

Summary
The entire genome is replicated in a programmed manner with specific regions undergoing DNA synthesis at different times in S phase. Active genes generally replicate in early S phase, while repressed genes replicate late, and for some loci this process is developmentally regulated. Using a nuclear microinjection system, we demonstrate that DNA sequences originally packaged into nucleosomes containing deacetylated histones during late S become reassembled with acetylated histones after undergoing replication in early S. Conversely, a change from early to late replication timing is accompanied by repackaging into nucleosomes containing deacetylated histones. This is carried out by differential cell cycle-controlled acetylation and deacetylation of histones H3 and H4. These studies provide strong evidence that switches in replication timing may play a role in the regulation of nucleosome structure during development.
Introduction
Although many studies have shown a close correlation between early replication timing and gene expression in animal cells (Consortium, 2007; White et al., 2004), the mechanistic relationship between these parameters has not yet been fully elucidated. It was previously suggested that replication timing may affect expression by influencing the re-establishment of gene structure following its disruption during DNA replication (Brown, 1984; Gottesfeld and Bloomer, 1982). Since it is hard to follow events that take place at the replication fork itself, we developed a nuclear microinjection system that allows one to observe chromatin packaging on “new” DNA molecules at different times in S phase and thus mimic the process of replication in vivo (Zhang et al., 2002). In addition, this approach serves to isolate the effect of replication timing from many other factors that affect nucleosome structure.
In these studies, DNA injected into early-S cells adopts a relatively active and stable chromatin conformation characterized by acetylated histones H3 and H4. In contrast, naked DNA exposed to late S phase nuclei undergoes assembly with nucleosomes containing deacetylated histones. Similar results were obtained on actively replicating molecules in this same system and these results were also confirmed for endogenous chromosomal replicating DNA, as well (Zhang et al., 2002). Taken together, these findings suggest that the microinjection system provides a reliable way for studying the effect of replication timing on gene structure.
While most gene regions replicate at a fixed time in S phase in all cell types, there are also a large number of genes that replicate in a developmentally regulated manner with DNA synthesis taking place during late S phase in inactive cell types, while being early replicating in cells that express these gene regions (Goren and Cedar, 2003). In a similar manner, a number of early embryonic genes have been shown to switch from early to late replication as a function of differentiation (Hiratani et al., 2004; Perry et al., 2004). According to the model described above, these changes in replication timing should have a critical effect on gene structure.
Results
Microinjection
We previously developed a microinjection technique for studying the effect of replication timing on nucleosome reassembly. In this system, Rat-1 cells containing the EF1 and EF2 replication factors (Ohe et al., 1995) are synchronized by mitotic shake-off and resulting cells then grown in culture for various times and injected with an origin-containing BPV vector (O-S16-LacZ). About 70% of the injected cells remained viable and divided normally in culture. Injected cells were found to have a cell cycle of 19 hr (8 hr G1, 9 hr S, 2 hr G2), as determined by BrdU labeling (Zhang et al., 2002). In this procedure, over 90% of the exogenously-introduced plasmid molecules get packaged into a nucleosomal structure very soon after they are injected (Zhang et al., 2002), and can then undergo multiple replication cycles at any time during S phase (Gilbert and Cohen, 1987). Since the injected DNA is of bacterial origin, it is initially digestible by DpnI (GmATC), but becomes resistant following replication in animal cells. By this criterion, at least 30–40% of the plasmid molecules injected either into early or late S phase undergo replication.
Switch from late to early replication
In order to test the dynamics of nucleosome reassembly, we generated a microinjection protocol that could pick up changes in histone modification when reporter genes are switched from “early to late” or “late to early” replication. In the first experiment, cells were injected in late S phase (15 hr after shake-off) and grown for an additional time period to allow them to complete the next early S phase. BrdU was added to the medium toward the end of G1 (10 hr) (Figure 1A). This protocol allows one to specifically label molecules that have undergone DNA replication during early S phase (BrdU) as opposed to late-injected DNA that has not undergone further replication in early S (unlabeled), and therefore maintains its initial histone modification pattern (Zhang et al., 2002).
Figure 1. Late to early replication switch.
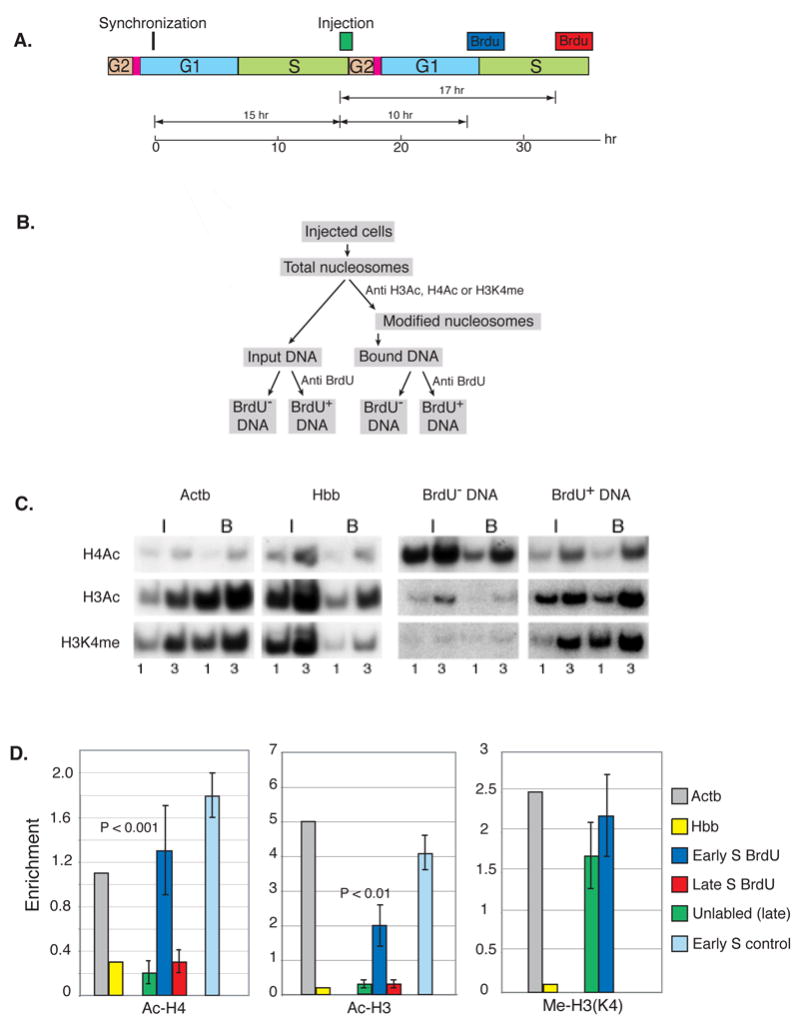
A. Replication competent Rat-1 cells were synchronized by mitotic shake off, injected (about 1000 cells per experiment) after 15 hr (late S) with O-S16–LacZ (0.75 ng/ml) and labeled with BrdU (10−5 M) either after 10 hr during early S or after 17 hr in late S phase of the following cycle (see diagram). B. After 3 hr, nucleosomes were prepared and immunoprecipitated with anti Ac-H4, anti Ac-H3 or anti Me-H3(K4). DNA from both input and bound samples was then separated into BrdU-labeled (37%) and unlabeled fractions by immunoprecipitation with anti-BrdU antibodies. C. These fractions were subjected to semi-quantitative PCR at several different concentrations (1X, 3X) using primers specific for the injected DNA in the presence of radioactive dCTP. Resulting products were separated by PAGE, visualized by autoradiography and then quantitated by scanning. The quantities used for analysis were adjusted to ensure they remain in the linear range. Positive (Actb) and negative (Hbb) controls were also analyzed. The results shown in this example were all derived from a single injection experiment. D. Histone enrichment levels (B/I) for both BrdU labeled (early replicating, blue; late replicating, red) and unlabeled (green) DNA are shown in the bar graph together with positive Actb (grey) and negative Hbb (yellow) ChIP controls. As a control (light blue), 0-S16-LacZ was injected directly into early S phase, and after 3 hr, nucleosomes were isolated and subjected to ChIP analysis. Replicating molecules were singled out by digestion with DpnI prior to PCR analysis. B/I levels were then normalized against the positive and negative controls. The ChIP results for Ac-H4 and Ac-H3 are presented with their standard deviations. P values (two-tailed student T test) for the difference between unreplicated (green) and replicated (blue) molecules are shown. These results were calculated as a composite from 3 individual injection experiments (as in C) after being normalized to the positive control.
Following this protocol, cells were harvested and subjected to ChIP analysis using antibodies against Ac-H3 or Ac-H4 (Figure 1B & C). DNA extracted from both the input and bound fractions was then immunoprecipitated by antibodies to BrdU to separate the replicated from non-replicated DNA. The efficiency of this ChIP reaction was determined by measuring the enrichment of positive (Actb) and negative (Hbb) endogenous controls. As expected, those molecules of plasmid DNA initially injected into late S phase cells that did not undergo further replication (unlabelled) in early S, are packaged with deacetylated H3 and H4 histones (green). In contrast, DNA molecules that underwent a round of replication in early S phase (BrdU labeled, blue) become re-assembled with acetylated histones to the same extent as replicating DNA directly injected into early S-phase nuclei (light blue). This experiment suggests that molecules originally associated with deacetylated histones become dynamically repackaged with acetylated histones following replication in early S phase. In contrast, replication timing has no effect on the modification of histone H3 lysine 4, which is apparently methylated in a constitutive manner during nucleosome reassembly (Figure 1). It should be noted that over 40% of the plasmid DNA becomes labeled with BrdU in this experiment (legend to Figure 1C & D), indicating that the early replication-dependent change in histone modification occurs on a substantial fraction of the injected molecules.
As a control, we carried out this exact same protocol, but instead of adding BrdU during early S, we allowed the cells to continue further through the cycle and added BrdU as they entered late S phase (17 hr). We then compared histone structure on molecules that underwent replication in late S of the subsequent cycle (BrdU labeled) as opposed to those that did not replicate during this labeling period. Following this protocol, both late replicated (red) and non-late replicated (green) molecules were found to be assembled with un-acetylated histones (Figure 1D). This experiment shows that late-assembled DNA remains packaged with deacetylated histones if it subsequently replicates in late S phase, thus mimicking what normally happens to endogenous late-replicating DNA during consecutive cell cycles. In addition, this control indicates that while new replication is indeed required to switch histones into an acetylated state, this only occurs if DNA synthesis and chromatin reassembly take place during early S phase.
Switch from early to late replication
In order to further understand the role of replication-time switching in the control of chromatin assembly, we next designed a converse experiment in which microinjected DNA is initially packaged in early S phase (8 hr after synchronization), but then undergoes replication in late S (4 hr) (Figure 2A). As expected, plasmid DNA molecules injected into early S phase that did not undergo any further replication (unlabeled) remained packaged with acetylated histones H3 and H4 (green)(Figure 2B & C). Those molecules that underwent subsequent replication during late S (BrdU labeled), however, were now found to be packaged with nucleosomes containing completely un-acetylated histone H4 (red) in a manner similar to replicating DNA directly injected into late S nuclei (light red).
Figure 2. Early to late replication switch.
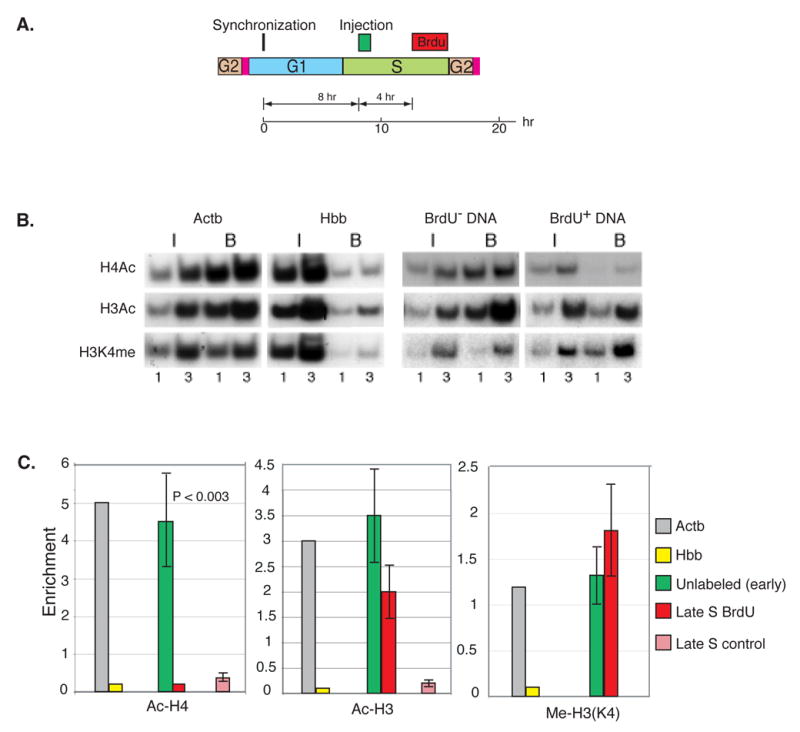
A. Replication-competent Rat-1 cells were synchronized by mitotic shake off, injected (about 1000 cells per experiment) after 8 hr (early S) with O-S16–LacZ (0.75 ng/ml) and labeled with BrdU (10−5 M) in late S (beginning 4 hr after injection). Nucleosomes were prepared 3 hr after the start of labeling and immunoprecipitated with anti Ac-H4, anti Ac-H3 or anti Me-H3(K4). DNA from both input and bound samples was then separated into BrdU-labeled (red) (41%) and unlabeled (green) fractions by immunoprecipitation with anti-BrdU antibodies. B. These samples were analyzed by semi quantitative PCR. C. Histone enrichment levels (B/I ± SEM) for both BrdU labeled (replicated) and unlabeled (unreplicated) DNA are shown in the bar graph together with the enrichment levels of the positive (Actb, grey) and negative (Hbb, yellow) controls and the P value for each experiment. As a control (light red), O-S16-LacZ was injected directly into late S phase and after 3 hr nucleosomes were isolated and subjected to ChIP analysis. Replicating molecules were singled out by digestion with DpnI prior to PCR analysis. The ChIP results from 2 individual injection experiments have been normalized against the positive and negative controls. P values were calculated by the two-tailed students T test.
In contrast, replication in late S (red) had only a marginal effect on the pre-existing acetylation pattern of histone H3 (green), even though replicating DNA directly injected into late S nuclei normally adopts an unacetylated structure (light red)(Figure 2C). Since newly replicated DNA is packaged by a combination of both old and new histones, this striking result strongly suggests that previously acetylated nascent histone H4 molecules must undergo active de-acetylation following replication in late S. There does not, however, seem to be a parallel process for forcibly de-acetylating histone H3 or for affecting methylation on lysine 4 (Figure 4C). Taken together, these result show that early packaged DNA undergoes a change in nucleosome structure when it subsequently undergoes replication and reassembly in late S.
Figure 4. Model for histone acetylation during replication in early and late S.
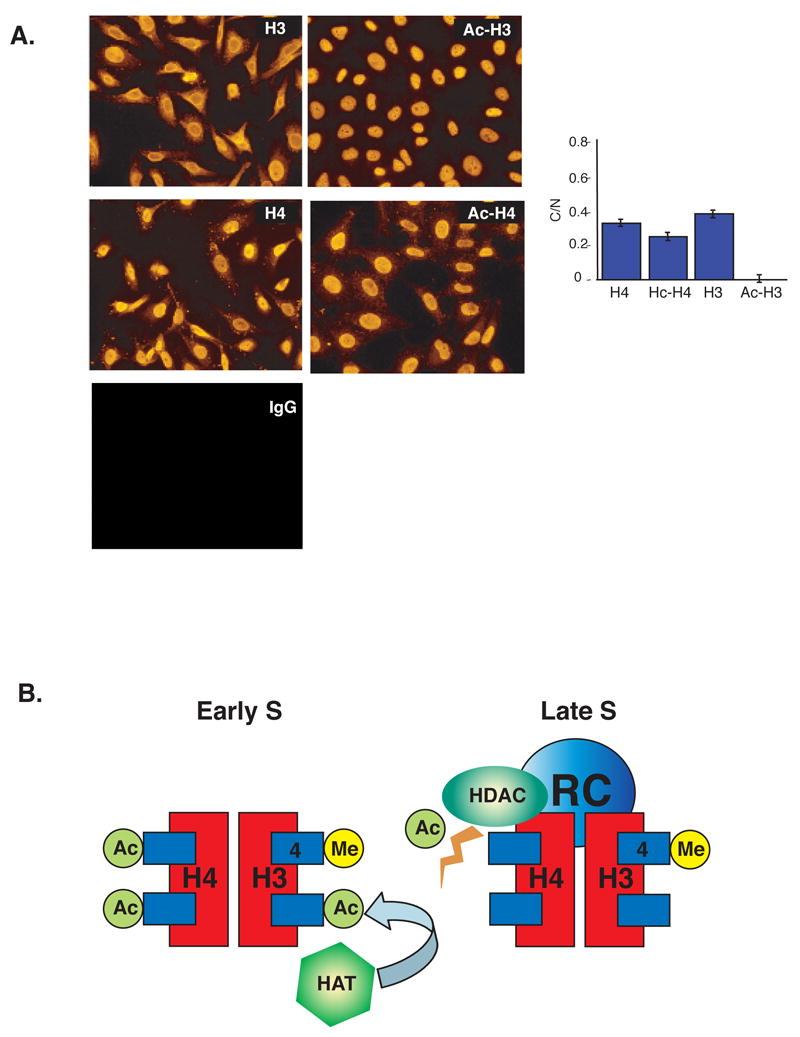
A. Localization of histone acetylation. Unsynchronized HeLa Cells were fixed, permeabilized and stained with various histone antibodies as described (Experimental Procedures). The panels show results using anti-H3, anti-H4, anti-AcH3, anti-AcH4 and an IgG control. The histogram shows micrometric quantification (± SD) of total intensity in the cytoplasm relative to the nucleus (C/N). This analysis was carried out over the linear range of intensities. Background was determined by carrying out the same procedure using IgG instead of the primary antibodies. In most cases, the level of background fluorescence was negligible (< 1%) as compared to the levels obtained with specific antibodies. In the case of AcH3, we obtained an average specific intensity reading of 0.42 for the cytoplasm and 26.1 for the nucleus, with a background level of 0.18. Since the average area of the cytoplasm is about 2 fold greater than that of the nucleus, the relative intensity (C/N) is calculated as 2•(0.42 – 0.18)/(26.1 – 0.18) = 0.019 with a standard deviation of ± 0.005. Note the low relative amount of AcH3 in the cytoplasm as compared to AcH4 (P<0.0001). B. In early S, histone H3 becomes actively acetylated by HATs prior to assembly on newly replicated DNA. Histone H4 is already pre-acetylated before entering the nucleus and is therefore not affected by this process. In late S, this acetylation machinery is not present, but histone H4 becomes actively deacetylated by HDACs that are probably present in the replication complex (RC) itself. Methylation of histone H3K4 is present in nucleosomes assembled both in early and late S phase.
Histone acetylation and deacetylation
The picture that emerges from these experiments is that the histone H4 used for replication-dependent nucleosome deposition is automatically acetylated either prior to or during the packaging process, and becomes subject to active de-acetylation only if DNA synthesis takes place in late S. This concept is consistent with cytochemical evidence linking the histone de-acetylase, HDAC2, exclusively to late replication foci (Rountree et al., 2000). Since pre-acetylated H3 does not undergo a similar deacetylation following a switch to late-S replication (Figure 2), it appears that cell-cycle control of its acetylation state must be carried out by an alternate molecular mechanism, perhaps involving active histone acetylation, specifically in early S-phase. In order to test this hypothesis, we designed an experiment to pinpoint the limiting components regulating the acetylation state of both H3 and H4 during nucleosome packaging. To this end, we challenged the system by micro-injecting a large excess (> 50 molecules/nucleus) of reporter plasmid into early or late S phase cells, and then examined the histone acetylation pattern of these templates after they became assembled with new nucleosomes (Figure 3A).
Figure 3. Differential histone H3 and H4 acetylation.
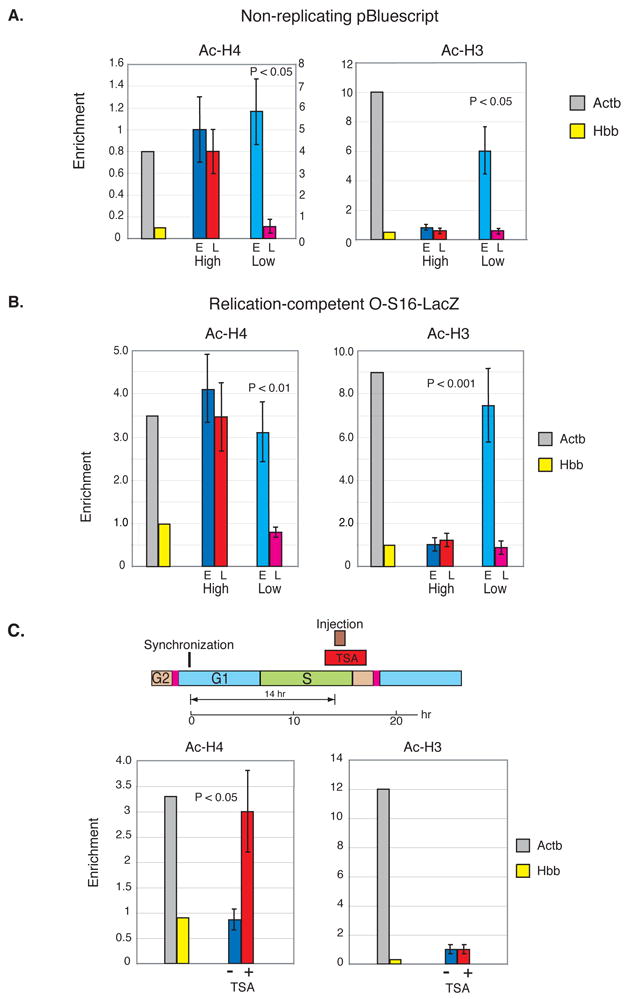
A. High (7.5 ng/ml) or low (0.75 ng/ml) concentrations of non-replicating DNA were injected into Rat-1 cells (500 each) during early (blue) and late (red) S phase after mitotic-shakeoff synchronization. We injected two different pBluescript plasmids, which differ from each other by a small deletion. In each experiment, one plasmid was injected into cells in early S, while the other was injected into late S cells. Nucleosomes from both cell populations were collected 3 hr after injection and then combined for ChIP analysis using anti-Ac-H4, anti-Ac-H3 or anti Me-H3(K4). Enrichment (B/I ± SEM) levels and P values (two-tailed students T test) for the results of 2 individual injection experiments are shown in the bar graph. Actb (grey) serves as a positive and Hbb (yellow) as a negative control. For Ac-H4, these controls were taken from the high concentration experiment. In the low concentration experiment, Actb had an enrichment level of 4 as compared to that of Hbb (0.8), and for this reason the results are shown with its own scale (right y-axis). For Ac-H3, the Actb control showed an enrichment of 10 in both the high and low experiments.
B. High (7.5 ng/ml) or low (0.75 ng/ml) concentrations of O-S16-LacZ DNA were injected into replication-competent Rat-1 cells (500 each) during early (blue) or late (red) S phase after mitotic-shakeoff synchronization. Nucleosomes were collected 3 hr after injection and subjected to ChIP analysis using Anti-Ac-H4 or anti-Ac-H3. Replicated molecules were assayed by digestion with DpnI before PCR. Actb (grey) serves as a positive and Hbb (yellow) as a negative control in each injection experiment and the results were then adjusted by equalizing the relative enrichment of Actb to the early S high concentration injection. Enrichment levels (B/I ± SEM) for the results of 2 individual injection experiments are shown together with the P value for the difference between low-concentration early and late injected DNA (students T-test). C. Rat-1 cells were synchronized by mitotic shake-off and then injected (700 cells) with pBluescript (0.75 ng/ml) in late S in the presence or absence of the HDAC inhibitor TSA (50 ng/ml) that was added for an hour prior to injection (Diagram). Nucleosomes were prepared from cells after 4 hr and immunoprecipitated with anti Ac-H4 or Ac-H3. Untreated (blue) and TSA treated (red) enrichment levels (B/I ± SEM) from 2 individual injection experiments as determined by semiquantitative PCR on input and bound fractions are shown in the bar graph together with positive Actb (grey) and negative Hbb (yellow) controls from untreated cells. P values were calculated using the two-tailed students T test. In TSA treated cells, the ratio of Actb/Hbb enrichment was reduced to about 50% of that shown in the figure. It should be noted that a 24 hr treatment at this concentration of TSA has been shown to bring about extensive histone acetylation at endogenous loci (Hashimshony et al., 2003).
As usual, DNA inserted in low copy number underwent packaging in a cell-cycle specific manner, with both histones H3 and H4 being acetylated on DNA injected in early S phase, but un-acetylated on late-injected templates. When the reporter DNA is present in the nucleus in excess, packaging in early S phase is still associated with acetylated histone H4, but late-injected molecules undergo abnormal deposition, with H4 now showing an acetylated pattern. This result is consistent with the idea that the limiting component for setting up differential H4 acetylation involves active de-acetylation at late replication foci, and this system is apparently unable to keep up with the presence of excess DNA. Overloading the nucleus with reporter plasmid also affects the acetylation pattern of histone H3, but in this case, the defect occurs during nucleosome assembly in early S, where H3 fails to adopt its expected acetylated pattern (Figure 3A). Although indirect, this experiment implies that H3 is initially assembled in its un-acetylated state, and differential modification is mainly controlled by active acetylation that occurs specifically in early S phase. Similar results for H3 and H4 acetylation were also obtained with actively replicating template molecules, as well (Figure 3B), suggesting that this phenomenon is relevant to the process of normal nucleosome reassembly at the replication fork.
In keeping with their histone acetylation states, non-replicating early S phase-injected reporter plasmids are actively transcribed even several generations after entering the nucleus, while late S-injected templates are constitutively repressed (Zhang et al., 2002). This inhibition can be partially relieved by the transient addition of TSA at the time of injection during late S phase, presumably because this treatment prevents the action of HDACs associated directly with the packaging machinery or the histone pools that supply nucleosomes for newly replicating DNA. To test this idea, we micro-injected plasmid DNA into late S nuclei (14 hr after synchronization) in the presence or absence of TSA, and carried out ChIP analysis for Ac-H3 and Ac-H4 on these reporter templates. As expected, TSA treatment causes histone H4 to become abnormally acetylated on these late-injected plasmids. In contrast, this drug apparently has no effect on the acetylation state of H3 (Figure 3C), further suggesting that this histone is not regulated by the enzymatic activity of HDAC in late S phase. These studies thus provide independent support for the idea that H3 and H4 acetylation of newly assembled nucleosomes are regulated differentially.
Location of histone acetylation
Biochemical experiments previously demonstrated that newly synthesized histone H4 undergoes acetylation prior to its deposition on chromatin, while newly made H3 is largely unacetylated (Grunstein, 1997; Sobel et al., 1995). In an attempt to decipher the origins of this difference, we carried out immuno-cytochemistry on animal cells in culture using antibodies specific to Ac-H3 or Ac-H4 (Figure 4A). Through this approach, it is possible to directly visualize the relative amounts of these histone species in the nucleus as compared to the cytoplasm. Strikingly, histone H4 was found to be acetylated both in the nucleus and cytoplasm, as determined by quantitative micrometric analysis. In contrast, only trace amounts of histone H3 acetylation could be detected in the cytoplasm. Although this type of relative assay does not yield absolute values for the degree of acetylation, it strongly suggests that histone H3 and H4 are processed very differently as they relocate from the cytoplasm to the nucleus.
Discussion
When taken together, these molecular and biochemical experiments clearly indicate that while the reassembly-associated acetylation profile of both H3 and H4 core histones is regulated by replication timing, this is actually accomplished by two completely independent, yet highly-coordinated molecular mechanisms (Figure 4B). Histone H4 evidently enters the nucleus in a pre-acetylated state (Lee et al., 1993; Sobel et al., 1995), and then becomes subject to the action of histone de-acetylases (HDACs) specifically associated with late replication foci (Allshire and Bickmore, 2000; Rountree et al., 2000). In contrast, initially un-acetylated histone H3 molecules undergo active acetylation on nucleosomes (Loyola et al., 2006) specifically in early S phase, and this may be mediated by the presence of histone acetylases (HATs) preferentially associated with the packaging machinery in early S. In both cases, the handling of these histones undoubtedly involves histone chaperones associated with DNA replication (Groth et al., 2007).
The entire genome is organized into large sub-domains that are arranged into early and late replicating chromosomal bands (Hand, 1978). Early replicating regions are largely DNaseI sensitive (Kerem et al., 1984), packaged with acetylated histones (Consortium, 2007; Jeppesen and Turner, 1993) and contain many active genes. In contrast, late replicating bands are characterized by an overall closed conformation and include mostly unexpressed DNA sequences (Farkash-Amar et al., 2008; Jeon et al., 2005; MacAlpine et al., 2004; Schubeler et al., 2002; Woodfine et al., 2004). During the process of DNA synthesis these basic chromatin structures become partially disrupted, and must then be properly reassembled following replication. We have taken a closer look at this problem by asking whether the core histone component of nucleosomes destined for newly-synthesized DNA packaging is primarily programmed to be in an acetylated or de-acetylated state.
In order to study this problem, we have employed a new microinjection strategy that allows one to observe nucleosome re-assembly directly associated with changes in DNA replication timing. These experiments demonstrate that the basic decision about histone acetylation is actually bimodal and temporally controlled as an integral part of the replication machinery. Thus, DNA programmed to replicate during the first half of S phase automatically gets repackaged with acetylated core histones, while regions of the genome located in late replicating bands will acquire a deacetylated histone pattern. In this manner, overall re-assembly is accomplished by initially generating a simple two-state nucleosome platform that represents an approximate copy of the original band-like regional acetylation pattern (Jeppesen and Turner, 1993). Since, by it’s very nature, the replication and repackaging machinery moves along the DNA in a dynamic manner and thus does not remain anchored to the newly reassembled DNA template, it is likely that this primary histone acetylation profile then becomes subject to more stable post-replication adjustments that are directed by underlying epigenetic mechanisms such as local DNA methylation or sequence-tethered chromatin modification enzymes (Lande-Diner and Cedar, 2005). This may account for the finding that many gene sequences do not fit the general correlation between gene expression and replication timing (Azuara et al., 2003; Consortium, 2007; Gartler et al., 1999; White et al., 2004).
Replication-time switching often represents an integral part of developmental gene regulation. This includes a number of early-replicating gene regions that become late replicating and undergo inactivation during embryogenesis, as well as many examples of tissue specific genes or gene regions that become early replicating and active during somatic cell differentiation (Goren and Cedar, 2003; Hiratani et al., 2004; Hiratani et al., 2008; Perry et al., 2004). Our results strongly suggest that these programmed changes may actually play an active role in gene regulation by directly causing these regions to get reassembled with a completely new primary histone acetylation pattern. A good example of this paradigm is X-chromosome inactivation in female embryos, where a developmentally-regulated shift to late replication is one of the earliest events in this process, which is then followed by chromosome-wide histone deacetylation, gene repression (Keohane et al., 1996) and finally DNA methylation (Lock et al., 1987).
Experimental Procedures
Microinjections
Rat-1 cells were grown in DMEM medium, supplemented with 10% FCS, Pen-Strep and L-Glu. Cells (105) were seeded on 5.5-cm tissue culture plates containing gridded Cellocate disks (Eppendorf) 20 hr before injection. Under these conditions each disk contains about 150–200 well-spaced cells, each of which can be identified from markings on the disk. Nuclear injection (0.4–7.5 ng/μl DNA) was performed as described (Graessmann and Graessmann, 1983). Calibration of the system by using radioactive material showed that the average injection volume was 20 fl, containing 3–60 molecules of supercoiled plasmid DNA.
To measure the histone modification state of injected episomal DNA, cells were treated with Nocodozole (0.2 μM) and synchronized at M/G1 by mitotic shake-off (Brandeis and Hunt, 1996). More than 80% of these cells cycle normally. In our standard protocol, non-replicating pBluescript plasmid DNA is injected either into early or late S phase cells and nucleosomes harvested 3 hr later. In some experiments we employed two separate plasmids (I and II), which differ from each other by a small deletion that can be distinguished using differential primer pairs (Zhang et al., 2002). One plasmid was injected into cells in early S, while the other was injected into late S cells. Nucleosomes from both cell populations were then combined prior to ChIP analysis.
Histone modification analysis of replicating DNA
ChIP analysis was performed with anti-Ac-H4, anti-Ac-H3 or anti 2Me-H3(K4) (Upstate Biotechnology) to separate input from bound fractions as described (Hebbes et al., 1994). Because about 1% of the nucleosomes are precipitated by this procedure, the bound fraction was always compared with a 1:100 dilution of the input DNA in order to measure the degree of enrichment. In experiments in which BrdU was added, anti-BrdU antibody was used to separate the labeled and unlabeled portions of both input and bound DNA fractions in order to be able to calculate the histone modification enrichment for both unreplicated (unlabeled Bound/unlabeled Input) and newly replicated DNA (BrdU Bound/BrdU Input). For each experiment, the percent of BrdU DNA was calculated from the input fraction using Real-time PCR. Since these ChIP experiments were carried out on nucleosomal DNA, the Input fraction actually represents a true total histone control for H3 and H4 modifications. It should be noted that because the O-S16-LacZ plasmid can replicate multiple times in S phase (Gilbert and Cohen, 1987), BrdU-unlabeled molecules may have undergone replication prior to the labeling period.
PCR analysis
Semi-quantitative PCR analysis of DNA was performed on LacZ (5′-ACGGCATGGTGCCAATGAAT-3′; 5′-GACCAGATGATCACACTCGG-3′, 127 nucleotides (nt), 36 cycles), endogenous -Actb (5′-CGCCATGGATGACGATATCG-3′; 5′-CGAAGCCGGCCTTGCACATG-3′, 68 nt, 21 cycles or 32 cycles for ChIP), Hbb (5′-GCTTCTGACACAACTGTGTTC-3′; 5′-CTGAAGTTCTCAGGATCCACG-3′, 450 nt, 36 cycles) and pBluescript (primer I, 5′-GGGCTGCAGGAATTCGAT-3′,90 nt primer II, 5′-GGATCCCCCATCAAGCTTATCG-3′, 81 nt; T7, 5′-GTAATACGACTCACTATAGGG-3′, 40 cycles) in the presence of radioactive dCTP and products separated by PAGE. Quantification was performed by scanning autoradiograms or by Phosphorimager analysis and averaging the results for two or three different concentrations of DNA. It should be noted that we attempted to reproduce our results with Real Time PCR, but amplification was found to require a large number of cycles and yield erratic results, probably because of the low number of injected molecules in each sample. In contrast, we succeeded in obtaining highly reproducible values using the semi-quantitative gel electrophoresis technique, which allows one to visually distinguish between the real amplicon and non specific PCR products. All injection experiments were carried out 2–3 times and the results combined by normalizing the data for each point using the positive control (Actb) for comparison. Where possible (e.g. Fig. 1), the results were analyzed by calculating the standard deviation (SD) for each data point individually. In other cases we employed the standard error of the mean (SEM = 27%) which was derived by combining results from all injection experiments. Although there was some variability in the numbers derived from PCR experiments, the degree of Actb enrichment was usually about 5–10 fold over the negative control (Hbb). P values were calculated using the two-tailed student T test.
In situ analysis of histone acetylation
HeLa cells were grown in DMEM medium, supplemented with 10% FCS, Pen-Strep and L-Glu. For immunostaining, cultured HeLa cells were permeabilized with 0.2% triton in cytoskeletal buffer (100 mM NaCl, 300 mM sucrose, 10 mM PIPES and 3 mM MgCl2), and fixed with 2% paraformaldehyde, as described (Green and Almouzni, 2003). Cells were incubated with primary antibodies diluted in 5% BSA in PBST overnight at room temperature, washed with PBST, and incubated with secondary antibodies for 1 hr and counterstained with DAPI. Primary antibodies were rabbit anti-histone H3 (Abcam ab1791), rabbit anti-histone H4 (Abcam ab7311), rabbit anti-AcH3 (Upstate Biotechnologies #06-599) and rabbit anti-AcH4 (Upstate Biotechnologies #06-598). Secondary antibodies conjugated to CY3 were from Jackson Immunoresearch (1:200). Images were taken using a Nikon 90i C1 fluorescent microscope, mounted with a DXM1200 (Nikon) camera. Micrometric calculations of nuclear versus cytoplasmic intensities were performed on non-saturated images (> 20 for each primary antibody) within the linear dynamic range after background correction using NIS Elements software (Nikon).
Acknowledgments
This research was supported by grants from the N.I.H., the Israel Science Foundation and the Israel Cancer Research Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allshire R, Bickmore W. Pausing for thought on the boundaries of imprinting. Cell. 2000;102:705–708. doi: 10.1016/s0092-8674(00)00058-1. [DOI] [PubMed] [Google Scholar]
- Azuara V, Brown KE, Williams RR, Webb N, Dillon N, Festenstein R, Buckle V, Merkenschlager M, Fisher AG. Heritable gene silencing in lymphocytes delays chromatid resolution without affecting the timing of DNA replication. Nature Cell Biol. 2003;5:668–674. doi: 10.1038/ncb1006. [DOI] [PubMed] [Google Scholar]
- Brandeis M, Hunt T. The proteolysis of mitotic cyclins in mammalian cells persists from the end of mitosis until the onset of S phase. EMBO J. 1996;15:5280–5289. [PMC free article] [PubMed] [Google Scholar]
- Brown DD. The role of stable complexes that repress and activate eucaryotic genes. Cell. 1984;37:359–365. doi: 10.1016/0092-8674(84)90366-0. [DOI] [PubMed] [Google Scholar]
- Consortium TEP. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkash-Amar S, Lipson D, Polten A, Goren A, Helmstetter C, Yakhini Z, Simon I. Global organization of replication time zones of the mouse genome. Genome Res. 2008;18:1562–1570. doi: 10.1101/gr.079566.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartler SM, Goldstein L, Tyler-Freer SE, Hansen RS. The timing of XIST replication: dominance of the domain. Hum Mol Genet. 1999;8:1085–1089. doi: 10.1093/hmg/8.6.1085. [DOI] [PubMed] [Google Scholar]
- Gilbert DM, Cohen SN. Bovine papilloma virus plasmids replicate randomly in mouse fibroblasts throughout S phase of the cell cycle. Cell. 1987;50:59–68. doi: 10.1016/0092-8674(87)90662-3. [DOI] [PubMed] [Google Scholar]
- Goren A, Cedar H. Replicating by the clock. Nature Rev Mol Cell Biol. 2003;4:25–32. doi: 10.1038/nrm1008. [DOI] [PubMed] [Google Scholar]
- Gottesfeld J, Bloomer LS. Assembly of transcriptionally active 5S RNA gene chromatin in vitro. Cell. 1982;28:781–791. doi: 10.1016/0092-8674(82)90057-5. [DOI] [PubMed] [Google Scholar]
- Graessmann M, Graessmann A. Microinjection of tissue culture cells. Methods Enzymol. 1983;101:482–492. doi: 10.1016/0076-6879(83)01033-2. [DOI] [PubMed] [Google Scholar]
- Green CM, Almouzni G. Local action of the chromatin assembly factor CAF-1 at sites of nucleotide excision repair in vivo. EMBO J. 2003;22:5163–5174. doi: 10.1093/emboj/cdg478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, Almouzni G. Regulation of replication fork progression through histone supply and demand. Science. 2007;318:1928–1931. doi: 10.1126/science.1148992. [DOI] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Hand R. Eucaryotic DNA: Organization of the genome for replication. Cell. 1978;15:317–325. doi: 10.1016/0092-8674(78)90001-6. [DOI] [PubMed] [Google Scholar]
- Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H. The role of DNA methylation in setting up chromatin structure during development. Nature Genet. 2003;34:187–192. doi: 10.1038/ng1158. [DOI] [PubMed] [Google Scholar]
- Hebbes TR, Clayton AL, Thorne AW, Crane-Robinson C. Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. EMBO J. 1994;13:1823–1830. doi: 10.1002/j.1460-2075.1994.tb06451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Leskovar A, Gilbert DM. Differentiation-induced replication-timing changes are restricted to AT-rich/long interspersed nuclear element (LINE)-rich isochores. Proc Natl Acad Sci USA. 2004;101:16861–16866. doi: 10.1073/pnas.0406687101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon Y, Bekiranov S, Karnani N, Kapranov P, Ghosh S, MacAlpine D, Lee C, Hwang DS, Gingeras TR, Dutta A. Temporal profile of replication of human chromosomes. Proc Natl Acad Sci USA. 2005;102:6419–6424. doi: 10.1073/pnas.0405088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen P, Turner BM. The inactive X chromosome in female mammals is distinguished by a lack of histone H4 Acetylation, a cytogenetic marker for gene expression. Cell. 1993;74:281–289. doi: 10.1016/0092-8674(93)90419-q. [DOI] [PubMed] [Google Scholar]
- Keohane AM, O’Neill LP, Belyaev ND, Lavender JS, Turner BM. X-Inactivation and histone H4 acetylation in embryonic stem cells. Dev Biol. 1996;180:618–630. doi: 10.1006/dbio.1996.0333. [DOI] [PubMed] [Google Scholar]
- Kerem BS, Goitein R, Diamond G, Cedar H, Marcus M. Mapping of DNase-I sensitive regions on mitotic chromosomes. Cell. 1984;38:493–499. doi: 10.1016/0092-8674(84)90504-x. [DOI] [PubMed] [Google Scholar]
- Lande-Diner L, Cedar H. Silence of the Genes – mechanisms of long term repression. Nature Rev Genet. 2005;6:648–654. doi: 10.1038/nrg1639. [DOI] [PubMed] [Google Scholar]
- Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- Lock LF, Takagi N, Martin GR. Methylation of the HPRT gene on the inactive X occurs after chromosome inactivation. Cell. 1987;48:39–46. doi: 10.1016/0092-8674(87)90353-9. [DOI] [PubMed] [Google Scholar]
- Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol Cell. 2006;24:309–316. doi: 10.1016/j.molcel.2006.08.019. [DOI] [PubMed] [Google Scholar]
- MacAlpine DM, Rodriguez HK, Bell SP. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 2004;18:3094–3105. doi: 10.1101/gad.1246404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohe Y, Zhao D, Saijo N, Podack ER. Construction of a novel bovine papillomavirus vector without detectable transforming activity suitable for gene transfer. Hum Gene Ther. 1995;6:325–333. doi: 10.1089/hum.1995.6.3-325. [DOI] [PubMed] [Google Scholar]
- Perry P, Sauer S, Billon N, Richardson WD, Spivakov M, Warnes G, Livesey FJ, Merkenschlager M, Fisher AG, Azuara V. A dynamic switch in the replication timing of key regulator genes in embryonic stem cells upon neural induction. Cell Cycle. 2004;3:1645–1650. [PubMed] [Google Scholar]
- Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nature Genet. 2000;25:269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- Schubeler D, Scalzo D, Kooperberg C, Van Steensel B, Delrow J, Groudine M. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nature Genet. 2002;32:438–442. doi: 10.1038/ng1005. [DOI] [PubMed] [Google Scholar]
- Sobel RE, Cook RG, Perry CA, Annunziato AT, Allis CD. Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc Natl Acad Sci USA. 1995;92:1237–1247. doi: 10.1073/pnas.92.4.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EJ, Emanuelsson O, Scalzo D, Royce T, Kosak S, Oakeley EJ, Weissman S, Gerstein M, Groudine M, Snyder M, Schubeler D. DNA replication-timing analysis of human chromosome 22 at high resolution and different developmental states. Proc Natl Acad Sci USA. 2004;101:17771–17776. doi: 10.1073/pnas.0408170101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfine K, Fiegler H, Beare DM, Collins JE, McCann OT, Young BD, Debernardi S, Mott R, Dunham I, Carter NP. Replication timing of the human genome. Hum Mol Genet. 2004;13:191–202. doi: 10.1093/hmg/ddh016. [DOI] [PubMed] [Google Scholar]
- Zhang J, Feng X, Hashimshony T, Keshet I, Cedar H. Establishment of transcriptional competence in early and late S-phase. Nature. 2002;420:198–202. doi: 10.1038/nature01150. [DOI] [PubMed] [Google Scholar]