

Abstract
Senile plaques associated with Alzheimer's disease contain deposits of fibrils formed by 39- to 43-residue β-amyloid peptides with possible neurotoxic effects. X-ray diffraction measurements on oriented fibril bundles have indicated an extended β-sheet structure for Alzheimer's β-amyloid fibrils and other amyloid fibrils, but the supramolecular organization of the β-sheets and other structural details are not well established because of the intrinsically noncrystalline, insoluble nature of amyloid fibrils. Here we report solid-state NMR measurements, using a multiple quantum (MQ) 13C NMR technique, that probe the β-sheet organization in fibrils formed by the full-length, 40-residue β-amyloid peptide (Aβ1–40). Although an antiparallel β-sheet organization often is assumed and is invoked in recent structural models for full-length β-amyloid fibrils, the MQNMR data indicate an in-register, parallel organization. This work provides site-specific, atomic-level structural constraints on full-length β-amyloid fibrils and applies MQNMR to a significant problem in structural biology.
A variety of peptides and proteins form amyloid fibrils, including those involved in amyloid diseases such as Alzheimer's disease, type II diabetes, and spongiform encephalopathies (1, 2), as well as proteins that are studied primarily as models for the elucidation of fundamental aspects of protein biophysics (3, 4). Compared with monomeric or oligomeric forms of peptides and proteins, relatively little is known definitively about the molecular structure and supramolecular organization of amyloid fibrils. All amyloid fibrils exhibit similar morphologies in electron micrographs (Fig. 1A), despite their diversity of amino acid sequences and sequence lengths. X-ray diffraction measurements on oriented amyloid fibril bundles show a characteristic “cross-β” pattern (2, 5–8), implying an extended β-sheet structure with polypeptide chains running roughly perpendicular to the long axis of the fibril and interchain hydrogen bonds roughly parallel to this axis. This β-sheet structure is supported by recent cryo-electron microscopy measurements (9). Finer structural details are generally not well established because amyloid fibrils are not amenable to structural characterization by single-crystal diffraction and high-resolution, liquid-state NMR techniques.
Figure 1.

(A) Electron micrographs of negative-stained Aβ1–40 fibrils adsorbed to carbon films from an Aβ1–40 solution after incubation at 24°C and pH 7.4 for 3 days. Typical amyloid fibrils are observed, appearing as single filaments or bundles of filaments with overall diameters ranging from 8 to 20 nm and with twist periodicities between 40 and 150 nm. The same solution, in which the Aβ1–40 peptides were labeled with 13C at the methyl carbon of Ala-21, subsequently was lyophilized for MQNMR measurements shown in Fig. 2. (B) Aβ1–40 fibrils are believed to have a predominantly β-sheet structure with peptide chains (blue arrows) approximately perpendicular to and hydrogen bonds approximately parallel to the long axis of the fibril (green arrow). Four candidates for the supramolecular organization of the fibrils are shown. These can be distinguished experimentally by incorporating 13C labels (red dots) at a single site in the peptide and measuring 13C multiple quantum NMR spectra, because observation of an n-quantum signal requires that at least n 13C nuclei be close enough in space to have significant magnetic dipole-dipole couplings.
Amyloid fibrils that form senile plaques in Alzheimer's disease are comprised of β-amyloid (Aβ) peptides that range in length from 39 to 43 residues (10–12). Various shorter fragments of the full-length Aβ peptides are also known to fibrillize in vitro (13–21). A number of structural models for full-length Aβ fibrils (8, 22–25) and fibrils formed by Aβ fragments (13–16, 21) have been proposed, most (16, 21–26), but not all (13–15), of which are based on an antiparallel β-sheet organization. Here we present solid-state 13C NMR data, obtained with multiple quantum (MQ) NMR techniques based on time-reversal principles (27–32), which support an extended parallel β-sheet organization for fibrils formed by the full-length, 40-residue Aβ peptide (Aβ1–40). These data have implications for the development of an understanding of the physical principles that govern fibril formation and also may contribute to the development of therapeutic agents by structure-based approaches. Our data provide structural constraints on full-length Aβ fibrils from solid-state NMR and demonstrate the utility of MQNMR in structural biology.
The conceptual basis for MQNMR measurements on Aβ1–40 fibrils is depicted in Fig. 1B. After the isotopic labeling scheme introduced by Lynn, Meredith, Botto, and coworkers (13–15), we introduce a single 13C label into the peptide chain. Given that Aβ1–40 fibrillizes in a predominantly β-sheet structure, as supported by fiber diffraction (7, 8), cryo-electron microscopy (9), and infrared spectroscopy (7, 20), the positions of the 13C labels depend strongly on the supramolecular organization within the β-sheets. In an in-register, parallel β-sheet structure, the labels would form a nearly linear chain with internuclear distances of approximately 4.8 Å. In an antiparallel β-sheet structure, the labels would form a nearly planar zigzag pattern with nearest-neighbor distances that greatly exceed 4.8 Å. If the β-sheets were organized as an antiparallel packing of parallel dimers, as might conceivably occur if certain intermolecular interactions favor a parallel dimerization whereas other interactions favor antiparallel alignment of the dimers, the labels would be grouped as pairs with a 4.8-Å internuclear distance and with larger distances between pairs. If the β-sheets were constructed from parallel trimers packed in an antiparallel fashion, the labels would occur in linear groups of three. Roughly speaking, a 13C MQNMR spectrum displays signals that arise from NMR transitions (more precisely, coherences) in which a net number n of 13C nuclear spins flip simultaneously in the magnetic field of the spectrometer, where n is the MQ order (33). For an n-quantum signal to be observable, groups of at least n spins must be linked by a network of nuclear magnetic dipole-dipole couplings, whose strengths are given by coupling constants d = γ2ħ/2πR3, where γ is the nuclear magnetogyric ratio and R is the internuclear distance. Moreover, the radio-frequency (rf) pulse sequence used to excite the MQ coherences must be applied for a time τMQ on the order of 1/d. Because d = 70 Hz for 13C nuclei with r = 4.8 Å, when τMQ ≈ 15 ms one might expect only one-quantum signals in the antiparallel β-sheet, two-quantum signals in the dimerized β-sheet, three-quantum signals in the trimerized β-sheet, and higher-order MQNMR signals in the in-register, parallel β-sheet. These qualitative expectations are made more rigorous in our data analyses, which proceed by quantitative comparisons of experimental MQNMR signal amplitudes with numerical simulations that include couplings among all 13C labels as well as contributions from natural-abundance 13C nuclei.
Methods
Peptide Synthesis and Fibrillization.
Peptides with the human Aβ1–40 amino acid sequence Asp-Ala-Glu-Phe-Arg-His-Asp-Ser-Gly-Tyr-Glu-Val-His-His-Gln-Lys-Leu-Val-Phe-Phe-Ala-Glu-Asp-Val-Gly-Ser-Asn-Lys-Gly-Ala-Ile-Ile-Gly-Leu-Met-Val-Gly-Gly-Val-Val were synthesized on an Applied Biosystems model 433A peptide synthesizer using standard fluorenylmethoxycarbonyl (FMOC) protocols with H-benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (HBTU) activation. 13C-labeled alanine was obtained from Cambridge Isotopes Laboratories, Cambridge, MA and FMOC-protected by Midwest Biotech, Fishers, IN. Peptide purity after cleavage from the synthesis resin with a solution of phenol, ethanedithiol, and thioanisole in 95% trifluoracetic acid and precipitation in t-butylmethylether was approximately 60%, as estimated from electrospray mass spectrometry. Purification to a level of 90 ± 5% was carried out with preparative reverse-phase HPLC, using a Vydac C18 column and a water/acetonitrile gradient with 0.1% trifluoroacetic acid. After lyophilization of the HPLC eluent, purified peptides were fibrillized by incubation of unbuffered solutions at 1 mM peptide concentration, 24°C, and pH 7.4 for 5–20 days. Disappearance of monomeric Aβ1–40 was monitored by periodic measurements of the UV absorption spectrum of aliquots of the incubating solutions after ultrafiltration (Millipore Centricon, 30-kDa cutoff). Typically, the absorbance peak at 274 nm due to tyrosine residues dropped to less than 10% of its initial value after 3 days of incubation. Concomitantly, the solutions became viscous. Fibrillized solutions then were lyophilized for MQNMR measurements. Approximately 10 μmol of purified, fibrillized Aβ1–40 was obtained from a 0.1-mmol scale synthesis. Portions of the final MQNMR samples were taken for Fourier transform-IR, Raman, and optical microscopy measurements. Lyophilized, purified material was used without incubation for control measurements on unfibrillized Aβ1–40.
Evidence for Full Fibrillization.
Because solid-state NMR signals include contributions from all Aβ1–40 molecules in the sample, it is important to establish that fibrillized samples do not contain significant quantities of unfibrillized material. Full fibrillization is supported by the following observations: (i) Electron micrographs of material from incubated Aβ1–40 solutions that were subsequently lyophilized for MQNMR measurements show no structures other than fibrils. (ii) Optical polarizing microscope images of material from fibrillized MQNMR Aβ1–40 samples show strong birefringence from more than 90% of the sample volume after staining with Congo red, including the green birefringence that is characteristic of amyloid. Material from unfibrillized Aβ1–40 did not stain or show birefringence. (iii) Fourier transform-IR spectra of material from the Ala-30-labeled MQNMR sample show a strong amide I band at 1,636 cm−1 with a width less than 20 cm−1, as previously reported for undeuterated Aβ fibrils (20), and Raman spectra show a strong amide I band at 1,668 cm−1, also with a width less than 20 cm−1. (iv) Conventional, one-dimensional solid-state 13C NMR spectra of Aβ1–40 samples, obtained with cross-polarization, high-power proton decoupling, and magic-angle spinning at 10 kHz, show characteristic and pronounced changes upon fibrillization. Spectral features generally become significantly sharper, including a reduction in the widths (full widths at half maximum) of the methyl carbon signal at 20 ppm, from a 6.7 ppm to a 2.3 ppm width (in the case of Aβ1–40 labeled at the methyl carbon of Ala-30), and of the natural-abundance carbonyl signal at 172 ppm, from a 5.7 ppm to a 3.7 ppm width. The natural-abundance α-carbon signal develops two distinct peaks at 58.5 ppm and 52.4 ppm upon fibrillization. We interpret these spectral changes empirically as signatures of fibrillization. Samples that do not show these signatures also do not show strong high-order MQ signals.
NMR Spectroscopy.
NMR experiments were carried out on a Varian/Chemagnetics Infinity-400 spectrometer, operating at a 13C NMR frequency of 100.4 MHz. A Varian/Chemagnetics 3.2-mm magic-angle spinning probe was used for all measurements, although the MQNMR experiments were performed without magic-angle spinning. MQNMR measurements used the time-reversible rf pulse cycle
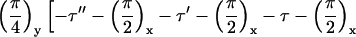 |
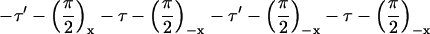 |
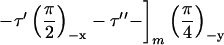 |
described by Suter et al. (30) for generation of a single-quantum effective dipole-dipole coupling Hamiltonian during MQ preparation and mixing periods, with cycle time τc = 4.8 ms, total excitation time
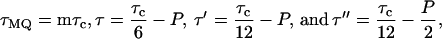 |
where P is the π/2 pulse length. To average out chemical shifts, four π pulses were inserted in each τ interval and two π pulses in each τ′ and τ′′ interval as described (31, 32). Phases of π pulses followed the sequence −x, −x, x, x. The 13C rf amplitude during the multiple pulse cycle was nominally 41.7 kHz, but the lengths of the π, π/2, and π/4 pulses were carefully adjusted to maximize the amplitude of 10-, 11-, and 12-quantum signals from polycrystalline l-methionine-methyl-13C before Aβ1–40 data were acquired. The multiple pulse cycle was incorporated into a double-resonance technique, with cross-polarization and proton decoupling, as described (31, 32). Proton decoupling fields during the MQ preparation and mixing periods were 140 kHz. 13C NMR signals were digitized with a 50-kHz spectral width. To separate signals from different MQ orders, 13C pulses during the preparation period were phase-shifted in 32 increments of 11.25°. MQNMR measurements were carried out on 10–15 mg of 13C-labeled Aβ1–40 fibrils. For τMQ = 14.4 ms, signals were acquired for approximately 100 h with a recycle delay of 1 s. Extensive block averaging was used to prevent distortions of MQ amplitudes due to drifts in rf amplitudes, probe tuning, or other factors. Data were processed as described (31, 32).
NMR Simulations.
MQNMR excitation spectra were simulated with Fortran programs written specifically for this purpose. These programs calculated the quantum mechanical evolution of the nuclear spin density operator that describes the state of the spin system under the time-averaged effective dipole-dipole coupling Hamiltonian ideally created by the rf pulse sequence during MQ preparation and mixing periods, using 512 × 512 matrix representations for a nine-spin system, and produced as output the total NMR signal amplitude arising from each order of MQ coherence present at the end of the preparation period. Averaging more than 1,024 magnetic field directions and natural-abundance 13C configurations (see below) was performed. As controls, simulations that included the experimental rf pulse sequence explicitly as a time-dependent Hamiltonian term also were performed and gave essentially identical results. Simulations of effects of rf inhomogeneity, using the full pulse sequence, indicated that inhomogeneities of ± 10% have negligible effects on the relative MQNMR amplitudes. Simulations for linear chains of five, six, and seven spins (plus four, three, and two natural-abundance spins, respectively) revealed no significant changes in the relative MQNMR amplitudes for the τMQ values used in the experiments. Simulated MQNMR amplitudes for a six-spin chain with nuclear spin polarization initially on the central two spins were not significantly different from simulated amplitudes with all spins initially polarized. These control simulations indicate the adequacy of finite-spin-system simulations for quantitative analysis of the experimental data.
Electron Microscopy.
Thin carbon films were evaporated from a carbon rod source onto freshly cleaved mica placed on white filter paper in an Edwards Auto 306 coating system, floated off in deionized water, and picked up on lacy Formvar/carbon films (EM Science) supported on 200 mesh copper grids. Grids were glow-discharged in air immediately before application of 5 μl of fibrillized Aβ1–40 solution at a concentration of 0.5 μg/μl. After allowing 2 min for adsorption, grids were washed in deionized water and negatively stained by passing through two drops of 1% uranyl acetate. Excess fluid was blotted off, and grids were dried in air. Transmission electron micrographs were recorded by using a Philips/FEI CM120 electron microscope and Gatan GIF100 imaging filter equipped with a cooled slow scan charge-coupled device camera.
Results
MQNMR spectra of 13C-labeled Aβ1–40 fibrils are shown in Fig. 2 A and B for samples labeled at the methyl carbons of alanine residues at positions 21 and 30 in the peptide chain (Ala-21 and Ala-30). The MQNMR spectra of the two fibrillized samples are strikingly similar, both exhibiting two-, three-, and four-quantum signals detectable above the noise with amplitudes that increase relative to the one-quantum signal as τMQ increases. The similarity of the MQNMR spectra of the two fibrillized Aβ1–40 samples is significant because Ala-21 is near the midpoint of the 40-residue peptide, whereas Ala-30 is shifted toward the C terminus. Of the structural models in Fig. 1B, only the parallel β-sheet leads to similar distances (i.e., similar dipole-dipole couplings) among all 13C labels for the two different labeled sites. Thus, even a rudimentary inspection of the MQNMR spectra strongly favors a parallel β-sheet organization over an antiparallel organization.
Figure 2.

13C MQNMR spectra of fibrillized and unfibrillized Aβ1–40 samples, shown in order of increasing MQ excitation time τMQ. Each MQ spectrum is displayed as a series of subspectra for MQ orders from 1 to 6, with a spectral window from −15 kHz to + 15 kHz in each subspectrum. Vertical scales are adjusted so that one-quantum peaks are clipped at 25% of their maximum values. In the fibrillized samples (A and B), the amplitudes of two-, three-, and four-quantum signals increase with increasing τMQ. Spectra of samples with 13C labels at methyl carbons of Ala-21 and Ala-30 are nearly identical. In unfibrillized samples (C), the three-quantum amplitude is small and no four-quantum signal is observed.
Control MQNMR spectra of an unfibrillized sample (Fig. 2C) exhibit much weaker MQ signal amplitudes under identical experimental conditions. In Fig. 2C, the two-, three-, and four-quantum amplitudes at τMQ = 14.4 ms (relative to the one-quantum amplitude) are 0.13, 0.01, and 0.00, respectively, in contrast to the values 0.23, 0.05, and 0.02 in Fig. 2B. The two-quantum and weak three-quantum signals in the unfibrillized sample are due primarily to couplings of 13C labels to natural-abundance 13C nuclei and couplings among natural-abundance 13C nuclei.
To analyze the MQNMR data in Fig. 2 A and B quantitatively, MQNMR spectra of nine-spin systems were simulated numerically for the values of τMQ used in the experiments. In these simulations, six of the spins were placed at positions appropriate for 13C labels in the parallel, antiparallel, dimeric, or trimeric β-sheet models in Fig. 1B, with coordinates determined from an examination of typical β-sheets in protein crystal structures. Specifically, in simulations for Ala-30-labeled Aβ1–40 fibrils, the six 13C labels were assigned coordinates (xi, yi, zi) = (32.0ki, 4.8i, 1.5ki) in units of Å, where i = 1, 2, … , 6. In simulations for Ala-21-labeled Aβ1–40 fibrils, the coordinates were (1.7ki, 4.8i, 1.5ki). The parallel, antiparallel, dimeric, and trimeric β-sheet models were represented by (k1, k2, k3, k4, k5, k6) = (0, 0, 0, 0, 0, 0) (−1, 1, −1, 1, −1, 1), (−1, 1, 1, −1, −1, 1), (−1, −1, −1, 1, 1, 1), respectively. The x, y, and z coordinates represent displacements along the direction of the peptide chains, the direction of hydrogen bonding between chains, and the direction perpendicular to the β-sheet, respectively. To account for couplings of the 13C labels to nearby natural-abundance 13C nuclei (present at all carbon sites at a 1.1% level), the remaining three spins were positioned randomly within a rectangular box around the six labels with a volume of 1.5 × 104 Å3, based on an estimated average volume per natural-abundance aliphatic 13C of 5,000 Å3. Simulated MQ signal amplitudes were averaged over the magnetic field direction relative to the labels and over the random configuration of the three natural-abundance spins. Natural-abundance 13C nuclei that are far from the 13C labels also contribute to the experimental MQNMR signals. Simulations and experiments on unlabeled Aβ1–40 samples indicate that these distant natural-abundance 13C nuclei contribute significantly only to the one- and two-quantum signals, with the ratio of one-quantum to two-quantum amplitudes being roughly 25:1, 13:1, and 9:1 for τMQ values of 4.8 ms, 9.6 ms, and 14.4 ms.
Because the experimental MQ amplitudes are not measured on an absolute scale, scaling factors Csim and Cnat were adjusted to minimize the total squared deviation between experiments and simulations
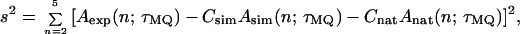 |
where Aexp(n;τMQ) is the experimental n-quantum signal amplitude for a given τMQ; Asim(n;τMQ) is the n-quantum signal amplitude from the nine-spin simulations, and Anat(n;τMQ) is the estimated contribution from distant natural-abundance 13C nuclei (nonzero only for n = 1 and n = 2). One-quantum amplitudes were not included in s2 because of uncertainty in the Anat(1;τMQ):Anat(2;τMQ) ratios that would otherwise dominate the minimization.
Fig. 3 shows comparisons of the experimental MQ signal amplitudes with simulations for each β-sheet model in Fig. 1B, i.e., the quantities Aexp(n;τMQ) and CsimAexp(n;τMQ) + CnatAnat(n;τMQ). Only the parallel β-sheet model adequately describes all of the experimental data, in particular the amplitudes of three- and four-quantum signals relative to the two-quantum signal at all values of τMQ for both labeling sites. This conclusion is insensitive to the details of the definition of s2 or the treatment of natural-abundance 13C signals. In the case of Ala-30-labeled Aβ1–40 fibrils, the trimeric β-sheet model leads to a significantly poorer fit than the parallel β-sheet model, indicating that the parallel organization of the β-sheets extends over at least four peptide chains. The parallel organization may extend to much longer length scales, but this cannot be established from our measurements without using much larger values of τMQ, which would lead to lower overall signal levels and would require excessive signal acquisition times.
Figure 3.

Comparison of experimental MQNMR amplitudes (black) with simulations for parallel (red), trimeric (green), dimeric (blue), and antiparallel organizations of β-sheets in Aβ1–40 fibrils, for samples labeled with 13C at methyl carbons of Ala-21 and Ala-30. Experimental MQNMR amplitudes are normalized to a one-quantum amplitude of 100. A logarithmic vertical scale is required because the amplitudes vary over 2 orders of magnitude. The parallel β-sheet model fits all of the experimental data most closely. Experimental amplitudes were determined from MQNMR spectra in Fig. 2 by integrating each subspectrum over the interval from −2 kHz to +3 kHz. Uncertainties in the experimental amplitudes, evaluated as the rms noise integrated over a 5-kHz-wide interval, are ±0.11, ±0.14, and ±0.14 for the Ala-21-labeled Aβ1–40 fibril data, and ±0.15, ±0.17, and ±0.24 for the Ala-30-labeled Aβ1–40 fibril data, for τMQ = 4.8 ms, 9.6 ms, and 14.4 ms, respectively.
13C MQNMR spectra of a fibrillized Aβ1–40 sample labeled at the carbonyl carbons of both Val-24 and Gly-25 also were obtained (data not shown). For this sample, the relative two-, three-, and four-quantum signal amplitudes are 0.28, 0.05, and 0.03 at τMQ = 9.6 ms. Simulated amplitudes are 0.27, 0.05, and 0.03, from simulations for six carbonyl labels on three peptide chains in a parallel β-sheet, including effects of the carbonyl chemical shift anisotropy and the full rf pulse sequence. Thus, MQNMR data on this doubly labeled Aβ1–40 sample also support a parallel β-sheet organization.
MQNMR spectra of a fibrillized Aβ1–40 sample labeled at the methyl carbon of Ala-2 (data not shown) are nearly identical to the spectra of the unfibrillized, Ala-30-labeled sample in Fig. 2C. No four-quantum signals are observed. In addition, the methyl carbon linewidth in the 13C magic-angle spinning NMR spectrum of the Ala-2-labeled sample is 11 ppm, which is significantly greater than the methyl carbon linewidths in fibrillized Ala-21- and Ala-30-labeled samples. These results indicate that, unlike Ala-21 and Ala-30, Ala-2 is not located in an in-register, parallel β-sheet and may be structurally disordered. Approximately the first 10 residues of Aβ1–40 are not required for in vitro fibrillization (20) and are proteolyzed in fibrils in vivo (34). These N-terminal residues are therefore likely to extend outside the β-sheet fibril structure, in agreement with our MQNMR data.
Discussion
Of the models for the supramolecular organization of β-sheets in Aβ1–40 fibrils in Fig. 1B, only the in-register, parallel model is consistent with the MQNMR data and simulations presented above. Although acceptable alternative models might conceivably exist, any such models must place Ala-21 and Ala-30 methyl carbons in groups of at least four with internuclear distances less than approximately 5.5 Å, i.e., d > 45 Hz. In an out-of-register, parallel β-sheet, the internuclear distances would depend on the extent of displacement of hydrogen-bonded peptide chains but would exceed 6.0 Å and therefore be inconsistent with the MQNMR data. Structural models for amyloid fibrils commonly invoke lamination of several β-sheet layers (2, 4–7, 14, 21, 25), with an average spacing between peptide backbones in adjacent laminae of approximately 9 Å in accordance with fiber diffraction results (7, 8). Coupling constants between methyl carbons in adjacent laminae then are expected to be much less than 30 Hz unless the laminae are unusually close together at the labeled site. Given the similarity of the experimental MQNMR spectra of Ala-21- and Ala-30-labeled Aβ1–40 fibrils (Fig. 2), omission of possible couplings between 13C labels in different laminae in our simulations of MQ signal amplitudes is justified. Couplings between laminae alone cannot possibly account for the observed four-quantum signal amplitudes.
Several molecular-level structural models for full-length Aβ fibrils have been proposed (22–26). Although very different in detail, all of these models invoke an antiparallel supramolecular organization of β-sheets and are not supported by our MQNMR data. In the Aβ fibril model of Tjernberg et al. (22), Ala-21 and Ala-30 are located in an intermolecular antiparallel β-sheet and an intramolecular β-hairpin, respectively, leading to distances between 13C labels that would exceed 9 Å in our experiments. In the model of Li et al. (25), the β-sheet is constructed from antiparallel intermolecular hydrogen bonding of β-hairpins with a turn between residues 25 and 28, leading to a minimum 13C-13C distance within one β-sheet of approximately 20 Å. In the model of Chaney et al. (24), Ala-30 is located in an antiparallel β-sheet, leading to a minimum 13C-13C distance of approximately 9.6 Å, and Ala-21 is contained in a dimeric globular domain outside the β-sheet core. In the two-chain, antiparallel β-helix model of Lazo and Downing (23), 13C pairs with a separation of roughly 5 Å may be possible, but longer chains of 13C labels are not present.
Evidence for antiparallel β-sheets in full-length Aβ fibrils comes principally from infrared absorption spectra, which show a strong amide I band at roughly 1,630 cm−1 and a weak band at roughly 1,690 cm−1 that has been interpreted to be characteristic of antiparallel, but not parallel, β-sheets (7, 17, 19, 20, 35). Fourier transform-IR spectra of our fibrillized MQNMR samples also show these spectral features, suggesting that proper interpretation of the infrared data may be more subtle than previously assumed. Definitive resolution of this issue requires further study, but the following observations may be relevant. First, as discussed below, we do not claim that β-sheets in all amyloid fibrils are parallel. Second, our analyses of MQNMR data are based on numerical simulations of the nuclear spin dynamics, which can be carried out accurately and without reference to model compounds or other empirical data. In contrast, the analyses of infrared data are based on experimental spectra (36) or normal mode calculations (37) for model systems that are significantly less complex than β-amyloid peptides. Third, our MQNMR data do not rule out the possibility, suggested by sequence analysis (21), structural models (22–26), liquid-state NMR measurements (38, 39), and circular dichroism spectra (20), that fibrillized Aβ peptides may adopt a conformation that includes intramolecular β-hairpins (20, 22, 23, 25) or other secondary structure elements that may account for the infrared results.
Solid-state NMR measurements by Lynn, Meredith, Botto, and coworkers (13–15) have established a parallel β-sheet structure for fibrils formed by Aβ10–35, i.e., residues 10–35 of Aβ1–40. Our experimental MQNMR spectra of Aβ10–35 fibrils labeled with 13C at the methyl carbon of Ala-21 (data not shown) are nearly identical to those of Ala-21- and Ala-30-labeled Aβ1–40 fibrils in Fig. 2, supporting the conclusions of Lynn and coworkers (13, 14) and providing additional evidence that the parallel β-sheet organization in Aβ10–35 fibrils extends beyond dimers. Lansbury, Griffin, and coworkers (16) have reported solid-state NMR measurements on fibrillized Aβ34–42 that support an antiparallel structural model. In addition, our MQNMR and rotational-echo double resonance NMR data on amyloid fibrils formed by the peptide N-acetyl-Lys-Leu-Val-Phe-Phe-Ala-Glu-NH2, i.e., Aβ16–22 with capping groups at the N and C termini, indicate an antiparallel β-sheet structure (40). Thus, it appears that amyloid fibrils can exhibit a variety of supramolecular organizations, depending on the specific details of the amino acid composition and sequence.
A common feature of the Aβ1–40 and Aβ10–35 sequences is the existence of hydrophobic segments (residues 17–21 and 29–40) that are not symmetrically disposed about the midpoint of the peptide chain. As depicted in Fig. 4, an in-register, parallel β-sheet organization juxtaposes the hydrophobic segments of neighboring peptide chains, whereas an antiparallel organization does not. An in-register, parallel β-sheet organization also juxtaposes charged residues, which might create electrostatic repulsions that destabilize the fibril, but unfavorable electrostatic interactions could be overcome by incorporation of counterions into the fibrils or possibly by interactions between β-sheet laminae. In contrast, Aβ16–22 and Aβ34–42 contain central hydrophobic sequences with a positive charge at the N terminus and a negative charge at the C terminus (i.e., the positively charged Lys and negatively charged Glu side chains in capped Aβ16–22, and the positive amino and negative carboxylate groups at the ends of uncapped Aβ34–42). These two Aβ fragments therefore resemble simple electric dipoles when in extended conformations. An antiparallel β-sheet organization then may simultaneously juxtapose hydrophobic segments and minimize the electrostatic energy. Simple considerations such as these based on amino acid composition and sequence may help explain the supramolecular organizations adopted by different Aβ peptides, although the full explanations are undoubtedly more subtle. Additional atomic-level structural constraints from solid-state NMR will further elucidate the details of amyloid fibril structures and the physical basis for these structures.
Figure 4.

The amino acid sequence of Aβ1–40 contains hydrophobic (black) and nonhydrophobic (red) residues. Ala-21 and Ala-30, sites that are shown by the MQNMR data to participate in parallel β-sheets, are colored green and blue, respectively. An in-register, parallel alignment of neighboring peptide chains in a β-sheet (Upper) juxtaposes the hydrophobic segments of neighboring chains, whereas an antiparallel alignment (Lower) does not. Hydrophobic interactions may dictate the β-sheet organization in Aβ1–40 fibrils.
The MQNMR methods used in this work represent a general approach to investigations of supramolecular organization in amyloid fibrils. Although 13C labeling of alanine methyl sites is advantageous because of the relatively small chemical shift anisotropy (CSA) of these sites, the same MQNMR methods are applicable to other sites with larger CSA (32). It is worth noting that the β-sheet helix model for transthyretin fibrils developed by Blake and Serpell (5) is depicted with parallel β-sheets. However, as discussed by Blake and Serpell (5), the supramolecular organization of β-sheets in this model cannot be established unambiguously from their synchrotron x-ray diffraction data.
Although Aβ1–40 accounts for the majority of Aβ production in vivo, Aβ1–42, with an additional two hydrophobic residues at the C terminus, is the predominant species in senile plaques and is believed to nucleate plaque formation (11, 41, 42). In light of the small difference in the two peptide sequences and experimental evidence that Aβ1–40 and Aβ1–42 can cofibrillize (41–43), it seems likely that Aβ1–42 and Aβ1–40 fibrils have the same supramolecular organization.
Acknowledgments
We are grateful to Drs. P. McCarthy, H. Wang, and I. W. Levin for carrying out Fourier transform-IR and Raman measurements on Aβ fibril samples and to Dr. L. K. Pannell for mass spectrometry. Simulations were executed on the SGI Origin 2000 computer in the National Institutes of Health Center for Information Technology. O.N.A. was supported by a fellowship from the Swedish Foundation for International Cooperation in Research and Higher Education. J.R. was supported by the Whitaker Foundation through the National Institutes of Health Biomedical Engineering Summer Internship Program.
Abbreviations
- MQ
multiple quantum
- Aβ
β-amyloid
- rf
radio frequency
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.230315097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.230315097
References
- 1.Sipe J D. Annu Rev Biochem. 1992;61:947–975. doi: 10.1146/annurev.bi.61.070192.004503. [DOI] [PubMed] [Google Scholar]
- 2.Sunde M, Blake C C F. Q Rev Biophys. 1998;31:1–39. doi: 10.1017/s0033583598003400. [DOI] [PubMed] [Google Scholar]
- 3.Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson C M. Proc Natl Acad Sci USA. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jimenez J L, Guijarro J L, Orlova E, Zurdo J, Dobson C M, Sunde M, Saibil H R. EMBO J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blake C, Serpell L. Structure (London) 1996;4:989–998. doi: 10.1016/s0969-2126(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 6.Sunde M, Serpell L C, Bartlam M, Fraser P E, Pepys M B, Blake C C F. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 7.Fraser P E, Nguyen J T, Inouye H, Surewicz W K, Selkoe D J, Podlisny M B, Kirschner D A. Biochemistry. 1992;31:10716–10723. doi: 10.1021/bi00159a011. [DOI] [PubMed] [Google Scholar]
- 8.Malinchik S B, Inouye H, Szumowski K E, Kirschner D A. Biophys J. 1998;74:537–545. doi: 10.1016/S0006-3495(98)77812-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serpell L C, Smith J M. J Mol Biol. 2000;299:225–231. doi: 10.1006/jmbi.2000.3650. [DOI] [PubMed] [Google Scholar]
- 10.Glenner G G, Wong C W. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 11.Storey E, Cappai R. Neuropathol Appl Neurobiol. 1999;25:81–97. doi: 10.1046/j.1365-2990.1999.00164.x. [DOI] [PubMed] [Google Scholar]
- 12.Yankner B A. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 13.Gregory D M, Benzinger T L S, Burkoth T S, Miller-Auer H, Lynn D G, Meredith S C, Botto R E. Solid State Nucleic Magn Reson. 1998;13:149–166. doi: 10.1016/s0926-2040(98)00086-1. [DOI] [PubMed] [Google Scholar]
- 14.Benzinger T L S, Gregory D M, Burkoth T S, Miller-Auer H, Lynn D G, Botto R E, Meredith S C. Proc Natl Acad Sci USA. 1998;95:13407–13412. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benzinger T L S, Gregory D M, Burkoth T S, Miller-Auer H, Lynn D G, Botto R E, Meredith S C. Biochemistry. 2000;39:3491–3499. doi: 10.1021/bi991527v. [DOI] [PubMed] [Google Scholar]
- 16.Lansbury P T, Costa P R, Griffiths J M, Simon E J, Auger M, Halverson K J, Kocisko D A, Hendsch Z S, Ashburn T T, Spencer R G S, et al. Nat Struct Biol. 1995;2:990–998. doi: 10.1038/nsb1195-990. [DOI] [PubMed] [Google Scholar]
- 17.Halverson K, Fraser P E, Kirschner D A, Lansbury P T. Biochemistry. 1990;29:2639–2644. doi: 10.1021/bi00463a003. [DOI] [PubMed] [Google Scholar]
- 18.Huang T H J, Fraser P E, Chakrabartty A. J Mol Biol. 1997;269:214–224. doi: 10.1006/jmbi.1997.1050. [DOI] [PubMed] [Google Scholar]
- 19.Fraser P E, McLachlan D R, Surewicz W K, Mizzen C A, Snow A D, Nguyen J T, Kirschner D A. J Mol Biol. 1994;244:64–73. doi: 10.1006/jmbi.1994.1704. [DOI] [PubMed] [Google Scholar]
- 20.Hilbich C, Kisterswoike B, Reed J, Masters C L, Beyreuther K. J Mol Biol. 1991;218:149–163. doi: 10.1016/0022-2836(91)90881-6. [DOI] [PubMed] [Google Scholar]
- 21.Kirschner D A, Inouye H, Duffy L K, Sinclair A, Lind M, Selkoe D J. Proc Natl Acad Sci USA. 1987;84:6953–6957. doi: 10.1073/pnas.84.19.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tjernberg L O, Callaway D J E, Tjernberg A, Hahne S, Lilliehook C, Terenius L, Thyberg J, Nordstedt C. J Biol Chem. 1999;274:12619–12625. doi: 10.1074/jbc.274.18.12619. [DOI] [PubMed] [Google Scholar]
- 23.Lazo N D, Downing D T. Biochemistry. 1998;37:1731–1735. doi: 10.1021/bi971016d. [DOI] [PubMed] [Google Scholar]
- 24.Chaney M O, Webster S D, Kuo Y M, Roher A E. Protein Eng. 1998;11:761–767. doi: 10.1093/protein/11.9.761. [DOI] [PubMed] [Google Scholar]
- 25.Li L P, Darden T A, Bartolotti L, Kominos D, Pedersen L G. Biophys J. 1999;76:2871–2878. doi: 10.1016/S0006-3495(99)77442-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.George A R, Howlett D R. Biopolymers. 1999;50:733–741. doi: 10.1002/(SICI)1097-0282(199912)50:7<733::AID-BIP6>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 27.Yen Y-S, Pines A. J Chem Phys. 1983;78:3579–3582. [Google Scholar]
- 28.Warren W S, Weitekamp D P, Pines A. J Chem Phys. 1980;73:2084. [Google Scholar]
- 29.Baum J, Munowitz M, Garroway A N, Pines A. J Chem Phys. 1985;83:2015–2025. [Google Scholar]
- 30.Suter D, Liu S B, Baum J, Pines A. Chem Phys. 1987;114:103–109. [Google Scholar]
- 31.Tycko R. J Magn Reson. 1999;139:302–307. doi: 10.1006/jmre.1999.1776. [DOI] [PubMed] [Google Scholar]
- 32.Antzutkin O N, Tycko R. J Chem Phys. 1999;110:2749–2752. [Google Scholar]
- 33.Weitekamp D P. In: Advances in Magnetic Resonance. Waugh J S, editor. Vol. 11. New York: Academic; 1983. pp. 111–274. [Google Scholar]
- 34.Roher A E, Lowenson J D, Clarke S, Wolkow C, Wang R, Cotter R J, Reardon I M, Zurcherneely H A, Heinrikson R L, Ball M J, Greenberg B D. J Biol Chem. 1993;268:3072–3083. [PubMed] [Google Scholar]
- 35.Lansbury P T. Biochemistry. 1992;31:6865–6870. doi: 10.1021/bi00145a001. [DOI] [PubMed] [Google Scholar]
- 36.Yamada N, Ariga K, Naito M, Matsubara K, Koyama E. J Am Chem Soc. 1998;120:12192–12199. [Google Scholar]
- 37.Bandekar J, Krimm S. Biopolymers. 1988;27:909–921. [Google Scholar]
- 38.Shao H Y, Jao S C, Ma K, Zagorski M G. J Mol Biol. 1999;285:755–773. doi: 10.1006/jmbi.1998.2348. [DOI] [PubMed] [Google Scholar]
- 39.Sticht H, Bayer P, Willbold D, Dames S, Hilbich C, Beyreuther K, Frank R W, Rosch P. Eur J Biochem. 1995;233:293–298. doi: 10.1111/j.1432-1033.1995.293_1.x. [DOI] [PubMed] [Google Scholar]
- 40.Balbach J J, Ishii Y, Antzutkin O N, Leapman R D, Rizzo N W, Dyda F, Reed J, Tycko R. Biochemistry. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]
- 41.Jarrett J T, Berger E P, Lansbury P T. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 42.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 43.Hasegawa K, Yamaguchi I, Omata S, Gejyo F, Naiki H. Biochemistry. 1999;38:15514–15521. doi: 10.1021/bi991161m. [DOI] [PubMed] [Google Scholar]