hitachi model F1000 fluorescence detector, using a Hi-Pore RP-318 (Bio-Rad) 250 × 10-mm column. Elution was carried out with a linear gradient between 10% and 50% methanol in 5 mM ammonium acetate solution, and the fluorescence at 530 nm of the eluant was monitored employing an excitation wavelength of 450 nm. The flow rate was 2 ml/min at room temperature.
Matrix-Assisted Laser Desorption Ionization-Time-of-Flight (MALDI-TOF) Mass Spectra.
A PerSeptive Biosystems Vestec Mass Spectrometry Products with Voyager RF BioSpectrometry Workstation was used for the measurement of MALDI-TOF mass spectra of both the extracted flavins from LOX and the R268K mutant enzyme and for the intact enzymes without flavin release. Either sinapinic acid for flavin extracts or α-cyano-4-hydroxycinnamic acid for protein-bound flavin was used as the matrix material. A 10 mg/ml Matrix solution was made in water/acetonitrile (2:1 and 1:1 ratio, respectively) in the presence of 0.1% TFA, and vortexed for 1 min. Equal volume of supernatant of this matrix solution was mixed with the samples, and 1-μL aliquots of the mixture were used for analysis. The spectral calibration was done by using neurotensin, angiotensin, or BSA as the external standard or by using the peaks of the matrix in flavin extract samples as the internal standard.
Other experimental methods used in this work were as described before (1, 2, 8).
Results
Absorption Spectra of Mutants.
All of the mutant enzymes were purified by the same method as for the wild type (1). Absorption spectra of enzyme-bound flavin of mutant forms in 10 mM imidazole buffer in the presence of 100 mM KCl are shown in Fig. 2. The peak position of the visible wavelength region is at 458 nm (ɛ458 = 11.2 × 103 M−1⋅cm−1) for R181M, and those for other mutant forms are at 456 nm, the same as that of the wild-type LOX (ɛ458 = 11.0 × 103 M−1⋅cm−1 both for wild type and R181K). The single mutations at Arg-181, R181K, or R181M, did not show any spectral change with time, but the mutant enzyme where Arg-268 was replaced with Lys (R268K) showed time-dependent spectral changes. The double mutant R181K/R268K showed quite similar changes of its spectrum with time (see later section for details).
Figure 2.

Absorption spectra of wild-type and mutant forms of lactate oxidase, and the effect of addition of pyruvate. The dotted line spectra in each box are those of the enzyme without pyruvate; the solid line spectra are those in the presence of saturating concentrations of pyruvate. Perturbations of the spectra caused by the binding of pyruvate are shown as difference spectra.
Spectral Perturbation by Pyruvate.
Fig. 2 also shows the effects of titration of the enzymes with pyruvate. Pyruvate addition to all of the oxidized enzyme forms results in the development of a slight but distinct shoulder around 490 nm. Red shifts of the flavin visible absorption by 2 nm, 2 nm, and 10 nm were observed in wild type, R181K, and R181M, respectively. On the other hand, neither R268K nor R181K/R268K showed any peak shift. Using the difference spectral data from titration with pyruvate, the dissociation constants of the oxidized enzyme–pyruvate complexes were determined as 21 mM, 30 mM, and 3 mM for wild type, R181K, and R181M, respectively.
Reductive Half Reaction of R181K and R181M.
The stopped flow traces at 456 nm of R181K and R181M show single exponential kinetics. From the l-lactate concentration dependence, limiting first order rate constants of 25.6 s−1 and 2.3 s−1 were estimated for R181K and R181M, respectively (Table 1). These values are considerably lower than the reduction rate constant for wild-type enzyme of 320 s−1 (9) In addition to a decrease in kred, the Kd for the oxidized enzyme-lactate complex was also increased significantly for the mutant enzymes. The ratio kred/Kd lactate is a measure of catalytic efficiency, numerically equal to the better known ratio of kcat/Km (9) and is decreased by a factor of 230 for R181K and 32,000 for R181M (Table 1).
Table 1.
Kinetic and thermodynamic parameters of l-lactate oxidase and the site-directed mutants
| Wild type | R181K | R181M | R268K | R181K/R268K | |
|---|---|---|---|---|---|
| kred | 320 s−1 | 26 s−1 | 2.3 s−1 | (210 M−1⋅s−1) | (0.11 M−1⋅s−1) |
| 180 M−1⋅s−1* | |||||
| 8.0 × 103 M−1⋅s−1 | ND | ||||
| 1.3 × 104 M−1⋅s−1* | |||||
| Kd lactate | 0.67 mM | 7.3 mM | 94 mM | — | — |
| kred/Kd lactate, M−1⋅s−1 | 8.1 × 105 | 3.5 × 103 | 25 | — | — |
| kox oxygen, M−1⋅s−1 | 1.8 × 106 | 9.0 × 105 | 8.0 × 104 | (3.6 × 105) | (2.9 × 105) |
| 1.3 × 105* | |||||
| 7.0 × 103 | (4.6 × 103) | ||||
| 8.0 × 103* | |||||
| kon sulfite, M−1⋅s−1 | 1.5 × 105 | 7.9 × 103 | 140 | (7.7 × 103) | 110 |
| 7.0 × 104 | 2.8 × 103 | ||||
| koff sulfite, s−1 | 7.6 × 10−2 | 2.5 × 10−2 | 3.0 × 10−3 | (3.6 × 10−3) | (6.8 × 10−3) |
| 5.3 × 10−4 | 3.2 × 10−3 | ||||
| Kd sulfite, M | 5.0 × 10−7 | 3.2 × 10−6 | 2.1 × 10−5 | (4.7 × 10−7) | (6.2 × 10−5) |
| 7.6 × 10−9 | 1.1 × 10−6 | ||||
| kcat, s−1 | 280 | 23.1 | 2.3 | ND | ND |
| Km lactate | 0.94 mM | 7.5 mM | 103 mM | ND | ND |
| Km oxygen | 160 μM | 29 μM | 30 μM | ND | ND |
| Kd Eox-pyruvate | 21 mM | 30 mM | 3 mM | ND | ND |
| Kd Ered-pyruvate | 3 mM | 56 mM | 160 mM | ND | ND |
| Em ox/red | −128 mV | −126 mV | −140 mV | ||
| Em ox/SQ | −95 mV | ||||
| Em SQ/red | −161 mV |
ND, not determinable. In the entries for R268K and R181K/R268K, the values in parentheses are for the major phase reactions of freshly prepared enzyme and presumably due to the normal FMN form. The corresponding values for aged enzyme are identified by asterisks. All measurements were carried out in 10 mM imidazole with 100 mM KCl at pH 7.0, 25°C.
Time-Dependent Changes in Properties of R268K and R181K/R268K Mutant Enzymes.
Freshly prepared enzyme in which Arg-268 had been replaced by Lys did not show any unusual characteristics in absorbance properties. However, unlike the other mutant enzyme forms, the kinetics of reduction by l-lactate showed biphasic kinetics. For example, the 456-nm traces of freshly prepared R268K decreased in a biphasic manner with ca. 10% occurring in a fast phase and ca. 90% in a slow phase. The fast phase of absorbance decrease at 456 nm correlates with an increase in absorbance at 395 nm and with a small monophasic increase at 550 nm. From the major slow phase traces at 456 nm, the reduction rate constant of R268K by l-lactate was calculated as 210 M−1⋅s−1 (Table 1). The small amplitude changes of the fast phase made it difficult to determine the associated kinetic parameters accurately, but the reduction rate constant was of the order of 8 × 103 M−1⋅s−1. The double mutant enzyme R181K/R268K also displayed biphasic kinetics, summarized in Table 1.
On long-term storage in ice, the spectrum of R268K gradually changed to one with wavelength maxima at 470 and 350 nm. Similar changes occurred with R181K/R268K, but not with wild-type enzyme or the other mutant forms. The aged enzyme displayed the same type of multiphasic kinetic patterns as the freshly prepared enzyme, except that now the fastest phase of reduction was associated with the major amplitude changes at 395, 456, and 550 nm. For this phase, the observed rate constants are directly proportional to lactate concentration, with a second order rate constant of 1.3 × 104 M−1⋅s−1. The second and third phases are manifest only in the 430- to 500-nm region, but because of the small amplitude changes, 17% and 28% the total at 456 nm, it is difficult to determine accurately the associated kinetic constants. Approximate values of 180 M−1·s−1 and 20 M−1⋅s−1 appear to be associated with these two phases. In the spectral regions 360–420 nm and 510–570 nm, >90% of the absorbance changes are associated with the fast phase of reduction.
Fig. 3 shows the spectra of the oxidized and fully reduced forms of the enzyme, freshly prepared and after aging. The unusual spectral properties of the oxidized and reduced aged enzyme are similar to ones reported by Edmondson (10) for 8-formyltetraacetyl riboflavin, where the intramolecular hemiacetal formation with the 5′ ribityl hydroxyl group found for underivatized 8-formyl riboflavin is prevented by the tetraacetyl substitution. Similar properties would be expected for 8-formyl-FMN, where the terminal ribityl phosphate would also prevent hemiacetal formation.
Figure 3.

Spectral properties of the fresh and aged forms of R268K. (A) Enzyme freshly purified. The spectra of the oxidized enzyme and that of the reduced enzyme after reduction by lactate are shown. (B) Enzyme after storage in ice for 3.5 mo in 10 mM imidazole/100 mM KCl buffer, pH 7.0. The spectrum identified with ● is that of the oxidized enzyme. The spectrum identified with ▴ is that formed maximally on irradiation with visible light under anaerobic conditions in the presence of 10 mM glycine and 1 μM 5-deazaflavin (11). The spectrum identified with □ is that of the fully reduced enzyme obtained after prolonged irradiation. The Inset shows the time course of the spectral changes at 425, 470, and 700 nm as a function of irradiation time. The spectrum of enzymed reduced by l-lactate was the same as that of the photochemically reduced form.
To test the possibility that the spectral changes observed on aging are because of conversion of the 8-methyl substituent of FMN to a formyl residue, the aged enzyme was denatured with TFA, and the extract was subjected to RP-HPLC and MALDI-TOF mass spectrometry. Control extracts from wild-type enzyme were run in parallel. The HPLC retention time for wild-type extract was 12.54 min, identical to that of authentic FMN, whereas the extract from R268K showed a major component at 9.95 min and a minor component at 12.54 min. MALDI-TOF analyses of the main HPLC fractions showed a molecular weight of 471 for the flavin from aged enzyme and one of 457 from wild-type enzyme. The latter value is exactly that of FMN, and the difference of 14 mass units for the flavin from aged enzyme R268K is that expected for conversion of a CH3 residue to a CHO residue. The same values of 471 and 457 for the flavins were also evident in the MALDI-TOF spectra of intact enzyme, showing that the 14 mass difference was not associated with release of the flavin from the protein.
Further confirmation of the nature of the modified flavin was obtained by comparison of spectra of the oxidized and reduced forms with those reported for 8-formyl tetraacetylriboflavin (10). Reduction was carried out photochemically under anaerobic conditions to yield spectra essentially identical to those reported for the model flavin (Fig. 4) and characterized by the unusually strong absorbance maxima at 390 and 520 nm for the reduced form. In the pH range around neutrality, lactate oxidase stabilizes both the neutral and anionic forms of FMN, with an observed pKa of 6.0 (1). Because of paucity of modified enzyme, we were not able to make a study of the effect of pH, but, at pH 7, the spectral changes on photochemical reduction (Fig. 3B) indicate the stabilization of both semiquinone forms of 8-formyl-FMN, characterized by the intense peak at 425 nm, presumably that of the anionic semiquinone, and the broad absorption band in the 600- to 800-nm region, characteristic of the neutral semiquinone forms of normal flavin and other modified flavins (12)
Figure 4.

Spectra of the oxidized and photochemically reduced form of the flavin extracted from aged R268K. The spectrum of the reduced flavin was obtained under anaerobic conditions by brief irradiation with visible light in the presence of 15 mM EDTA, pH 7.0.
Oxidative Half Reaction by Molecular Oxygen.
The two mutant forms where Arg-181 was replaced by Lys or Met were reoxidized by molecular oxygen in a single exponential fashion with observed rate constants directly proportional to the oxygen concentration. The second order rate constants of R181K and R181M were 9.0 × 105 s−1⋅M−1 and 8.0 × 104 s−1⋅M−1, 47% and 4% of that for wild-type enzyme. On the other hand, the mutant enzyme with Arg-268 replaced by Lys showed two phases, the major phase being the faster one in the case of freshly purified samples of the enzyme. Rate constants are listed in Table 1. On aging, the R268K enzyme again showed changes in the kinetic pattern of reoxidation by oxygen. Now the minor change in absorbance at 468 nm (≈22% total) was the fastest phase, with a second order rate constant of 1.3 × 105 M−1⋅s−1. The major change in absorbance (≈73% total) was associated with a rate constant of 8 × 103 M−1⋅s−1. The third phase (≈5% total) was not capable of consistent analysis. The major phase in aged enzyme is probably because of the 8-formyl-FMN form of the enzyme, because this form is present only in small amounts (≈15%) in freshly prepared enzyme and increases on long-term storage (see previous section).
Sulfite Reactivity.
Lactate oxidase, in common with many flavoprotein oxidases (13), forms an equilibrium flavin N5 adduct with essentially complete loss of the typical flavin absorbance in the 350- to 500-nm region. Thus, the kinetics of adduct formation (kon) can be monitored conveniently by stopped flow spectrophotometry, and that of adduct dissociation (koff) by competition with methylmethane thiosulfonate, which reacts very rapidly with free sulfite (14). All of the mutant forms were found to react much more slowly with sulfite than does wild-type enzyme. R181K and R181M forms showed single exponential reactions with sulfite. Similar simple behavior was found for sulfite release. The relevant kon and koff values are listed in Table 1. In common with all other properties, the freshly prepared R268K and R181K/R268K mutant forms showed complex behavior, with minor fast and major slow changes. The derived rate constants are listed in Table 1.
Steady State Kinetics.
The catalysis of oxidation of l-lactate by the mutant enzymes was determined by the method of enzyme-monitored turnover, in which the absorbance of the enzyme itself in the presence of excess l-lactate and limiting concentrations of oxygen was used to determine the rates of the enzyme-catalyzed reaction (15). In all cases, parallel Lineweaver-Burk plots of reciprocal turnover number versus reciprocal oxygen concentration were obtained at various concentrations of l-lactate. Secondary plots of the intercepts versus l-lactate concentration yield the kinetic constants kcat, Km lactate, and Km oxygen, as listed in Table 1. The results are consistent with a modified ping-pong mechanism as established for wild-type enzyme (1).
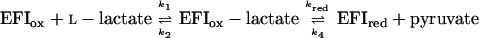 |
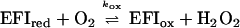 |
The kinetic constants obtained are in impressive agreement with those predicted from the individual rate constants determined in the separate reductive and oxidative half reactions. For the kinetic mechanism above, kcat = kred, Km lactate = k2 + kred/k1 and Km oxygen = kred/kox. Also in agreement with the observed apparent second order reactions of R268K and R181K/R268K with both l-lactate and oxygen, the steady state kinetic plots with these enzyme forms also gave parallel Lineweaver-Burk primary plots, but the secondary plots of intercepts versus reciprocal l-lactate concentration extrapolate to the origin.
Oxidation-Reduction Potentials.
The redox properties of R181M were determined by stopped flow experiments, where oxidized enzyme was mixed anaerobically with varied concentrations of l-lactate at a series of fixed concentrations of pyruvate. The degree of reduction of the enzyme flavin was monitored by the spectra of the equilibrium mixture of each reaction. The plots of log [pyruvate]/[lactate] versus log [Eox]/[Ered] yield values for the observed redox potential at the varied fixed pyruvate concentrations as shown in Fig. 5. These results are expected because of the very different values of the dissociation constants of pyruvate binding to oxidized enzyme (3 mM) and reduced enzyme (160 mM). The data may be analyzed by the following equation (16):
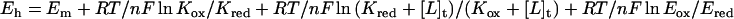 |
where [L]t = total pyruvate concentration, Kox = Kd for Eox-pyr complex, Kred = Kd for Ered-pyr complex, and Eox and Ered the concentrations of oxidized and reduced forms of the enzyme.
Figure 5.

Determination of the mid point potential at pH 7.0 of the R181M enzyme form and the effect of pyruvate concentration on the apparent Em value. The concentrations of oxidized (Eox) and reduced (Ered) enzyme forms were determined from the spectra at equilibrium on mixing enzyme under anaerobic conditions in a Hi-Tech model SF-61 stopped flow spectrophotometer at fixed pyruvate concentrations of 2.5 mM (⧫), 10 mM (▾), 20 mM (▴), and 40 mM (●). The Em apparent values were determined from the intercept values of log (Eox/Ered) and are shown in the Inset. The line through the points of the inset is a theoretical one for an Em value of −140 mV in the absence of pyruvate (see text for details).
The inset of Fig. 5 shows the plots of apparent midpoint potentials at different pyruvate concentrations and also shows the calculated values based on the above equation with the Kox value of 3 mM and Kred value of 160 mM. From this plot, the real redox midpoint potential of R268K mutant form is determined as −140 mV. With R181K, the Kd values for pyruvate binding to oxidized and reduced enzyme are similar; hence, pyruvate concentration should have very little effect on the measured redox potential. We therefore determined its redox potential by the same method as described above. In this case, as expected, a single linear plot of log [pyruvate]/[lactate] versus log [Eox]/[Ered] was obtained, independent of the total pyruvate concentration. The Em value at pH 7.0 was determined as −126 mV (Table 1).
With wild-type enzyme, we were not able to employ the above system, because no convenient ratios of pyruvate and lactate could be obtained that provided reliable and reproducible measurements of partially reduced enzyme. In this case, the midpoint potential was obtained by a combination of measurements of the 1-electron redox potential, Em,ox/SQ, and the semiquinone stability constant (17). The Em,ox/SQ potential was measured as −95 mV by the xanthine/xanthine oxidase method (18) using indigo disulfonate (Em = −116 mV) as standard. The semiquinone stability constant K was determined by following the formation and decay of the semiquinone, with its characteristic large extinction at 398 nm, as a function of time during reduction of the enzyme by the xanthine/xanthine oxidase system in the presence of benzyl viologen as 1 electron mediator. The value of K was determined as 12.4, which determines the value of the second one-electron couple, Em,SQ/red as being 66 mV more negative than that of Em,ox/SQ, resulting in a value of −161 mV for the Em,SQ/red couple and in a midpoint potential for the couple Eox/Ered of −128 mV (Table 1).
Discussion
The proposed roles of Arg-181 and Arg-268 in the oxidation of substrate were probed by conservative mutations, in which the original positive charge should be maintained. Thus, the two single mutant forms R181K and R268K, and the double mutant form R181K/R268K were prepared and their effects on enzyme properties determined. These three mutations had comparatively little effect on the reactivity of the reduced enzymes with oxygen, but affected greatly the rate constant of reduction of the enzyme by l-lactate (Table 1). Results with the R268K and double mutant enzymes are complicated by the gradual changes of the flavin, but with freshly prepared enzyme the reduction by l-lactate in both cases is effectively second order with rate constants of 210 M−1⋅s−1 and 0.11 M−1⋅s−1, respectively. This result indicates that not only was reduction more difficult, but that binding of the substrate was so weak that no enzyme substrate complex could be detected. The R181K enzyme showed saturation kinetics on reduction by l-lactate but with a 12-fold lower value of kred than wild-type enzyme and an 11-fold weaker binding of lactate. The binding of pyruvate to the R181K reduced enzyme form was almost 20-fold weaker. These results are consistent with both arginine residues playing a role in catalysis through their interaction with substrate and product.
Because the R181K enzyme retains appreciable catalytic activity, the positive charge of this arginine residue was removed by replacement with methionine. The reduction of R181M by l-lactate still showed saturation kinetics, but the rate was decreased by a further 11-fold from that of R181K and with a further 13-fold weaker binding of lactate. The reoxidation of the reduced enzyme also decreased 23-fold and 11-fold, respectively, from that of the wild-type and R181K enzymes. The R181M enzyme was also unusual in its binding of pyruvate to the oxidized enzyme form, where an unusually large spectral change was observed on binding (Fig. 2). The binding of pyruvate to the oxidized enzyme was much tighter than that found with wild-type and R181K enzymes, and was also unusual in binding much more tightly to the oxidized enzyme than to the reduced form (Table 1). Indeed, these properties appear to be the reason for the decreased reactivity of the enzyme with lactate, because the preferential binding of pyruvate to oxidized enzyme would result in a 50-mV decrease of the midpoint redox potential to a value close to that of the free pyruvate/lactate couple (−189 mV). This property should be contrasted with that of the R181K enzyme, where pyruvate binding is similar to both oxidized and reduced enzyme forms and so does not perturb the redox potential. On the other hand, with wild-type enzyme, pyruvate binding is 7-fold tighter to reduced enzyme than to oxidized enzyme, which would be predicted to result in a 25-mV increase in the flavin midpoint potential, making the thermodynamics of reduction by lactate even more favorable.
The major surprise with this study was the unexpected conversion of the normal FMN in R268K and R181K/R268K to 8-formyl-FMN. This is a slow process occurring over many months, but some of the modified flavin is present even in fresh preparations of the enzymes. A corresponding instability has been observed with an analogous mutant form of lactate monooxygenase, where the R293K enzyme appeared to form an equilibrium N5-flavin adduct with a protein thiol residue (19). Lehman and Thorpe (20) have reported previously on variable amounts of an unidentified flavin in preparations of the electron transfer flavoprotein from pig kidney. The spectral properties of this flavin are very similar to those reported here and in ref. 10. The mechanism of conversion of normal flavin to 8-formyl FMN has not yet been addressed experimentally. The conversion involves a four-electron oxidation, possibly initiated by proton abstraction from the 8-methyl substituent by the unprotonated ɛ-amino residue of the introduced lysine and subsequent reactions of the resulting 8-quinone methide, a very reactive flavin form involved in covalent flavin–protein attachment (21–23). In this respect, we plan to change Arg-268 to a methionine residue, where the conversion to 8-formyl FMN would not be expected to occur.
The properties of the 8-formyl FMN enzyme are consistent with the higher redox potential expected of 8-formyl-FMN. Based on the studies of Edmondson (10), the midpoint potential of 8-formyl-FMN at pH 7 would be expected to be ca. −90 mV, a value ca. 120 mV more positive than that of FMN. From the previously observed linear free energy relationship data, this increase in redox potential would be expected (9) to result in much faster reduction by lactate, much slower reoxidation of the reduced flavin by oxygen, and a considerably tighter binding of sulfite, in keeping with the results obtained (Table 1).
In conclusion, the present studies confirm a catalytic role for Arg-181 in addition to that of Arg-268, as expected from the crystal structures of flavocytochrome b2 (5) and glycolate oxidase (4) and the impressive conservation of active site residues in this whole family of enzymes (1, 14, 24). Similar mutations have been carried out with lactate monooxygenase (25, 26), mandelate dehydrogenase (27), and flavocytochrome b2 (6, 28). In each case, it is clear that the pairs of arginine residues are involved in substrate binding and catalysis. Although the results do not offer any real proof, they are consistent with the hypothesis that the two arginine residues serve the complementary roles of assisting in binding of the substrate carboxylate and in neutralizing the negative charge developed in the transition state on removal of the proton from the α-carbon position by the active site histidine base (2).
Acknowledgments
We thank Mr. Toshiyuki Watanabe (Asahi Chemical Company, Japan) for the help of mutant enzyme purification, the NEDO team from Novartis Pharma K.K (Tsukuba, Japan) for the help of the construction of the mutants, the Fujii-Otsuka Foundation (Tokushima, Japan) for travel support (to V.M.), Drs. Bette Jo Brown, Younus Meah, Sumita Chakraborty, and Colin Thorpe for valuable discussion, and Dr. Dale Edmondson for extensive discussion and critical reading of the manuscript. This work was supported in part by National Institutes of Health Grant GM-11106 (to V.M.).
Abbreviations
- LOX
l-lactate oxidase
- TFA
trifluoroacetic acid
- MALDI-TOF
matrix-assisted laser desorption ionization-time-of-flight
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.250472297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.250472297
References
- 1.Maeda-Yorita K, Aki K, Sagai H, Misaki H, Massey V. Biochimie. 1995;77:631–642. doi: 10.1016/0300-9084(96)88178-8. [DOI] [PubMed] [Google Scholar]
- 2.Yorita K, Janko K, Aki K, Ghisla S, Palfey A B, Massey V. Proc Natl Acad Sci USA. 1997;94:9590–9595. doi: 10.1073/pnas.94.18.9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindqvist Y, Branden C I. J Biol Chem. 1989;264:3624–3628. [PubMed] [Google Scholar]
- 4.Stenberg K, Lindqvist Y. Protein Sci. 1997;6:1009–1015. doi: 10.1002/pro.5560060506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia Z X, Mathews F S. J Mol Biol. 1990;212:837–863. doi: 10.1016/0022-2836(90)90240-M. [DOI] [PubMed] [Google Scholar]
- 6.Mowat C E, Beaudoin I, Durley R C E, Barton J D, Pike A D, Chen Z, Reid G A, Chapman S K, Mathews F S, Lederer F. Biochemistry. 2000;39:3266–3275. doi: 10.1021/bi9925975. [DOI] [PubMed] [Google Scholar]
- 7.Kunkel T A, Roberts J D, Zakour R A. Methods Enzymol. 1987;164:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 8.Yorita K, Aki K, Ohkuma-Soejima T, Kokubo T, Misaki H, Massey V. J Biol Chem. 1996;271:28300–28305. doi: 10.1074/jbc.271.45.28300. [DOI] [PubMed] [Google Scholar]
- 9.Yorita K, Misaki H, Palfey B A, Massey V. Proc Natl Acad Sci USA. 2000;97:2480–2485. doi: 10.1073/pnas.040559797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edmondson D E. Biochemistry. 1974;13:2817–2821. doi: 10.1021/bi00711a006. [DOI] [PubMed] [Google Scholar]
- 11.Massey V, Hemmerich P. Biochemistry. 1978;17:9–17. doi: 10.1021/bi00594a002. [DOI] [PubMed] [Google Scholar]
- 12.Mayhew S G, Ludwig M L. In: The Enzymes. Boyer P, editor. Vol. 12. New York: Academic; 1975. pp. 57–118. [Google Scholar]
- 13.Massey V, Muller F, Feldberg R, Schuman M, Sullivan P A, Howell L G, Mayhew S G, Matthews R G, Foust G P. J Biol Chem. 1969;244:3999–4006. [PubMed] [Google Scholar]
- 14.Ghisla S, Massey V. In: Chemistry and Biochemistry of Flavoenzymes. Muller F, editor. Vol. 2. Boca Raton, FL.: CRC; 1991. pp. 243–289. [Google Scholar]
- 15.Gibson Q H, Swoboda B E P, Massey V. J Biol Chem. 1964;239:3927–3934. [PubMed] [Google Scholar]
- 16.Clark W M. Oxidation-Reduction Potentials of Organic Systems. Baltimore: Williams & Wilkins; 1960. pp. 210–217. [Google Scholar]
- 17.Clark W M. Oxidation-Reduction Potentials of Organic Systems. Baltimore: Williams & Wilkins; 1960. pp. 184–188. [Google Scholar]
- 18.Massey V. In: Flavin and Flavoproteins. Curti B, Ronchi S, Zanetti G, editors. Berlin: Walter de Gruyter; 1991. pp. 59–66. [Google Scholar]
- 19.Muh U, Williams C H, Jr, Massey V. In: Flavin and Flavoproteins. Yagi K, editor. Berlin: Walter de Gruyter; 1994. pp. 201–210. [Google Scholar]
- 20.Lehman T C, Thorpe C. Arch Biochem Biophys. 1992;292:594–599. doi: 10.1016/0003-9861(92)90036-v. [DOI] [PubMed] [Google Scholar]
- 21.Brandsch R, Bichler V. J Biol Chem. 1991;266:19056–19062. [PubMed] [Google Scholar]
- 22.Mewies M, Packman L C, Mathews F S, Scrutton N S. Biochem J. 1996;317:267–272. doi: 10.1042/bj3170267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mewies M, Basran J, Packman L C, Hille R, Scrutton N S. Biochemistry. 1997;36:7162–7168. doi: 10.1021/bi970621d. [DOI] [PubMed] [Google Scholar]
- 24.Lederer F. In: Chemistry and Biochemistry of Flavoenzymes. Muller F, editor. Boca Raton, FL.: CRC; 1991. pp. 156–242. [Google Scholar]
- 25.Muh U, Williams C H, Jr, Massey V. J Biol Chem. 1994;269:7994–8000. [PubMed] [Google Scholar]
- 26.Sanders S A, Williams C H, Jr, Massey V. J Biol Chem. 1999;274:22289–22295. doi: 10.1074/jbc.274.32.22289. [DOI] [PubMed] [Google Scholar]
- 27.Lehoux I E, Mitra B. Biochemistry. 2000;39:10055–10065. doi: 10.1021/bi000161f. [DOI] [PubMed] [Google Scholar]
- 28.Reid G A, White S, Black M T, Lederer F, Mathews F S, Chapman S K. Eur J Biochem. 1988;178:329–333. doi: 10.1111/j.1432-1033.1988.tb14454.x. [DOI] [PubMed] [Google Scholar]