

Abstract
 The use of capsules and cavitands in physical organic chemistry is briefly reviewed, and their application to the study of salt bridges is introduced. Carboxylate/ammonium ion pairs are generated within an environment that more or less surrounds the functional groups within a synthetic fixed introverted solvent sphere. This is provided by cavitands that fold around amines and present them with a carboxylic acid function. Both organic and water-soluble versions were prepared, and their equilibrium affinities with quinuclidine bases were determined by NMR methods. The association constants range from approximately 103 M−1 in water to more than 105 M−1 in organic solvents. Studies of nitrogen inversion and tumbling of [2.2.2]-diazabicyclooctane within the introverted acids also illustrate the strength of the acid–base interactions. The barriers to in–out exchange of several amine guests were determined to be in the range from 15 to 24 kcal mol−1. Some parallels with enzymes are drawn: the receptor folds around the guest species; presents them with inwardly directed functionality; and provides a generally hydrophobic environment and a periphery of secondary amide bonds.
The use of capsules and cavitands in physical organic chemistry is briefly reviewed, and their application to the study of salt bridges is introduced. Carboxylate/ammonium ion pairs are generated within an environment that more or less surrounds the functional groups within a synthetic fixed introverted solvent sphere. This is provided by cavitands that fold around amines and present them with a carboxylic acid function. Both organic and water-soluble versions were prepared, and their equilibrium affinities with quinuclidine bases were determined by NMR methods. The association constants range from approximately 103 M−1 in water to more than 105 M−1 in organic solvents. Studies of nitrogen inversion and tumbling of [2.2.2]-diazabicyclooctane within the introverted acids also illustrate the strength of the acid–base interactions. The barriers to in–out exchange of several amine guests were determined to be in the range from 15 to 24 kcal mol−1. Some parallels with enzymes are drawn: the receptor folds around the guest species; presents them with inwardly directed functionality; and provides a generally hydrophobic environment and a periphery of secondary amide bonds.
Introduction
Molecular encapsulation offers a tool for probing functional group interactions in isolation. By temporarily confining a guest to a fixed space isolated from solution, a host molecule restricts the motions and conformations of its occupant and provides surroundings of ordered functionality. The host imposes an environment on the guest that is both physically and chemically distinct from the exterior solution. Many examples of encapsulation come from synthetic receptor design, and a growing literature describes the unique ability of hosts to control the properties of their guests.1 Shifted equilibria of guest conformations2 have been observed, and in some cases, guests have adopted previously unknown conformations, such as the coiled helical conformation of alkanes in shape-mismatched hosts.3-5 New forms of isomerism6 have arisen from the coencapsulation of multiple guests in the same host,7 a consequence of the guests’ positional arrangement or orientation or both.8 Host molecules that more or less surround their guests can stabilize reactive species9-12 by compartmentalization and geometric constraint,13-19 but they can also increase the reactivity of guests20-27 or change the rates28,29 and selectivities of reactions that occur.30-32
Because the orientation, conformation, and motion of guests are subject to the constraints of the host, functional groups can be positioned in close proximity to the guest without the rapid exchange of partners that occurs in bulk solution. One approach involves the coencapsulation of multiple guests: hydrogen-bond-mediated functional group interactions have been studied in the context of enantioselection through coencapsulation.33 The other approach, used in this work, is to affix the host molecule with inwardly directed functionality. Guest molecules are then selectively bound via complementary functional group recognition, and the interactions between functional groups of host and guest may be examined.
Here, we study the ability of host cavitands to isolate a carboxylic acid and an amine in a semirigid amphiphilic pocket of some 170 Å3. Deep cavitands are open-ended vase-shaped hosts made up of an ordered arrangement of eight benzene groups and crowned with a hydrogen-bonded rim of up to eight secondary amides (Figure 1).34-36 They are appropriately thought of as persistent solvent cages for which the entropic cost of organization has been paid during a covalent synthesis, and they reproduce some of the properties of the ordered hydrophobic interiors of proteins. Synthetic modifications of deep cavitands have placed functional groups on the upper rim; the ability of the resulting hosts to selectively bind guests and control their properties has been recently reviewed.37 Of interest here are those cavitands, derived from Kemp's triacid, that direct a carboxylic acid rigidly toward the depths of their cavities.38 Because of the architecture of these cavitands H2, any guest that is bound is forced to confront the carboxylic acid. The acid provides a complement to Lewis basic guests, but the structured environment surrounding the resulting acid–base pair has consequences in terms of energetics and dynamics. The cavitands H2a and H2b and their water-soluble counterpart H2c are examined as tools for studying acid–base interactions in isolation.
FIGURE 1.
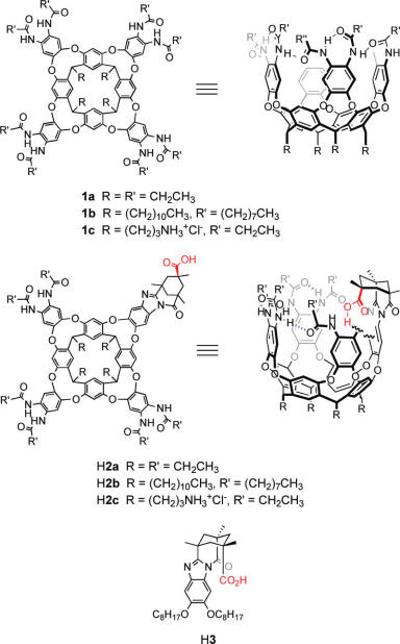
Deep cavitands 1, introverted acid cavitands H2, and control molecule H3.
Results and Discussion
Cavitand Synthesis and Structure
The cavitands used in this study are synthetic extensions of the original “deep cavitands”, which were prepared by multiple SNAr reactions between a resorcin[4]arene and 1,2-difluoro-4,5-dinitrobenzene.34 The best synthesis (in our hands) of hydrophobic introverted acid cavitands H2a,b was recently reported,39 as has that for the water-soluble derivative H2c.40 While the introverted acid cavitands synthesized and used in this study are racemic, it is possible to resolve the enantiomers by HPLC using a chiral column ((3S,4R) Pirkle 1J Regiscolumn, 4.6 × 250 mm, EtOH: CH2Cl2 (3:97), 1.0 mL/min). The water-soluble introverted acid host H2c was synthesized in a similar manner beginning from a Boc-protected amine-footed resorcin[4]arene.40,41 Control acid H3 was synthesized by the condensation of Kemp's triacid chloride anhydride with 1,2-dioctyloxy-4,5-diaminobenzene.42
The slow evaporation of a solution of H2a in chloroform resulted in the growth of crystals suitable for X-ray diffraction (Figure 2). In the crystalline state, the cavitand H2a adopts a distorted vase conformation. The resorcinarene framework adopts a nearly perfect crown conformation: the dihedral angles between the opposing resorcinol ring differ only by 5.3°. Three cavitand walls are nearly perpendicular to the main plane of the macrocycle (dihedral angles 89.9, 92.8, and 84.0°), whereas the fourth wall is tilted toward the cavity (the dihedral angle 72.4°). Four intramolecular hydrogen bonds are formed between the adjacent amide units; the other two involve the introverted carboxylic acid group. The C–O distances and the pattern of hydrogen bonds clearly indicate that the acid hydrogen atom is bound to oxygen O9. Neighboring molecules of H2a are associated through C=O–H–N hydrogen bonds (d(O13–N6) = 3.1 Å). In this way, 1D hydrogen-bonded molecular chains are formed in the crystal. While idealized model structures derived from molecular mechanics energy minimization (MM+ or AMBER) suggest an internal volume of 170 Å3, the tilting of a wall of H2a reduces the cavity volume; this is an example of induced-fit binding.43,44 The cavity volume in the X-ray structure, 145 Å3, provides a snug fit for chloroform, with a packing coefficient of 0.51.45,46 This value is close to the “ideal” of 0.55, typical for a wide array of hosts.47 The distances between opposing walls are 5 and 6.5 Å, and the acid group is 5 Å from the bottom of the accessible cavity space. Such available space and host flexibility is suitable for the binding of one small molecule up to a maximum volume of around 120 Å3. Chloroform bound inside H2a is oriented with its relatively polar C–H directed toward the aromatic ring of a cavitand wall. The CH–π interactions, as well as the stronger cation–π interactions, are major enthalpic contributors to the guest binding energy of cavitands of the types 1 and 2,48 and this is consistent with the orientation observed here. One chlorine atom is situated in the middle of the resorcinarene socket, forming close contacts with two resorcinol rings (3.4, 3.5 Å). Strong disorder of solvent molecules and high mosaicity of the crystal are responsible for high final R values.
FIGURE 2.
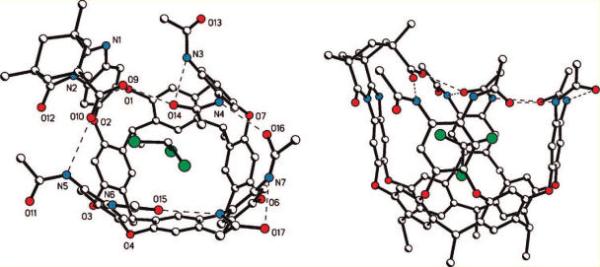
Side and top views of the X-ray crystal structure of introverted acid H2a with bound chloroform. Ethyl groups have been shown as methyl groups for clarity. Hydrogen bonds are shown as dashed lines.
The crystal structure reveals the strongly interactive nature of the carboxylic acid. Because chloroform offers a poor ability to make favorable intermolecular interactions with the acid, the cavitand's amide groups adapt to fill this role. The X-ray structure of C4-symmetric deep cavitand 1b shows the eight amide groups arranged in a ring of eight hydrogen bonds with a continuous clockwise or anticlockwise orientation.36 In contrast, two of the corresponding interamide hydrogen bonds of H2a have been broken so that an NH is dedicated entirely to a hydrogen bond with the acid carbonyl. The acid OH participates in a hydrogen bond to an amide carbonyl on the wall opposite the NH donor; this carbonyl maintains a second hydrogen bond to another amide group of the upper rim. When a basic guest is present, the acid OH is otherwise occupied (vide infra), but it is worthwhile to mention that the donation of a hydrogen bond to the carboxylic acid is expected to increase the acidity.
While the X-ray structure described here is that of a cavitand with ethyl groups as “feet”, the results presented in this study as well as our experience with related cavitands clearly indicates that structural variation of these feet does not perturb the nature of the cavitand's binding pocket. Instead, the feet of the cavitand offer a site for modifications that can be used to control the solubility, attach fluorescent labels, or anchor the cavitand to a solid support.41,49,50
Amine Binding: Structure and Thermodynamics
Simple deep cavitands 1 bind small molecules of appropriate size and shape, including lactams and 1-substituted adamantanes, with binding energies that are typically on the order of ΔG° = 2−4 kcal mol−1 in p-xylene-d10.34 Small amines such as triethylamine and quinuclidine are not particularly favored occupants of the cavity and are not bound appreciably in chloroform, which competes to occupy the inner space. When the cavitand is fitted with an introverted acid, as in H2, these amines are bound so tightly that the acid–base complex is stable to silica gel chromatography: the binding constants are Ka > 105 M−1. The structures of several complexes of H2 and an amine guest have been studied previously using NMR.38,42
Numerous attempts to grow diffraction-quality crystals of complexes of H2a with DABCO and quinuclidine were unsuccessful. In order to reveal the geometrical features of inclusion complexes of these amines, model complexes with the more symmetrical octaamidocavitand 1a were studied by single-crystal X-ray analysis.
Slow evaporation of a solution of 1a and DABCO in mesitylene-d12 gave colorless crystals that were suitable for single-crystal X-ray analysis (Figure 3a), although they were unstable in the absence of the mother liquor. In the crystalline state, cavitand 1a adopts a nearly ideal vase conformation: the dihedral angles between opposite pairs of resorcinol rings are 72.8 and 77.6°. The eight secondary amide groups form a cyclic array of hydrogen bonds, stabilizing this conformation. The clockwise or counterclockwise arrangement of amide groups makes the structure chiral. The unit cell contains both enantiomers, related by the center of symmetry (space group P1). The internal cavity of 1a is occupied by one DABCO molecule, which takes a well-defined position. The dihedral angle between the planes of methylene groups adjacent to N11 and the rms plane of the four methine bridges of resorcinarene was found to be 33.7°. Of note is the absence of clear indications of strong, noncovalent interactions between host and guest. Only one short contact between a methylene carbon atom DABCO and a carbon atom of an aromatic cavitand wall (3.59 Å) indicates some weak noncovalent attraction. These observations are consistent with the observed weak binding of DABCO in solvents that compete to occupy the cavitand.
FIGURE 3.
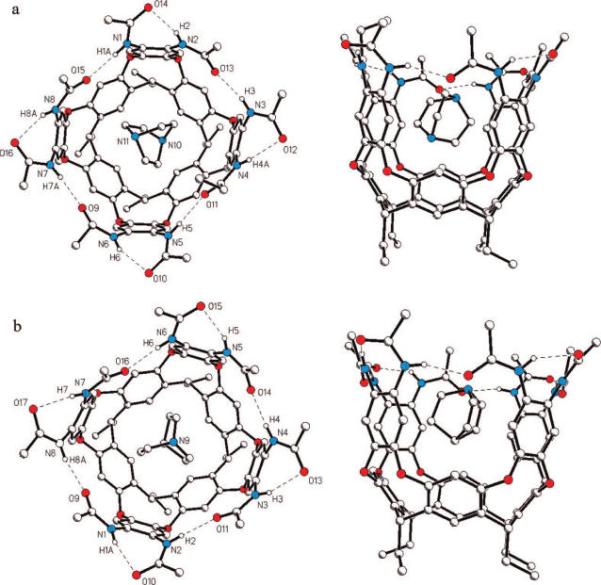
Top and side views of the X-ray crystal structures of octaamide cavitand 1a with bound DABCO (a) and with bound quinuclidine (b). Hydrogen bonds are shown as dashed lines, and methyl groups have been truncated from propionamides.
A similar slow evaporation of a solution of 1a and quinuclidine in mesitylene-d12 also afforded diffraction-quality crystals. Single-crystal X-ray analysis revealed that the structure of cavitand molecule 1a is nearly identical in both this complex and the complex of 1a and DABCO (Figure 3b). Furthermore, the elemental cell parameters and crystal packing are nearly identical. The main differences between these structures lie in the orientation of guest molecules in the cavity of 1a. One ordered quinuclidine molecule is situated in the cavity of 1a with its methine C–H bond pointing toward the resorcinarene socket. The dihedral angle between the planes of the methylene groups adjacent to N9 and the plane of the four resorcinarene methine bridges (9.5°) is about three times smaller than the analogous value for DABCO. Quinuclidine is naturally less tilted in the cavitand's binding site. The subtle differences in size and electrostatic potential of the guests have a well-pronounced effect on their orientations when bound.
1H,1H TOCSY NMR experiments on the complex of DABCO with H2a revealed 3JH,H coupling between quinuclidine methylenes and the proton shared by acid and base (Figures 4 and 5). This coupling indicates a bonding interaction between nitrogen and hydrogen, requiring a partial transfer of the proton to the base.51,52 Carboxylic acids and amines hydrogen bond in the gas phase, but the transfer of a proton is typical only in the liquid or solid states, in which the energy of solvation or the lattice energy stabilizes the charges.53 The observation of 3JH,H coupling shows that the cavitand has some ability to stabilize an ion pair, which is consistent with the observed high affinity of deep cavitands for protonated tertiary amines and quaternary ammonium salts. This direct observation of a stabilized, hydrogen-bonded salt bridge in a cavitand interior is reminiscent of the stabilization of salt bridges in the hydrophobic interior of some proteins.
FIGURE 4.
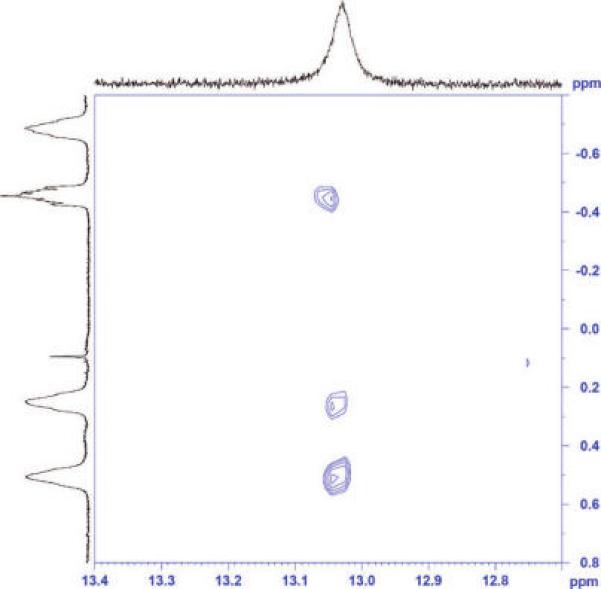
1H,1H TOCSY spectrum (CDCl3, 298 K) showing 3JH,H coupling between the DABCO methylene protons and the proton shared by the base and the acid of host H2a. This coupling reveals that the acid proton has been at least partially transferred to a nitrogen of DABCO.
FIGURE 5.
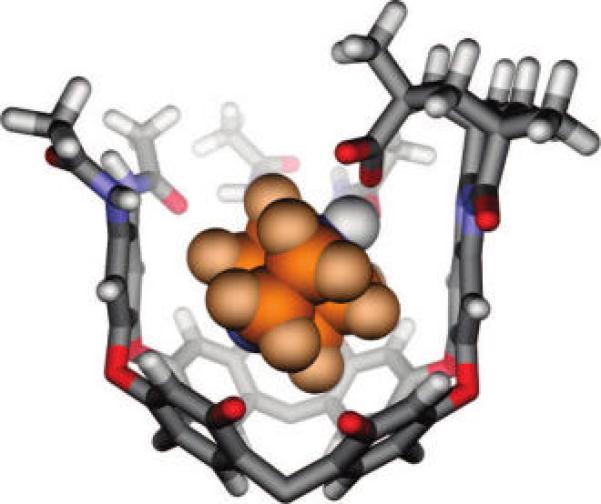
Modeled structure (HyperChem; MM+ force field) of DABCO bound by H2a. Ethyl groups have been truncated or shown as methyl groups, and one cavitand wall has been removed for clarity. The acid proton (shown in white, CPK style) has been transferred to DABCO, consistent with TOCSY measurements.
Competition experiments provide access to the interaction energy of the host acid and the guest amine, in a context suitable for the high association constants of amines and H2a in organic solvents (Figure 5). The unfunctionalized cavitand 1a binds N-methylquinuclidinium tetrafluoroborate more strongly than it binds quinuclidine HCl (equilibrium A, ΔG°A = −1.6 ± 0.2 kcal mol−1). This observation is consistent with reported findings that cation–π interactions of quaternary ammoniums are typically stronger than those of the corresponding protonated amines.54-56 In contrast, the introverted acid cavitand salt Na2a binds quinuclidine HCl more strongly than N-methylquinuclidinium tetrafluoroborate (equilibrium B, ΔG°B = −3.1 ± 0.1 kcal mol−1). Moreover, when unfunctionalized cavitand 1a and introverted acid salt Na2a compete for quinuclidinium hydrochloride, Na2a binds it more tightly by ΔG°D = −6.8 ± 0.5 kcal mol−1 (equilibrium D). The equilibrium lies too far to the side of quinuclidinium 2a complex to measure directly by NMR and had to be calculated (Figure 6; eq 3). This value shows the increase in binding energy of quinuclidinium when a carboxylate is affixed to the host, and we interpret this energy as an approximate measure of the strength of the acid–base interaction.
FIGURE 6.
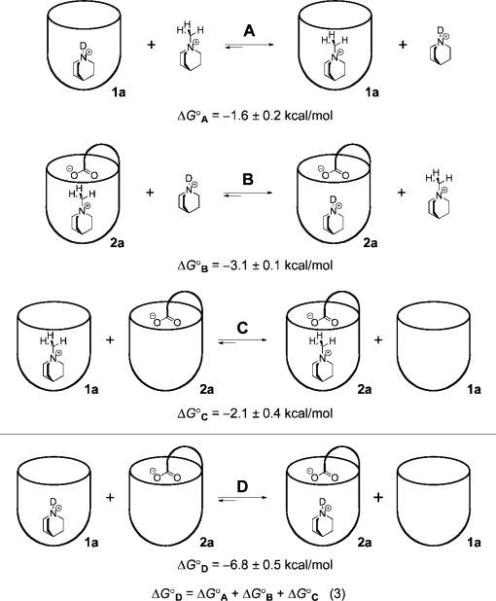
Competitive binding experiments elucidate the guest binding preference of each host (A and B) and the preferred host selection of each guest (C and D). Equilibrium D provides an estimate of the interaction energy of the quinuclidinium–carboxylate pair, ΔG°D = 6.8 ± 0.5 kcal mol−1. ΔG° values for A, B, and C were determined by 1H NMR titration in 5% CD3OD in CDCl3, and ΔG°D was calculated according to the eq 3. Free sodium cations, tetrafluoroborate anions, and chloride anions are not shown.
A similar competitive binding experiment, offering both 1a and Na2a to N-methylquinuclidinium tetrafluoroborate, revealed that host Na2a was preferred by ΔG°C = −2.1 ± 0.4 kcal mol−1 (equilibrium C). The presence of the introverted acid functionality resulted in a significant increase in binding energy, but compared to the protonated quinuclidinium (equilibrium D), the difference was much smaller: ΔΔG° = 4.7 ± 0.9 kcal mol−1. We cannot rule out that the larger size of the N-methyl guest causes repulsive interactions, but these cavitands have been shown to have a substantial ability to distend their walls and thereby accommodate larger occupants.43 The other side of this flexibility is seen in the crystal structure of chloroform bound in H2a (Figure 1), where the walls of the host tilt inward in response to the relatively small size of the guest. It is more likely that the presence of the proton contributes to the binding energy through a hydrogen-bond-mediated salt bridge, a suggestion consistent with the observed 3JH,H 1H NMR coupling.
It is of interest to compare the behavior of introverted acid H2a in organic solution with that of H2c in aqueous solution. The conformational behavior of H2c in D2O is somewhat different from that of the organic-soluble versions. In the absence of an appropriate guest, Na2c adopts an open kite conformation with the aromatic walls of the host splayed outward.57 Broadened, ill-defined proton resonances suggested any number of conformations, and even molecular aggregates in the free host that exchange at intermediate rates on the NMR time scale. Evidently, water is not a good enough guest, and the upper rim hydrogen bonds are not strong enough to hold the vase structure together in the competing aqueous solvent. The addition of a cationic guest, such as quinuclidinium hydrochloride, shifted the host equilibrium from the kite to the vase structure, through induced-fit recognition.40 Similar guest-templated folding was observed previously for host 1c in D2O.41
Receptor Na2c bound quinuclidinium hydrochloride with an affinity of 1300 M−1 in buffered D2O, an appropriate range to measure by NMR titration. In contrast, the nonfunctionalized receptor 1c bound the same guest with a feeble association constant of 12 M−1. A strongly favorable acid–base interaction exists in the complex formed with Na2c. By taking the difference between the affinities of the introverted acid receptor Na2c and the octaamide receptor 1c, an estimate of the buried salt bridge interaction is obtained: −2.7 ± 0.1 kcal mol−1 (cf. equilibrium D in Figure 6). While this method reasonably corrects for the interaction of the quinuclidinium cation with the host walls, as well as the desolvation of quinuclidinium, it does not account for the desolvation of the introverted carboxylic acid of the receptor, which occurs during the folding process. To address this problem, a “double mutant” cycle analysis58-62 was performed (Figure 7). In complex A, the interaction between the introverted acid and the protonated amine guest is present. Complexes B and C represent the single mutants, whereby the interacting protonated amine has been methylated and the introverted acid has been removed, respectively. In order to prevent a possible charge–charge interaction in single mutant B, the measurement was carried out at pD 1.4 to protonate the carboxylic acid. Complex D represents the double mutant in which both interacting sites have been removed. The electrostatic interaction in the complex formed between 2c− and the quinuclidinium cation was accordingly determined to be −3.0 ± 0.4 kcal mol−1 using eq 4 (Figure 7).58
FIGURE 7.
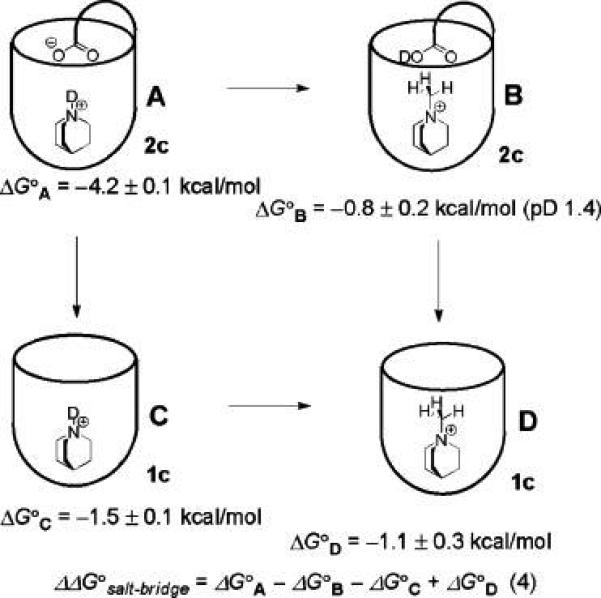
Free energy cycle in deuterated water. ΔG° values refer to the binding energy of each host–guest complex as shown schematically. Measurements were performed at 300 K in 10 mM sodium phosphate buffer, pD 5.25, except where indicated.
Amine Guest Exchange Kinetics and Host–Guest Complex Dynamics
In most examples, the nature of guest exchange of supramolecular complexes is determined predominately by the structural features of the host. Several mechanisms have been observed and are described in a recent review.63 For deep cavitands of the types 1 and H2, the process of guest exchange is intrinsically tied to the processes of cavitand folding and unfolding. The “folded” cavitand is that which has its walls oriented as in the crystal structure (Figures 1 and 3). The unfolding process is an inversion of some of the nine-membered diether rings with concomitant breaking of interwall (i.e., interamide) hydrogen bonds; the result is a flattened conformation termed “kite”.57 While the two conformations, vase and kite, are well-known, recent studies by Diederich reveal that a hybrid structure is also possible.64 In the early studies of deep cavitands 1 in organic solvents, it was found that the guest exchange energy, ΔG‡ = 17 kcal mol−1, had little dependence of the nature of the guest and was equal to the activation energy for cavitand unfolding.34 In contrast, the rate of guest self-exchange in water-soluble cavitands does show a dependence on guest size.4 The coupling of unfolding–folding to guest exchange is no surprise when one considers the alternative: a supramolecular SN1 mechanism in which the guest is withdrawn through the cavitand's opening and a new guest (or a solvent molecule) enters by the reverse of this mechanism. Such a process necessarily entails desolvation of the entire inner surface of the cavitand and the creation of a transient vacuum of some 170 Å3; this mechanism is expected only at high temperature and in systems that have no alternative.65
We performed EXSY NMR experiments on the diazabicyclo[2.2.2]octane (DABCO) complex of cavitand H2a and the N-ethyl-N-methyl-N-isopropylamine complex of H2b. In mesitylene-d12 solution, DABCO exchange in 1 operates by the unfolding–folding mechanism with nearly the usual activation energy, ΔG‡ = 14.6 ± 0.4 kcal mol−1 at 300 K, as measured by EXSY NMR. This barrier is lower than the typical 17 kcal mol−1 because the high local concentration of the DABCO amine at the cavitand opening (or excess DABCO in solution) can facilitate the breaking of the interamide hydrogen bonds to “catalyze” unfolding. When bound by the introverted acid H2, exchange of DABCO could not be observed by EXSY NMR experiments because this process is too slow. An alternative method, in which we preformed a 1:1 complex of DABCO and H2a in mesitylene-d12 and then introduced 1 equiv of quinuclidine (which is a better guest), showed that the quinuclidine displaced the DABCO with t1/2 = 27 min at 295 K, corresponding to ΔG‡ = 22.0 ± 0.3 kcal mol−1.66 If the 5 K temperature difference is neglected, the presence of the introverted acid raised the barrier to exchange by 7.4 ± 0.5 kcal mol−1, a value within experimental error of the 6.8 ± 0.5 kcal mol−1 change in thermodynamic binding energy described above (Figure 6, equilibrium D).
In the case of the N-ethyl-N-methyl-N-isopropylamine complex in CDCl3, integration of the NOE cross-peaks provided the activation energy ΔG‡ = 17 ± 0.5 kcal mol−1 for guest exchange, identical to that found for unfunctionalized cavitands 1. This seemingly counterintuitive observation is explained if cavitand unfolding and the interruption (or exchange) of the acid–base interaction are not coupled in CDCl3; that is, their energies are not additive. The cavitand unfolds like the unfunctionalized cavitand in a rate-limiting step, after which the acid is free to exchange partners rapidly.
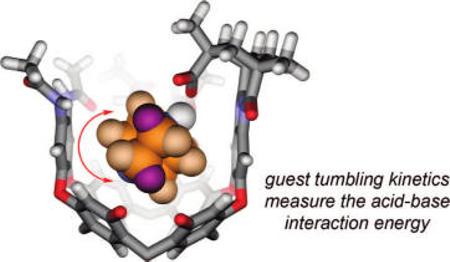
During the exchange studies of DABCO, we noted the presence of NOE cross-peaks that correspond to chemical exchange between the DABCO methylene groups closer to the acid and those deeper in the cavity (Figure 8). This corresponds to “tumbling” of the guest. The EXSY measurements in either CDCl3 or mesitylene-d12 showed that the barrier to tumbling was ΔG‡ = 18 ± 0.5 kcal mol−1 at 300 K (see Figure S1 in the Supporting Information). The nature of the solvent did not appreciably influence this process; it appears to take place inside the cavitand. Typically, tumbling is much more facile in unfunctionalized cavitands, and the introverted acid functionality must control tumbling through its interaction with the guest. By cooling an NMR sample of DABCO bound to 1a, a guest 1H signal coalescence was observed at 230 K. Such coalescence is explained by either (1) slow racemization of the cavitand (the clockwise or counterclockwise arrangement of interamide H bonds; Figures 1 and 3) or (2) tumbling becomes slow on the NMR time scale. Option (1) is more likely because a similar coalescence is observed when quinuclidine is the guest and in this case tumbling does not occur. Therefore, the coalescence temperature for tumbling is at most 230 K, and the maximum barrier to tumbling is ΔG‡ = 11.6 ± 0.1 kcal mol−1 in the unfunctionalized cavitand 1. The difference between the DABCO tumbling process in 1 and H2 is clearly attributable to the acid–base interaction. We propose that the difference in these barriers to tumbling, ΔΔG‡ > 6.5 ± 0.5 kcal mol−1, provides a minimum estimate for the interaction energy in the acid–base pair. This measurement is within experimental error of the ΔG° = 6.8 ± 0.5 kcal mol−1 measured in the thermodynamic cycle (Figure 6) and is consistent with the ΔΔG‡ = 7.4 ± 0.5 kcal mol−1 value obtained from DABCO guest exchange observations.
FIGURE 8.
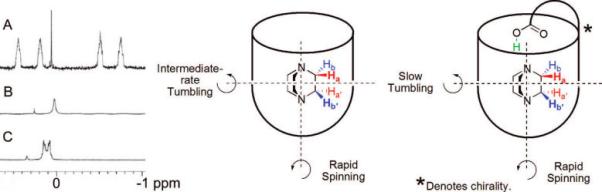
In the unfunctionalized cavitand (center), DABCO spins rapidly about the vertical axis and tumbles at a slower rate about a horizontal axis. The introverted acid cavitand H2 has no appreciable effect on spinning, but tumbling is dramatically slowed. The 1H NMR spectrum of DABCO bound by 2 (A) shows four distinct signals for the guest, the further upfield signals Ha′ and Hb′ corresponding to the methylenes deeper in the cavitand. DABCO bound by 1 shows only a single guest peak at 300 K (B), but separate signals are seen at 220 K (B); the coalescence temperature was 230 K.
The strength of this interaction energy profoundly affects the dynamics of the acid–base pair in H2. We reported the inversion dynamics of prochiral N-ethyl-N-methyl-N-isopropylamine bound by H2b and compared this process to that observed in the presence of simple carboxylic acids.42 In CD2Cl2, control acid H3 has the same influence as pivalic acid on N-ethyl-N-methyl-N-isopropylamine inversion. The barrier to inversion/rotation67 of the amine is ΔG‡ = 10.5 ± 0.5 kcal mol−1 in the presence of pivalic acid and ΔG‡ = 10.3 kcal mol−1 in the presence of H3. Introverted acid H2, on the other hand, formed complexes with N-ethyl-N-methyl-N-isopropylamine that had barriers to inversion/rotation that were too high to measure by EXSY NMR experiments, even at 323 K. Instead, the diastereomeric complexes interconvert by the lower barrier mechanism of guest exchange: the barrier to inversion/rotation inside the cavitand is greater than 24 kcal mol−1! While a significant part of this raised barrier must come from the strength of the acid–base interaction, the cavitand provides a geometric barrier to the conformational changes intrinsic to this amine inversion. Together these properties are sufficient to suppress the inversion dynamics inside the cavitand.
Conclusions
The fusion of Kemp's triacid to deep cavitands results in host molecules with functionality trained on the guests inside. Convergent functions are not new in molecular recognition studies, and they can show high selectivities.68,69 Even more effective would be assemblies such as capsules that completely surrounded the target and a functional molecule or two.70,71 These introverted acids selectively bind amines of appropriate size and shape with binding constants on the order of Ka = 103 in water and Ka > 105 in organic solvents. While cation–π, CH–π, and electrostatics contribute, a substantial component of binding energy comes from the acid–base interaction energy: −3 kcal mol−1 in water or −7 kcal mol−1 in organic solvents. The difference of 4 kcal mol−1 probably results from partial solvation of the acid–base interaction at the open top of the cavitand. Hydrogen bonding between acid and base stabilizes and influences the motions of the amine guest. For roughly spherical guests such as DABCO, the barrier to tumbling in the cavitand is raised by at least 6.5 kcal mol−1. For the prochiral amine N-ethyl-N-methyl-N-isopropylamine, nitrogen inversion is not observed in the cavity. In some cases, the exchange of amine guests is slowed markedly by the acid: exchange of DABCO was slowed in mesitylene-d12, and it was necessary to wash several times with 5 M HCl to completely extract quinuclidine from H2a. Together these results provide measurements of the interaction energy of acid–base pairs in isolation.
Experimental Section
X-ray Crystal Structure Determination
Crystallographic data were collected on a Siemens SMART72 diffractometer equipped with a CCD area detector using graphite monochromatized Mo KR radiation [λ(Mo KR) = 0.71073 Å] radiation. Data in the frames corresponding to an arbitrary hemisphere of data were integrated using SAINT.72 Data were corrected for Lorentz and polarization effects and were further analyzed using XPREP. An empirical absorption correction based on the measurement of redundant and equivalent reflections and an ellipsoidal model for the absorption surface was applied using SADABS. The structure solution and refinement were performed using SHELXTL73 (refining on F2).
Introverted acid H2a × 4.5 CHCl3: measurements at 293 K, crystal size 0.4 × 0.3 × 0.3 mm, monoclonic, P2(1)/c, a = 12.507(3)Å, b = 31.341(8) Å, c = 26.083(6)Å, β = 94.522(5)°, V = 10192(4)Å3, Z = 4, ρcalcd = 1.346 g cm−3, 2Θmax = 55.92°, μ = 0.431 mm−1, F(000) = 4226, 364 parameters, R1 = 0.3179, wR2 = 0.6075 (for 7841 refl. I > 2σ(I)), R1 = 0.4608, wR2 = 0.6557 (for 22 154 unique reflections), S = 1.619, Δρ (min/max) = −0.721/0.933 e Å−3.
1a · DABCO × 4C9H12: measurements at 173 K crystal size 0.4 × 0.3 × 0.2 mm, triclinic, , a = 15.413(1)Å, b = 18.268(1)Å, c = 22.201(2)Å, α = 91.5670(1)°, β = 107.567(1)°, γ = 114.4950(10)°, V = 5339.0(7) Å3, Z = 2, ρcalcd = 1.200 g cm−3, 2Θmax = 56.04°, μ = 0.072 mm−1, F(000) = 2046, 1383 parameters, R1 = 0.1123, wR2 = 0.2793 (for 17 655 refl. I > 2σ(I)), R1 = 0.1468, wR2 = 0.3040 (for 25 099 unique reflections), S = 1.105, Δρ (min/max) = −0.516/−1.049 e Å−3.
1a · quinuclidine × 4C9H12: measurements at 173 K, crystal size 0.3 × 0.2 × 0.2 mm, triclinic, , a = 15.501(2)Å, b = 18.249(3) Å, c = 22.303(3) Å, α = 91.827(3)°, β = 107.458(3)°, γ = 114.319(3)°, V = 5396(1) Å3, Z = 2, ρcalcd = 1.187 g cm−3, 2Θmax = 56.04°, μ = 0.079 mm−1, F(000) = 2046, 1287 parameters, R1 = 0.1339, wR2 = 0.3283 (for 11 687 refl. I > 2σ(I)), R1 = 0.2204, wR2 = 0.3855 (for 22 927 unique reflections), S = 1.077, Δρ (min/max) = −0.81/1.06 e Å−3.
Binding Constant Determination
Binding constants were determined by using an NMR titration that was optimized to show simultaneously resolved signals for cavitand, guest, and cavitand·guest complex. Solutions were prepared of the desired cavitand and guest at known concentrations in a deuterated NMR solvent. An initial spectrum of cavitand alone was recorded; the guest was then titrated, and new spectra were acquired until a point was reached where it was possible to accurately measure the integral area of peaks for the free cavitand, free guest, and the complex. The slow nature of guest exchange from these cavitands on the NMR time scale allows for these species to be resolved in the spectrum. Spectra with good resolution of species were re-acquired using a 30 s pulse delay to improve the accuracy of integration of the signals for any species that might have a longer relaxation time, particularly the free guest. Binding constants were then calculated using eq 1.
| (1) |
Competitive binding experiments were performed by preparing, for example, a solution of host molecule with one guest present in a deuterated NMR solvent. A second, competing guest was then titrated in, and 1H spectra were acquired until a significant portion of the first guest was displaced; this point is chosen somewhat arbitrarily but is placed in a range of good solubility and such that the relevant signals in the 1H spectrum can be integrated accurately. The relative binding affinity Krel is then calculated using eq 2.
| (2) |
EXSY, TOCSY, and ROESY Measurements
1H,1H EXSY, 1H,1H TOCSY, 1H,1H NOESY, and 1H,1H ROESY experiments were performed at the temperatures and in the solvents indicated using standard pulse sequences. Exchange rates were measured with the 2D EXSY pulse sequence by integrating the relevant peaks of the 2D NOESY spectrum using XWINNMR followed by processing with D2DNMR software.74
Acknowledgment
We are grateful to the Skaggs Institute and the National Institutes of Health (GM 27932) for financial support. B.W.P. is a Skaggs Predoctoral Fellow, A.S. is a Skaggs Postdoctoral Fellow, and S.M.B. is a Ruth L. Kirschstein NRSA Fellow (F32 GM73437). We also thank Drs. Laura Pasternack and Dee-Hua Huang for assistance with NMR experiments.
Footnotes
Supporting Information Available: General experimental, 2D ROESY spectrum showing exchange of DABCO in H2a, and a description of EXSY kinetic models. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hof F, Craig SL, Nuckolls C, Rebek J., Jr Angew. Chem., Int. Ed. 2002;41:1488–1508. doi: 10.1002/1521-3773(20020503)41:9<1488::aid-anie1488>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 2.Iwasawa T, Mann E, Rebek J., Jr J. Am. Chem. Soc. 2006;128:9308–9309. doi: 10.1021/ja062768a. [DOI] [PubMed] [Google Scholar]
- 3.Schramm MP, Rebek J., Jr Chem.—Eur. J. 2006;12:5924–5933. doi: 10.1002/chem.200600338. [DOI] [PubMed] [Google Scholar]
- 4.Hooley RJ, van Anda HJ, Rebek J., Jr J. Am. Chem. Soc. 2007;129:13464–13473. doi: 10.1021/ja0727058. [DOI] [PubMed] [Google Scholar]
- 5.Hooley RJ, Rebek J., Jr Org. Lett. 2007;9:1179–1182. doi: 10.1021/ol062782s. [DOI] [PubMed] [Google Scholar]
- 6.Timmerman P, Verboom W, van Veggel FCJM, van Duynhoven JPM, Reinhoudt DN. Angew. Chem., Int. Ed. Engl. 1994;33:2345–2348. [Google Scholar]
- 7.Heinz T, Rudkevich DM, Rebek J., Jr Angew. Chem., Int. Ed. 1999;38:1136–1139. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1136::AID-ANIE1136>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 8.Rebek J., Jr Angew. Chem., Int. Ed. 2005;44:2068–2078. doi: 10.1002/anie.200462839. [DOI] [PubMed] [Google Scholar]
- 9.Ziegler M, Brumaghim JL, Raymond KN. Angew. Chem., Int. Ed. 2000;39:4119–4121. doi: 10.1002/1521-3773(20001117)39:22<4119::aid-anie4119>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 10.Fiedler D, Leung DH, Bergman RG, Raymond KN. Acc. Chem. Res. 2005;38:349–358. doi: 10.1021/ar040152p. [DOI] [PubMed] [Google Scholar]
- 11.Kaanumalle LS, Gibb CLD, Gibb BC, Ramamurthy VJ. Am. Chem. Soc. 2005;127:3674–3675. doi: 10.1021/ja0425381. [DOI] [PubMed] [Google Scholar]
- 12.Brody MS, Shalley CA, Rudkevich DM, Rebek J., Jr Angew. Chem., Int. Ed. 1999;38:1640–1644. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1640::AID-ANIE1640>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 13.Yoshizawa M, Kusukawa T, Fujita M, Yamaguchi KJ. Am. Chem. Soc. 2000;112:6311–6312. [Google Scholar]
- 14.Kawano M, Kobayashi Y, Ozeki T, Fujita MJ. Am. Chem. Soc. 2006;128:6558–6559. doi: 10.1021/ja0609250. [DOI] [PubMed] [Google Scholar]
- 15.Dong VM, Fiedler D, Carl B, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2006;128:14464–14465. doi: 10.1021/ja0657915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Körner SK, Tucci FC, Rudkevich DM, Heinz T, Rebek J., Jr Chem.—Eur. J. 2000;6:187–195. doi: 10.1002/(sici)1521-3765(20000103)6:1<187::aid-chem187>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 17.Cram DJ, Tanner ME, Thomas R. Angew. Chem., Int. Ed. Engl. 1991;30:1024–1027. [Google Scholar]
- 18.Iwasawa T, Hooley RJ, Rebek J., Jr Science. 2007;317:493–496. doi: 10.1126/science.1143272. [DOI] [PubMed] [Google Scholar]
- 19.Hooley RJ, Iwasawa T, Rebek J., Jr J. Am. Chem. Soc. 2007;129:15330–15339. doi: 10.1021/ja0759343. [DOI] [PubMed] [Google Scholar]
- 20.Purse BW, Ballester P, Rebek J., Jr J. Am. Chem. Soc. 2003;125:14682–14683. doi: 10.1021/ja036595q. [DOI] [PubMed] [Google Scholar]
- 21.Purse BW, Gissot A, Rebek J., Jr J. Am. Chem. Soc. 2005;127:11222–11223. doi: 10.1021/ja052877+. [DOI] [PubMed] [Google Scholar]
- 22.Richeter S, Rebek J., Jr J. Am. Chem. Soc. 2004;126:16280–16281. doi: 10.1021/ja045167x. [DOI] [PubMed] [Google Scholar]
- 23.McCurdy A, Jimenez L, Stauffer DA, Dougherty DA. J. Am. Chem. Soc. 1992;114:10314–10321. [Google Scholar]
- 24.Stauffer DA, Barrans RE, Jr., Dougherty DA. Angew. Chem., Int. Ed. Engl. 1990;102:953–956. [Google Scholar]
- 25.Lee J-J, Stanger KJ, Noll BC, Gonzalez C, Marquez M, Smith BD. J. Am. Chem. Soc. 2005;127:4184–4185. doi: 10.1021/ja042208g. [DOI] [PubMed] [Google Scholar]
- 26.Heemstra JM, Moore JS. J. Am. Chem. Soc. 2004;126:1648–1649. doi: 10.1021/ja031842b. [DOI] [PubMed] [Google Scholar]
- 27.Hooley RJ, Rebek J., Jr J. Am. Chem. Soc. 2005;127:11904–11905. doi: 10.1021/ja052910s. [DOI] [PubMed] [Google Scholar]
- 28.Grotzfeld R, Branda N, Rebek J., Jr Science. 1996;271:487–489. doi: 10.1126/science.271.5248.487. [DOI] [PubMed] [Google Scholar]
- 29.Hooley RJ, Biros SM, Rebek J., Jr Angew. Chem., Int. Ed. 2006;45:3517–3519. doi: 10.1002/anie.200600405. [DOI] [PubMed] [Google Scholar]
- 30.Yoshizawa M, Tamura M, Fujita M. Science. 2006;312:251–254. doi: 10.1126/science.1124985. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, Rebek J., Jr Org. Lett. 2002;4:327–329. doi: 10.1021/ol0168115. [DOI] [PubMed] [Google Scholar]
- 32.Hooley RJ, Restorp P, Iwasawa T, Rebek J., Jr J. Am. Chem. Soc. 2007;129:15639–15643. doi: 10.1021/ja0756366. [DOI] [PubMed] [Google Scholar]
- 33.Scarso A, Shivanyuk A, Hayashida O, Rebek J., Jr J. Am. Chem. Soc. 2003;125:6239–6243. doi: 10.1021/ja029574p. [DOI] [PubMed] [Google Scholar]
- 34.Rudkevich DM, Hilmersson G, Rebek J., Jr J. Am. Chem. Soc. 1998;120:12216–12225. [Google Scholar]
- 35.Rudkevich DM, Rebek J., Jr Eur. J. Org. Chem. 1999:1991–2005. [Google Scholar]
- 36.Shivanyuk A, Rissanen K, Körner SK, Rudkevich DM, Rebek J., Jr HelV. Chem. Acta. 2000;83:1778–1790. [Google Scholar]
- 37.Purse BW, Rebek J., Jr Proc. Natl. Acad. Sci. U.S.A. 2005;102:10777–10782. doi: 10.1073/pnas.0501731102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renslo AR, Rebek J., Jr Angew. Chem., Int. Ed. 2000;39:3281–3283. doi: 10.1002/1521-3773(20000915)39:18<3281::aid-anie3281>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 39.Iwasawa T, Wash P, Gibson C, Rebek J., Jr Tetrahedron. 2007;63:6506–6511. doi: 10.1016/j.tet.2007.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butterfield SM, Rebek J., Jr J. Am. Chem. Soc. 2006;128:15366–15367. doi: 10.1021/ja0663374. [DOI] [PubMed] [Google Scholar]
- 41.Haino T, Rudkevich DM, Shivanyuk A, Rissanen K, Rebek J., Jr Chem.—Eur. J. 2000;6:3797–3805. doi: 10.1002/1521-3765(20001016)6:20<3797::aid-chem3797>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 42.Wash PL, Renslo AR, Rebek J., Jr Angew. Chem., Int. Ed. 2001;40:1221–1222. [PubMed] [Google Scholar]
- 43.Purse BW, Rebek J., Jr Proc. Natl. Acad. Sci. U.S.A. 2006;103:2530–2534. doi: 10.1073/pnas.0511149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koshland DE., Jr Proc. Natl. Acad. Sci. U.S.A. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 46.Nicholls A, Sharp KA, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 47.Mecozzi S, Rebek J., Jr Chem.—Eur. J. 1998;4:1016–1022. [Google Scholar]
- 48.Hof F, Trembleau L, Ullrich EC, Rebek J., Jr Angew. Chem., Int. Ed. 2003;42:3150–3153. doi: 10.1002/anie.200351174. [DOI] [PubMed] [Google Scholar]
- 49.Saito S, Rebek J., Jr Bioorg. Med. Chem. Lett. 2001;11:1497–1499. doi: 10.1016/s0960-894x(01)00138-x. [DOI] [PubMed] [Google Scholar]
- 50.Hauke F, Myles AJ, Rebek J., Jr Chem. Commun. 2005:4164–4166. doi: 10.1039/b506048f. [DOI] [PubMed] [Google Scholar]
- 51.Lyons BA, Montelione GT. J. Magn. Reson. 1993;101:206–209. [Google Scholar]
- 52.Braunschweiler L, Ernst RR. J. Magn. Reson. 1983;53:521–528. [Google Scholar]
- 53.Locke MJ, McIver RT., Jr J. Am. Chem. Soc. 1983;105:4226–4232. [Google Scholar]
- 54.Ngola SM, Kearney PC, Mecozzi S, Russell K, Dougherty DA. J. Am. Chem. Soc. 1999;121:1192–1201. [Google Scholar]
- 55.Rashkin MJ, Hughes RM, Calloway NT, Waters ML. J. Am. Chem. Soc. 2004;126:13320–13325. doi: 10.1021/ja0498538. [DOI] [PubMed] [Google Scholar]
- 56.Hughes RM, Waters ML. J. Am. Chem. Soc. 2005;127:6518–6519. doi: 10.1021/ja0507259. [DOI] [PubMed] [Google Scholar]
- 57.Moran J, Ericson J, Dalcanale E, Bryant J, Knobler C, Cram D. J. Am. Chem. Soc. 1991;113:5707–5714. [Google Scholar]
- 58.Carter PJ, Winter G, Wilkinson AJ, Fersht AR. Cell. 1984;38:835–840. doi: 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- 59.Kato Y, Conn M, Rebek J., Jr J. Am. Chem. Soc. 1994;116:3279–3284. [Google Scholar]
- 60.Park TK, Schröder J, Rebek J., Jr J. Am. Chem. Soc. 1991;113:5125–5127. [Google Scholar]
- 61.Vaughan CK, Harryson P, Buckle AM, Fersht AR. Acta Crystallogr. 2002;58:591–600. doi: 10.1107/s0907444902001567. [DOI] [PubMed] [Google Scholar]
- 62.Adams H, Carver FJ, Hunter CA, Morales JC, Seward EM. Angew. Chem., Int. Ed. Engl. 1996;35:1542–1544. [Google Scholar]
- 63.Palmer LC, Rebek J., Jr Org. Biomol. Chem. 2004;21:3051–3059. doi: 10.1039/B412510J. [DOI] [PubMed] [Google Scholar]
- 64.Gottschalk T, Juan B, Diederich F. Angew. Chem., Int. Ed. 2007;46:260–264. doi: 10.1002/anie.200603366. [DOI] [PubMed] [Google Scholar]
- 65.Tanner ME, Knobler CB, Cram DJ. J. Am. Chem. Soc. 1990;112:1659–1660. [Google Scholar]
- 66. Calculated from the Eyring equation.
- 67. The interconversion of the enantiomers of N-ethyl-N-methyl-N-isopropyl amine requires both the inversion of nitrogen and the rotation of a C-N bond. While the latter has a low activation energy in solution, this barrier may be raised considerably by geometric confines of the host.
- 68.Galan A, de Mendoza J, Toiron C, Bruiox M, Deslongchamps G, Rebek J., Jr J. Am. Chem. Soc. 1991;113:9424–9425. [Google Scholar]
- 69.Nowick JS, Ballester P, Ebmeyer F, Rebek J., Jr J. Am. Chem. Soc. 1990;112:8902–8906. [Google Scholar]
- 70.Shivanyuk A, Rebek J., Jr Chem. Commun. 2001:2424–2425. doi: 10.1039/b109464p. [DOI] [PubMed] [Google Scholar]
- 71.Shivanyuk A, Rebek J., Jr Chem. Commun. 2001:2374–2375. doi: 10.1039/b106793c. [DOI] [PubMed] [Google Scholar]
- 72.Siemens Industrial Automation, I. SMART, Area Detector Software Package. Siemens; Madison, WI: 1995. [Google Scholar]
- 73.Sheldrick G. Siemens Industrial Automation, I. Siemens; Madison, WI: 1993. [Google Scholar]
- 74.Abel EW, Coston TPJ, Orrell KG, Sik V, Stephenson D. J. Magn. Reson. 1986;70:34–53. [Google Scholar]