

Abstract
Cancer cells often exhibit increased reactive oxygen species generation and altered redox regulation. The current study was conducted to investigate the biochemical and molecular events associated with redox alterations during chemical-induced malignant transformation and to evaluate their potential roles in radiation sensitivity. Immortalized nonmalignant human bronchial epithelial cells were exposed to the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), and a clone of cells exhibiting malignant behaviors was isolated and characterized. This clone initially exhibited an increase in cellular superoxide that eventually decreased after a long-term culture in vitro, associated with altered expression of antioxidant molecules, including an increase in thioredoxin-1 and manganese superoxide dismutase, and a decrease in glutathione peroxidase-1. These cells also showed a significant decrease in sensitivity to ionizing radiation, as demonstrated by less cell death in acute apoptosis analyses and long-term cell proliferation assays. Using biochemical redox modulation and siRNA approach, we showed that the increase in thioredoxin-1 played a significant role in conferring resistance to IR. Although there was a substantial increase in cellular glutathione, inhibition of glutathione synthesis did not increase IR sensitivity. Our study showed complex redox alterations during NNK-induced malignant transformation, and identified Trx-1 as a radiosensitivity modulator.
INTRODUCTION
Reactive oxygen species (ROS) play an important role in cellular signal transduction and are essential in maintaining redox balance in biological systems. The balance between ROS generation and elimination determines the cellular redox state, which is often altered during malignant transformation. It has long been recognized that cancer cells exhibit increase in ROS generation compared to their normal counterparts, due in part to oncogenic signals, active metabolism, and mitochondrial dysfunction in cancer cells (6, 19, 27, 31). The oxidative stress in cancer cells is often associated with altered expression of molecules involved in ROS metabolism and change in sensitivity to various therapeutic modalities. Thus, detail analyses of the biochemical and molecular events associated with redox alterations during malignant transformation are important in understanding the carcinogenesis processes and may have significant therapeutic implication.
ROS play a central role in biological effects induced by ionizing radiation (IR), which is a major therapeutic modality in clinical treatment of cancer, either used alone or in combination with other approaches such as surgery and chemotherapy. When the high energy of radiation interacts with cellular water molecules, ROS, including hydroxyl radical (OH•), hydroxylion (OH−), superoxide (O2•−), and hydrogen peroxide (H2O2), are generated directly or indirectly through chain reactions. These molecules are chemically reactive and can cause damage to DNA, proteins, and lipid membranes, leading to cell death. Since the antioxidant systems in the cells play essential roles in elimination of ROS, the upregulation of antioxidant molecules in cancer cells is thought to contribute to radiation resistance (8, 14, 17, 26). However, the detail molecular events in antioxidant upregulation during malignant transformation and the mechanistic links to radioresistance are not fully understood.
Cellular antioxidant systems include enzymes such as superoxide dismutases (SOD), catalases, and various peroxidases, and small antioxidant molecules such as NAD(P)H, glutathione, and thioredoxins (Trx). Among these antioxidant molecules, Trx are small thiol proteins that play an important regulatory role in keeping cellular redox balance (15). Two forms of Trx have been identified. Trx-1 is a 12-kDa protein predominantly present in the cytosol, while Trx-2 is predominantly found in the mitochondria (16). The basic redox biochemistry of Trx-1 and Trx-2 appears similar. Most work on the roles of thioredoxins so far has mainly focused on Trx-1, whereas the biological functions of Trx-2 are less known. One main function of Trx-1 as an antioxidant is to act as a cofactor for peroxiredoxins (Prx) to scavenge hydrogen peroxide (23). Trx-1 is also known to selectively activate the DNA binding of certain transcription factors (20). This includes interactions with nuclear factor-κB (NF-κB), that is involved in cellular response to various stimuli, apoptosis, and tumorigenesis, and with activator protein-1 (AP-1), that plays important role in stress response and regulation of cell growth. Reduced Trx-1 exhibits an anti-apoptotic effect by inhibiting apoptosis signal-regulating kinase 1 (ASK1), which activates the c-Jun N-terminal kinase (JNK)/p38 mitogen-activated protein kinase (MAPK) pathway (24). It has been reported that Trx-1 levels are elevated in many human primary cancers, including lung cancer (13, 18), and that overexpression of Trx-1 is associated with poor prognosis in patients with non-small cell lung cancer (10). Widespread expression of thioredoxin and thioredoxin reductase has been observed in non-small cell lung carcinoma, with both cytoplasmic and nuclear localization, depending on the tumor grades (25). Interestingly, hypoxia seems to cause elevated expression of Trx and Prx I in human lung cancer tissues (11). Moreover, Trx-1 has been suggested to play a role in chemoresistance (2, 12, 29).
In the present study, we investigated the alterations of ROS and expression of antioxidant molecules during chemical-induced malignant carcinogenesis in vitro, and evaluated the role of these redox alterations in radioresistance. Nonmalignant human bronchial epithelial cells were exposed to the tobacco carcinogen NNK to induce malignant transformation in vitro. A clone of cells with malignant behaviors was identified and isolated for further evaluation of changes in ROS and expression of antioxidant molecules. We found that this clone of cells initially exhibited a significant increase in cellular superoxide, which eventually decreased after long-term culture in vitro, associated with profound changes in expression of multiple antioxidant molecules and reduced sensitivity to IR. We further showed that among these alterations, increased Trx-1 appears to be a significant radiosensitivity-modulating molecule in these cells.
METHODS
Chemicals and reagents
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) was purchased from ChemSyn Laboratories (Lenexa, KS). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), DL-buthionine-(S,R)-sulfoximine (BSO), and β-actin antibody were purchased from Sigma-Aldrich (St. Louis, MO). 5-(and-6)-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) and hydroethidine (HEt) were purchased from Invitrogen (Carlsbad, CA). Anti-CuZn-SOD (SOD1) antibody was purchased from Calbiochem (San Diego, CA). Anti-MnSOD (SOD2), glutathione peroxidase-1 (GPx-1), and peroxiredoxin III (Prx III) antibodies were purchased from Lab Frontier (Seoul, Korea). Anti-catalase antibody was purchased from Abcam (Cambridge, MA). Anti-thioredoxin-1 (Trx-1) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-thioredoxin-2 (Trx-2) antibody was purchased from R & D Systems (Minneapolis, MN). The siGENOME SMARTpool targeting Trx-1 and siCONTROL Non-Targeting siRNA Pool were purchased from Dharmacon RNA Technologies (Lafayette, CO).
Cells and cell culture
The immortalized human bronchial epithelial cells (BEAS-2B) described previously (21) were cultured in supplemented Keratinocyte-SFM medium (Invitrogen) at 37°C in a humid atmosphere with 5% CO2. NNK was used as a carcinogen to induce malignant transformation of BEAS-2B cells, which were exposed to the compound continuously at 1 mM for 14 days. NNK was then washed off, and the cells were plated at a low density in fresh medium without NNK. After 2 weeks, several large colonies, indicative of fast growing cells, were isolated and expanded for further characterization. One of the clones, designated as BEAS-NNK cells, exhibited the ability to form colony foci in soft agar in an anchorage-independent assay. BEAS-NNK cells no longer require the supplemented Keratinocyte-SFM medium for growth, and can be maintained in regular RPMI 1640 medium (Mediatech, Herndon, VA) containing 10% fetal bovine serum.
Anchorage-independent colony formation assay
The anchorage-independent colony formation assay was performed using the following conditions: BEAS-2B and BEAS-NNK cells (2,500 cells/well) were suspended in appropriate media containing 0.2% agar and plated in 6-well plates, whose well bottom had been pre-covered with a basal layer of 0.6% agar containing the same media. After a 14-day incubation at 37°C in a humidified incubator containing 5% CO2, the plates were photographed.
Gap-filling assay
BEAS-2B and BEAS-NNK cells were grown to 90% confluence in 6-well plates, and a gap was created by scratching the cell growth surface with a P-200 pipette tip in the mid area of each well. The rates of cell growth and migration to fill the gaps were observed and photographed every 24 h.
Assays for cellular sensitivity to irradiation (IR)
Cells were irradiated with appropriate doses of γ-radiation using Gammacell 1000 Elite (MDS Nordion, Ottawa, Canada). Three different assays were used to assess the effect of IR on cell viability. (a) Acute apoptotic cell death after irradiation was determined by flow cytometric analysis of the cells double-stained with annexin V-FITC (BD Pharmingen, San Diego, CA) and propidium iodide (PI; Sigma-Aldrich). Briefly, 72 h after irradiation, cells were collected and suspended in annexin V binding buffer, and stained with annexin V-FITC conjugate for 15 min in the dark at room temperature. After washing with binding buffer, the cells were stained with PI. A FACSCalibur (BD Biosciences, San Jose, CA) flow cytometer equipped with CellQuest Pro software (BD Biosciences) was used for analyses. For combination treatment with BSO, cells were incubated with BSO at 37°C for 24 h prior to irradiation. (b) Long-term cell survival was evaluated by a modified MTT-based assay, as described previously (28). Briefly, 1 h after irradiation, cells were collected, counted, and seeded to 24-well plates in appropreate densities. After 7-day incubation, MTT was added to each well (final concentration 0.5 mg/ml) and incubated for 4 h. After removal of the supernatant, 350–500 µl DMSO (equal volumes were used for all samples of the same cell line) was added to each well to lyse the cells. Two hundred µl of the cell lysates were transferred to 96-well plates and the optical absorbance density at 570 nm was determined using a MultiSkan microplate reader (LabSystems, Helsinki, Finland). The effect of IR on cell survival was expressed as percent of the control cells. (c) Colony formation assay was also used to assess long-term cell survival capacity. Briefly, 1 h after irradiation, cells were collected, counted, and seeded to 6-well plates in appropreate densities as indicated. After 14-day incubation, the colonies were fixed with a solution containing 50% methanol and 5% acetic acid, stained with Giemsa stain (Sigma-Aldrich), and counted.
Analyses of cellular ROS contents
Fluorescent dyes HEt and CM-H2DCFDA were used as chemical probes for superoxide and general ROS, respectively, as described previously (28). Cells were incubated with either dye at 37°C for 1 h. The cells were collected, washed with PBS twice, and analyzed by FACSCalibur flow cytometer using CellQuest Pro software for quantitative analyses. To test the effect of IR on cellular ROS, irradiation was performed immediately before the cells were collected for analysis.
Western blot analysis
Proteins in whole cell lysates were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with proper primary antibodies for the proteins of interest at 4°C overnight, washed, and then incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. The blots were then developed using the SuperSignal West Pico chemiluminescent reagents (Pierce Biotechnology, Rockford, IL). Chemiluminescent signals were revealed using X-ray film.
Assay of cellular glutathione
A glutathione assay kit (Cayman Chemical, Ann Arbor, MI) was used to quantify total cellular GSH. Cell extract was prepared by sonication and deproteination using the conditions recommended by the manufacturer. Total GSH was detected by measuring the product of glutathionylated 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) by UV spectrophotometer (Ultraspec, Amersham Pharmacia Biotech, Piscataway, NJ) at 405 nm. Cellular GSH content was calculated using the standard curve generated under the same conditions in parallel experiments. To determine the effect of BSO on cellular glutathione, BSO was added to the culture medium 24 h prior to the cells were harvested for analysis.
GPx activity assay
Glutathione peroxidase activity was assayed using a continuous spectrophotometric monitoring of the enzyme-catalyzed oxidation rates, as described previously (28). Briefly, whole cell extract was added to a reaction mixture containing 2 mM GSH, 0.1 U glutathione reductase, and 0.2 mg/ml NADPH. Reaction was initiated by adding hydrogen peroxide (0.001%), and the kinetics of NADPH oxidation were monitored for 5 min at 340 nm. GPx activity (units/105 cells) was calculated by subtracting the slope of the reaction from that of spontaneous oxidation in the control sample with reaction components but without cell extracts. One unit of enzyme activity was defined as oxidation of 1.0 µmole substrate per minute at pH 7.0 at 25°C.
siRNA knockdown of MnSOD and Trx-1
Suppression of MnSOD expression in BEAS-NNK cells was accomplished using the method described previously (9). Briefly, the DNA sequence coding for shRNA targeting Mn-SOD was subcloned into the pBabe-puromycinr retroviral vector under the control of the U6 promoter. Retroviruses were used for infecting BEAS-NNK cells at 40% confluence in medium containing 4 µg/ml polybrene (Sigma-Aldrich). Twenty-four h after infection, the cells were selected in the medium containing puromycin (1 µg/ml). The surviving colonies were pooled and cultured in the medium without puromycin. Inhibition of MnSOD expression was verified by Western blotting. To suppress Trx-1 expression, BEAS-NNK cells were seeded in 6-well plates or 35-mm2 dishes to obtain 30–50% confluence at the time of transfection. Lipofectamine 2000 (Invitrogen) was used to transfect siGENOME SMART-pool targeting Trx-1 or siCONTROL Non-Targeting siRNA Pool as a control, using the procedures recommended by the manufacturer. Twenty-four hours after the initial transfection, the medium was replaced with fresh medium and incubated for the duration indicated. Inhibition of Trx-1 expression was confirmed by Western blot analysis.
Statistical analysis
Student’s t test was used to analyze the statistical significance of the difference between two samples of interest under comparison. A p value of <0.05 was considered statistically significant.
RESULTS
Exposure of bronchial epithelial cells to NNK induced malignant cell behaviors and complex redox alterations
To evaluate possible changes in ROS generation and alterations in expression of antioxidant molecules during the carcinogenesis processes, we used a known tobacco carcinogen NNK to induce malignant transformation in the immortalized, nonmalignant human bronchial epithelial BEAS-2B cells. After a 2-week exposure of BEAS-2B cells to NNK as described under Methods, the cells were plated and cultured in NNK-free fresh medium for 2 weeks. Several large colonies containing fast-growing cells were isolated and expanded for further characterization. One clone of the cells, designated as BEAS-NNK, exhibited the ability to form colonies in an anchorage-independent assay, whereas the parental BEAS-2B cells were unable to form colonies under the same conditions (Fig. 1A). Unlike the parental cells that required supplemented keratinocyte-SFM medium for growth, BEAS-NNK cells were able to grow in regular RPMI 1640 medium + 10% FBS. Furthermore, when cellular mobility was evaluated using a gap-filing assay, BEAS-NNK cells exhibited a greater migration rate than the parental BEAS-2B cells, as evidenced by the ability of BEAS-NNK cells to fill the gap surface in a shorter time (Fig. 1B). These data together suggest that the BEAS-NNK cells have acquired the aggressive behavioral characteristic of malignant cells, and confirm the ability of NNK to induce malignant transformation (7).
FIG. 1. BEAS-NNK cells derived from human bronchial epithelial cells exhibited malignant cell behaviors.
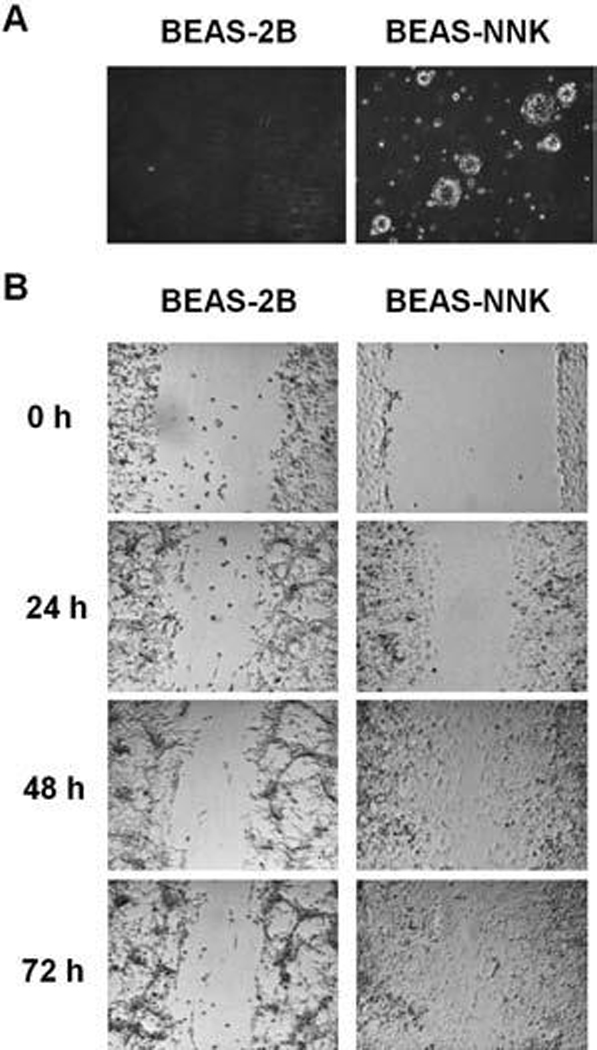
(A) BEAS-NNK cells formed colonies in soft agar, while the parental cells (BEAS-2B) did not. (B) BEAS-NNK cells showed increase of cell migration compared to BEAS-2B cells in a gap-filling assay as described in Materials and Methods.
Since cancer cells derived from primary tumors or established by oncogenic transformation often exhibit alterations in ROS generation and redox regulation, we then tested if such alterations might also occur during chemical-induced transformation. Cellular ROS levels in both cell lines were measured using fluorescence probes HEt and CM-H2DCFDA. HEt is relatively specific for superoxide detection (30), where CM-H2DCFDA detects various reactive species including hydrogen peroxide, hydroxyl radical, and nitric oxide. As shown in Fig. 2A, BEAS-NNK cells in early passages (up to passage 28) exhibited a moderate increase in superoxide content detected by HEt, and a decrease in CM-H2DCFDA-reactive ROS such as hydrogen peroxide, suggesting a possible decrease in conversion of superoxide to hydrogen peroxide in BEAS-NNK cells. However, analysis of expression of SOD, the enzyme that catalyzes this ROS conversion, showed an increase of both CuZn-SOD and MnSOD in BEAS-NNK cells at passages 6 and 28 (Fig. 2D). Thus, it appeared that superoxide generation in these cells was increased, leading to an upregulation of SOD. The reason why CM-H2DCFDA-reactive ROS such as hydrogen peroxide decreased in these cells was likely due to the upregulation of other antioxidant molecules. As the cells were maintained in culture for long term, the superoxide content in the late passage BEAS-NNK cells eventually decreased to a level moderately lower than the parental BEAS-2B cells, while the CM-H2DCFDA-reactive ROS in BEAS-NNK cells remained lower than the parental cells (Fig. 2B). These data suggest that there were complex alterations in ROS regulation during NNK-induced transformation, and that the transformed cells continue their adaptation to maintain a favorable redox state.
FIG. 2. Alterations of ROS contents and antioxidant molecules in BEAS-NNK cells in comparison with the parental BEAS-2B cells.
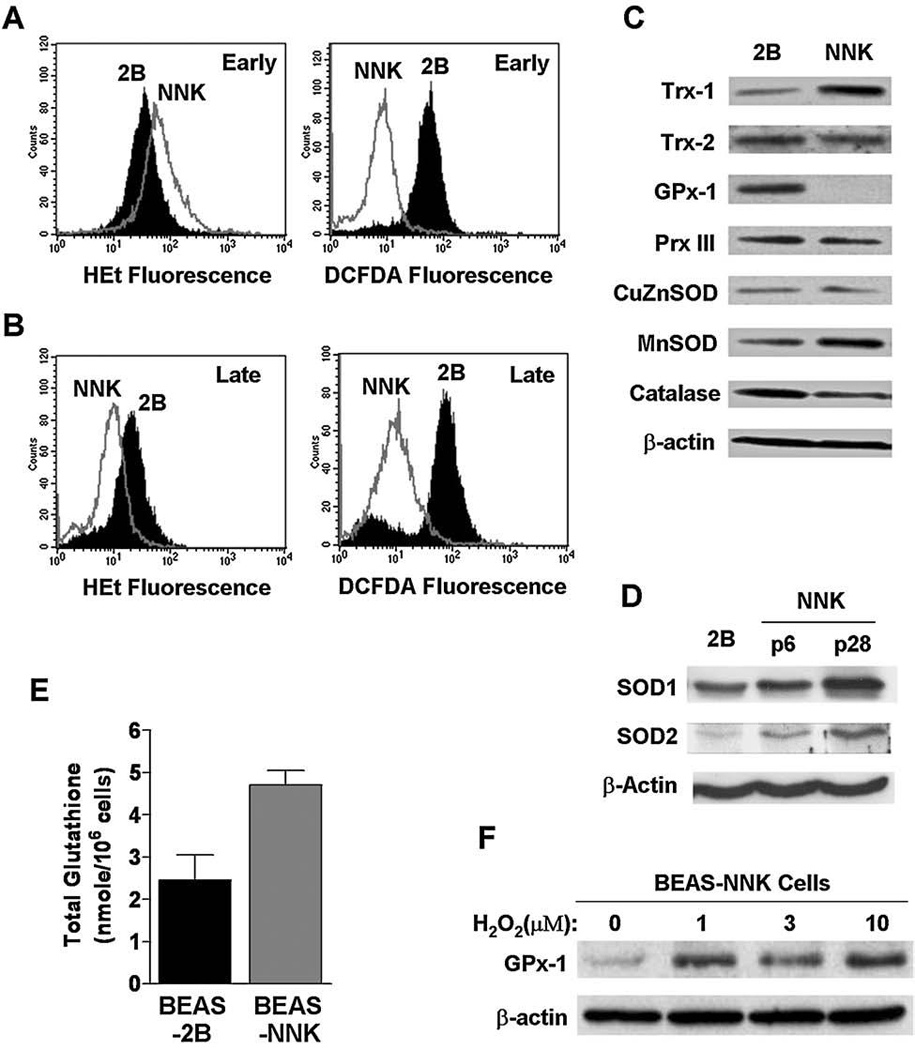
(A) ROS contents in early-passage BEAS-NNK cells detected by incubation with hydroethidine (100 ng/ml) or CM-H2DCFDA (3 µM) for 1 h, followed by analysis using flow cytometer. (B) ROS contents in late-passage BEAS-NNK cells detected by incubation with hydroethidine (100 ng/ml) or CM-H2DCFDA (3 µM) for 1 h, followed by analysis using flow cytometer. (C) Expressions of antioxidant proteins by western blotting. Whole cell lysates from the parental BEAS-2B cells and BEAS-NNK cells (later phase) were subjected to electrophoresis on SDS-PAGE, and the proteins of interest were detected using respective antibodies as indicated. β-Actin was used as a loading control. (D) Upregulation of CuZnSOD and MnSOD in BEAS-NKK cells early passages (p6 and p28). The expression of SOD was detected using respective antibodies. β-Actin was also probed as a loading control. (E) Total cellular glutathione levels in BEAS-2B and BEAS-NNK cells. (F) Induction of GPx-1 expression by incubation with exogenous hydrogen peroxide for 72 h at the indicated concentrations.
To investigate the possible molecular mechanisms that might contribute to the redox alteration observed above, we used Western blot analysis to compare the expression of a panel of molecules important for ROS metabolism in BEAS-2B and BEAS-NNK cells. As shown in Fig. 2C, there was a substantial increase in expression of Trx-1 and MnSOD in BEAS-NNK cells. Furthermore, the cellular glutathione level in BEAS-NNK cells was also significantly higher (twofold of the control BEAS-2B cells, Fig. 2D). The significant increase in expression of MnSOD, a mitochondrial enzyme that converts superoxide to hydrogen peroxide, in the late passage BEAS-NNK cells may explain why these cells exhibited lower superoxide. Trx-1 and glutathione are important cellular antioxidant molecules that are used in coupled reactions to scavenger hydrogen peroxide and other ROS. Their significant increase may be responsible for the decrease of CM-H2DCFDA-reactive ROS observed in BEAS-NNK cells.
Interestingly, we observed a significant decrease of GPx-1 expression in BEAS-NNK cells (Fig. 2C). Because this enzyme is involved in scavenging of hydrogen peroxide, its decrease in expression is seemingly inconsistent with decreased ROS levels in the cells. One possible explanation would be that the decreased expression of GPx-1 was a consequence of high cellular glutathione and low ROS in BEAS-NNK cells. The lower ROS in BEAS-NNK cells (due to increased MnSOD and elevated Trx-1 and glutathione) might lead to a reduced expression of GPx-1. If this were the case, one would predict that addition of exogenous ROS should upregulate GPx-1 expression. To test this possibility, we exposed BEAS-NNK cells to hydrogen peroxide in culture, and indeed observed a significant induction of GPx-1 expression (Fig. 2E). These results suggest that the GPx-1 gene in BEAS-NNK cells is functional and responsive to changes in ROS levels.
BEAS-NNK cells exhibit radioresistance
Since ROS play a critical role in mediating the cytotoxic effect of IR, we postulated that the alterations in the levels of ROS and redox molecules in BEAS-NNK cells might cause a change in their sensitivity to IR. To test this possibility, we first employed annexin-V/PI double staining, followed by flow cytometric analysis, as an apoptotic assay to compare the acute cytotoxic effect of IR in BEAS-2B and BEAS-NNK cells. As shown in Figs. 3A and B, a 10-Gy radiation caused a massive cell death (apoptosis and secondary necrosis) in ~80% of the parental BEAS-2B cells. Under the same conditions, this dose of IR induced only a small percentage (25%) of cell death in BEAS-NNK cells, suggesting that BEAS-NNK cells were more resistant than BEAS-2B cells to IR. We then decided to use a different assay to confirm this observation. Colony formation assay, which is a gold standard for radiosensitivity assessment, could not be used due to the poor colony forming ability of the parental BEAS-2B cells. Thus, we adapted a modified 7-day MTT assay to compare the effect of IR on the long-term cellular proliferation capacity in BEAS-2B and BEAS-NNK cells. As shown in Fig. 3C, BEAS-NNK cells were much more resistant to IR than their parental cells.
FIG. 3. Decreased sensitivity of BEAS-NNK cells to ionizing radiation.
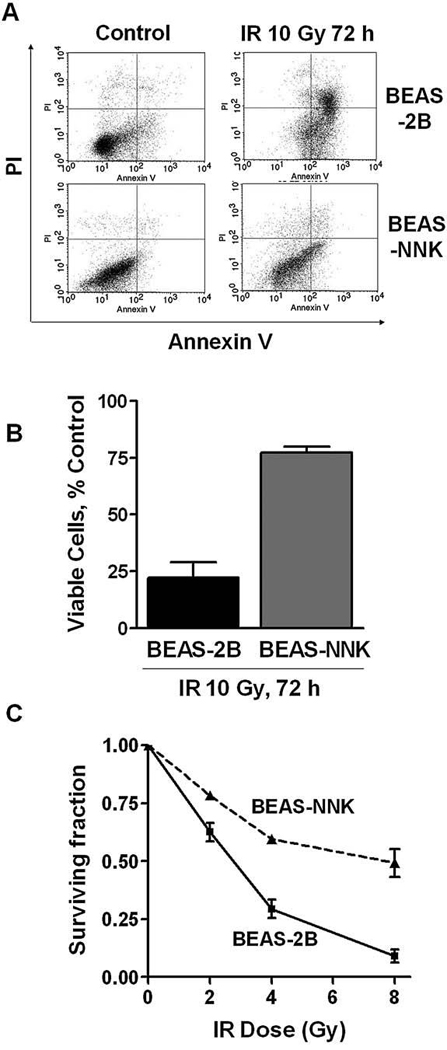
(A) BEAS-NNK or the parental BEAS-2B cells were irradiated at a dose of 10 Gy, and cell death was measured 72 h later using flow cytometry analysis after the cells were double-stained with annexin V and PI. (B) Quantitative comparison of cellular viability in BEAS-2B and BEAS-NNK cells 72 h after IR (10 Gy). Bars, mean ± SD of three experiments; p < 0.05. (C) Effect of IR on long-term cell proliferation in BEAS-2B and BEAS-NNK cells. The long-term (7 days) MTT assay was performed as described in Materials and Methods. The results were from three independent measurements.
We then used flow cytometric analysis to determine ROS levels in both cell lines before and after IR. As shown in Fig. 4, exposure to 10-Gy IR caused a significant increase of CM-H2DCFDA-reactive ROS in BEAS-2B cells, but not in BEAS-NNK cells. These results, together with the observations described above, suggest that BEAS-NNK cells might have acquired a powerful antioxidant system to effectively eliminate IR-induced ROS accumulation, leading to decreased sensitivity to IR.
FIG. 4. Comparison of ROS contents in BEAS-2B and BEAS-NNK cells before and after IR.
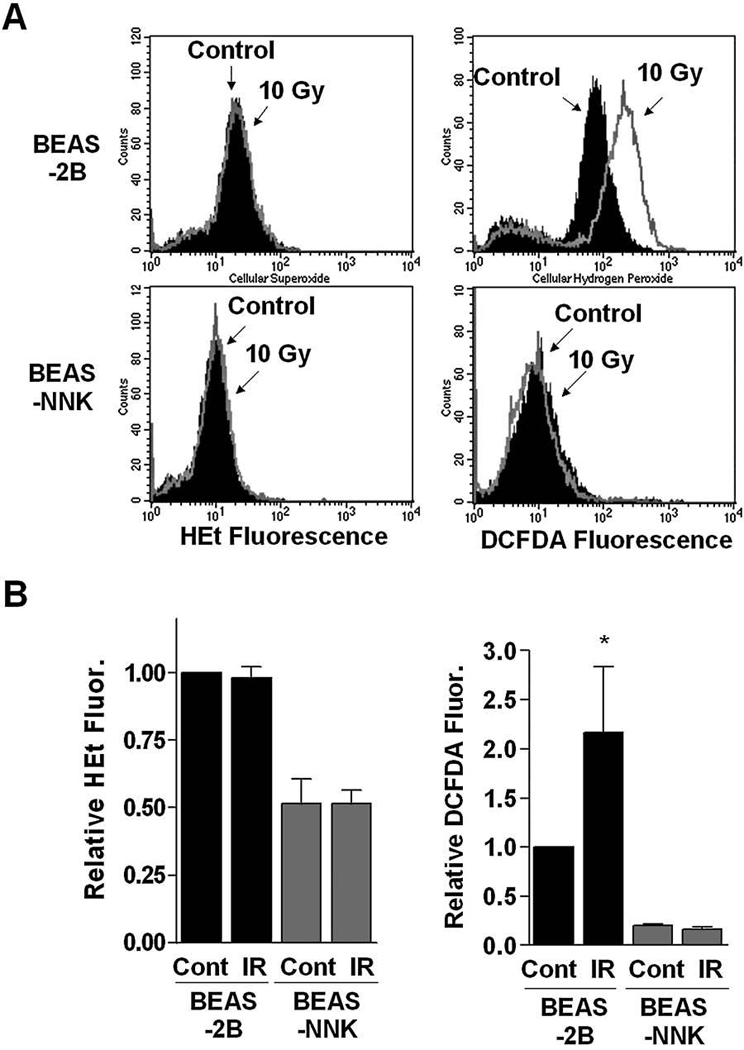
(A) Cells were treated with or without IR (10 Gy), and cellular ROS levels were measured by flow cytometry analysis, using dihydroethidine or DCFDA to stain for superoxide and hydrogen peroxide, respectively. (B) Quantitative analyses of cellular ROS in BEAS-2B and BEAS-NNK cells before and after IR. Each bar shows the mean ± SD of three independent experiments; *p < 0.05.
Increased Trx-1 expression significantly affect radiosensitivity in BEAS-NNK cells
Because there was a significant increase in cellular glutathione and an elevated expression of MnSOD and Trx-1 in BEAS-NNK cells (Fig. 2), we then examined the relative contributions of glutathione, MnSOD, and Trx-1 to the IR-resistant phenotype in these cells. Buthionine sulfoximine (BSO), a known inhibitor of glutathione synthesis, was first used to cause a depletion of glutathione in BEAS-NNK cells, and their sensitivity to IR was then tested. As shown in Fig. 5A, incubation of BEAS-NNK cells with 100 µM BSO for 24 h induced a decrease of total glutathione to ~30% of the control level. However, such glutathione depletion did not alter the sensitivity of BEAS-NNK cells to IR, as assessed by both annexin-V/PI staining (Fig. 5B) and colony formation assay (Fig. 5C). Thus, it appeared that the increase in glutathione level might not be the major reason for IR resistance in BEAS-NNK cells.
FIG. 5. Depletion of glutathione did not alter IR sensitivity in BEAS-NNK cells.
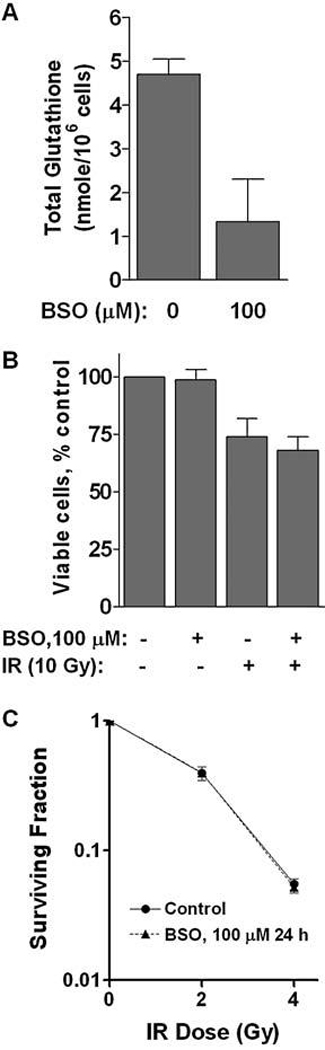
(A) BEAS-NNK cells were incubated with 100 µM BSO for 24 h, and cellular glutathione was measured as described in Materials and Methods. (B) Effect of BSO on the sensitivity of BEAS-NNK cells to IR. Cells were pretreated with 100 µM BSO for 24 h and then subjected to ionizing radiation (10 Gy). Cell viability was measured by annexin V/PI double staining 72 h after IR. (C) Effect of BSO on the sensitivity of BEAS-NNK cells to IR. Cells were pretreated with 100 µM BSO for 24 h and then subjected to ionizing radiation (10 Gy). Colony formation assay was performed to determine the ability of the surviving cells to form colonies during a 14-day incubation. The results were from three independent experiments.
We then used an shRNAi technique to test the potential role of elevated MnSOD expression in conferring IR-resistance in BEAS-NNK cells. MnSOD expression knockdown was achieved by using shRNAi expression vector as described previously (9). As shown in Fig. 6A, expression of MnSOD shRNAi specifically decreased MnSOD expression but not affect CuZnSOD expression, whereas the control vector did not affect either MnSOD or CuZnSOD. The significant decrease of MnSOD expression by shRNAi, however, did not alter the sensitivity of BEAS-NNK cells to IR (Fig. 6B).
FIG. 6. Suppression of MnSOD expression by siRNA did not affect radiosensitivity in BEAS-NNK cells.
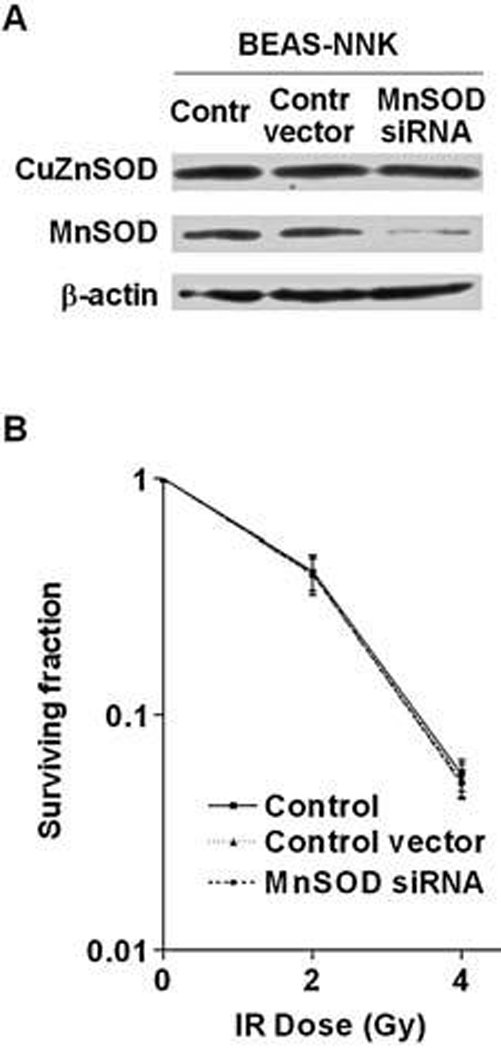
(A) BEAS-NNK cells were infected either with retroviral vector containing MnSOD-siRNA or with the empty vector as described in Materials and Methods. MnSOD levels were evaluated by Western blot analysis. (B) BEAS-NNK cells were infected either with retroviral vector containing MnSOD-siRNA or with the empty vector, and cellular sensitivity to IR was then determined by colony formation assay. Each error bar shows the standard deviation of three independent experiments.
Since the observations described above suggest that neither the increase of glutathione nor MnSOD expression played a major role in IR-resistance of BEAS-NNK cells, we then examined the role of Trx-1 in affecting radiosensitivity in these cells. As shown in Fig. 7A, transient transfection with 20–80 nM siRNA effectively suppressed the expression of Trx-1 protein, leading to an increase of sensitivity to IR (Fig. 7B). Although this increase in IR sensitivity was moderate, it was statistically significant (p < 0.01) at both 2 Gy and 4 Gy. Furthermore, the use of nonspecific siRNA (control oligomers) did not affect the expression of Trx-1 (Fig. 7A) nor altered the cellular sensitivity to IR. These data together suggest that the increase of Trx-1 expression in BEAS-NNK cells may be partially responsible for their decreased sensitivity to IR.
FIG. 7. Suppression of Trx-1 expression by siRNA enhanced radiosensitivity in BEAS-NNK cells.
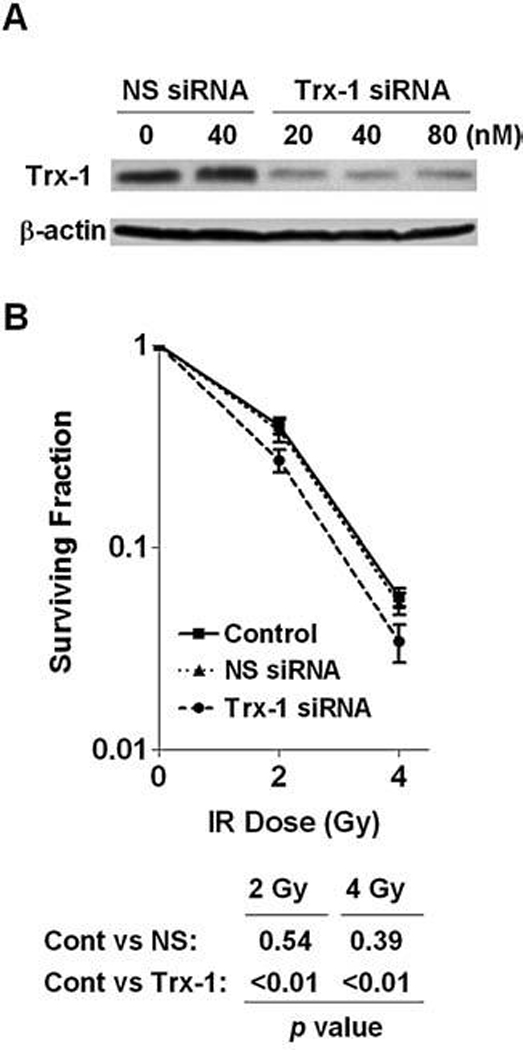
(A) BEAS-NNK cells were transfected with the indicated concentrations of MnSOD siRNA or nonspecific (NS) oligo. After 72 h, cells were collected for analysis of Trx-1 expression by Western blotting. (B) BEAS-NNK cells were transfected with the 40 nM MnSOD siRNA or nonspecific (NS) oligo. After 72 h, the cells were subjected to IR (2 and 4 Gy), and cell viability was measured by colony formation assay (14 day incubation in fresh medium). Each error bar shows the standard deviation of six measurements (three independent experiments in duplicates).
DISCUSSION
The transformation of normal cells to cancer cells is complex, multistep processes involving the interactions between the genetic factors and cellular environment. Both biological (viral) and chemical carcinogens often cause alterations in cellular genetic materials, leading to changes in gene expression, aberrant activation, or suppression of signaling pathways, and uncontrolled cell proliferation. Tobacco smoking is known to play an important role in the development of lung cancer and other human malignant diseases, and NNK is among the most potent tobacco carcinogens identified to date (1). Once NNK is metabolically activated by certain CYP450 enzymes, the active metabolites can then bind to DNA to form adducts, leading to mutations and tumor development. It is also known that NNK-induced carcinogenesis is associated with activation of several signaling pathways important for cell proliferation and cell survival, which include the ERK1/2, PKCα, PI3K/Akt, and MAPK pathways (1). However, little is known about the biochemical and redox alterations during malignant transformation induced by NNK. A recent study showed that NNK caused an increase in lipid peroxidation and oxidative DNA damage in lymphocytes, and that ROS generation and oxidative damage could be significantly enhanced by the combination of NNK and UV radiation in acute experiments within several hours (5). It is unclear if the cells that have survived NNK exposure might have any persistent alterations in ROS generation and expression of redox-regulating molecules when NNK is no longer present.
Our study showed that NNK-induced transformation of human bronchial epithelial cells was associated with complex redox alterations, involving an initial increase of superoxide generation, followed by a rather complex redox adaptation process with significant alterations of antioxidant enzyme expression and an elevation of cellular glutathione. Since NNK may cause DNA adduct formation in both nuclear DNA and mitochondrial DNA, it is unclear which gene(s) may have be mutated in BEAS-NNK cells, leading to the initial increase in superoxide generation. Because the mitochondrial respiratory chain is the major site of cellular superoxide production and mitochondrial DNA is more vulnerable to mutations induced by DNA-damaging agents due to the lack of histone protection and weak DNA repair capacity in the mitochondria (19), it is possible that NNK might cause mitochondrial mutations, leading to a dysfunction of the respiratory chain, where the increased leakage of electrons could be captured by molecular oxygen to form superoxide radicals. The association between mitochondrial mutations and increase superoxide generation has been observed previously in human leukemia cells (3). The increased expression of mitochondrial MnSOD (Fig. 2C), the key enzyme responsible for the elimination of mitochondrial superoxide, seems consistent with the possibility that BEAS-NNK cells might have elevated superoxide generation in their mitochondria. Previous studies showed that cells are able to upregulate their MnSOD expression in response to increased ROS stress to maintain redox balance (9). It is of interest to note that in early passages up to p28, both CuZnSOD and MnSOD were increased, most likely due to the cellular response to the stress of superoxide increase in the early passages. In the later phase, MnSOD remained high but CuZnSOD returned to the normal level (Figs. 2C and D), and cellular superoxide decreased. The most likely explanation is that the disturbance of mitochondrial superoxide generation was an initial event (possibly due to NNK-induced damage to mitochondrial DNA). Adaptation to this superoxide stress resulted in an increased expression of both CuZnSOD and MnSOD, which in turn led to the eventual decrease of superoxide. The sustained increased MnSOD might then cause a further decrease of superoxide in the late passage cells. In addition, the elevated expression of Trx1 and the increased cellular glutathione in BEAS-NNK cells likely reflect the adaptation to the initial oxidative stress, leading to a decrease of ROS after the cells had sufficient time to fully adapt to the initial stress. Interestingly, cellular MnSOD, Trx1, and glutathione remained high even after the cellular superoxide returned to the normal level. One possible explanation would be that the expression of MnSOD, Trx-1, and glutathione synthase might be upregulated epigenetically during the initial stress response.
In this study we identified only one subclone (BEAS-NNK) of cells that exhibited a radioresistant phenotype, and showed that the upregulation of Trx-1 may play a role in conferring radiation resistance. It is unclear, however, if this conclusion could be applied to other cell types, since our observations was made only in one subclone of cells in comparison with the parental cells. Thus, caution should be exercised in generalization of this conclusion. Interestingly, an early study show that in vivo photochemical oxidative stress to Emory mice resulted in a significant upregulation of Trx-1, but not Trx-2, in the lens (22). A recent study showed that nuclear Trx-1 seems to play an important role in protecting breast cancer cells from cisplatin-mediated apoptosis (4). Thus, Trx-2 may function as a survival factor and protect the cells from exogenous insults, including radiation, photochemical oxidative stress, and certain anticancer drugs. Since carcinogens with DNA-damaging properties such as NNK can induce multiple changes, including altered death mechanisms, MAP kinases activation, changes in Cyp450, p53, and DNA damage responses, it is unclear at the present time if BEAS-NNK cells might have alterations in these pathways, which require further studies.
It is worth noting that in Fig. 5–Fig. 7, the colony formation assay used to determine the effect of IR on cell viability was much more sensitive than the MTT assay and annexin-V/PI staining used in experiments shown in Fig. 3. The difference in survival fractions was most likely due to different assays. In the colony formation assay, any cells that failed to form colonies would be considered dead, even if the cells were still viable (but not proliferating to form countable colonies). In MTT and annexin/PI assays, the viable but cytostatic cells might still be detected as viable cells. It is conceivable that after radiation, many cells might still remain viable for days but not form colonies. This situation would lead to different survival fractions when different assays (colony formation, MTT, annexin/PI staining) were used.
These redox alterations in BEAS-NNK cells seem associated with reduced cellular sensitivity to IR. Due to the complex alterations involving multiple molecules, it is difficult to precisely identify the molecular event responsible for the radioresistant phenotype. Depletion of glutathione by BSO or knockdown of MnSOD by siRNA did not significantly restore of radiosensitivity, suggesting that other alterations such as increased expression of Trx-1 might be sufficient to confer a radioresistant phenotype. The finding that knockdown of Trx-1 expression by siRNA led to a partial restoration of radiosensitivity suggests that the increase in expression of Trx-1 may play a significant role in resistance to radiation. Since only partial restoration of radiosensitivity was observed, it is likely that other factors may also contribute to the resistance to radiation.
ACKNOWLEDGMENTS
This work was supported in part by Grants CA016672, CA085563, CA100428, and CA109041 from the National Cancer Institute, the National Institutes of Health. YD was supported in part by a Postdoctoral Fellowship for Study Abroad from the Uehara Memorial Foundation (Japan) and a Fellowship for International Exchange from Nakayama Foundation for Human Science (Japan).
ABBREVIATIONS
- AP-1
activator protein-1
- ASK1
apoptosis signal-regulating kinase 1
- GPx-1
glutathione peroxidase-1
- IR
ionizing radiation
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MnSOD
manganese superoxide dismutase
- NF-κB
nuclear factor-κB
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- ROS
reactive oxygen species
- Trx-1
thioredoxin-1
REFERENCES
- 1.Akopyan G, Bonavida B. Understanding tobacco smoke carcinogen NNK and lung tumorigenesis. Int J Oncol. 2006;29:745–752. [PubMed] [Google Scholar]
- 2.Arnold NB, Ketterer K, Kleeff J, Friess H, Buchler MW, Korc M. Thioredoxin is downstream of Smad7 in a pathway that promotes growth and suppresses cisplatin-induced apoptosis in pancreatic cancer. Cancer Res. 2004;64:3599–3606. doi: 10.1158/0008-5472.CAN-03-2999. [DOI] [PubMed] [Google Scholar]
- 3.Carew JS, Zhou Y, Albitar M, Carew JD, Keating MJ, Huang P. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: clinical significance and therapeutic implications. Leukemia. 2003;17:1437–1447. doi: 10.1038/sj.leu.2403043. [DOI] [PubMed] [Google Scholar]
- 4.Chen XP, Liu S, Tang WX, Chen ZW. Nuclear thioredoxin-1 is required to suppress cisplatin-mediated apoptosis of MCF-7 cells. Biochem Biophys Res Commun. 2007;361:362–366. doi: 10.1016/j.bbrc.2007.07.033. [DOI] [PubMed] [Google Scholar]
- 5.Chuang CH, Hu ML. Synergistic DNA damage and lipid peroxidation in cultured human white blood cells exposed to 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone and ultraviolet A. Environ Mol Mutagen. 2006;47:73–81. doi: 10.1002/em.20168. [DOI] [PubMed] [Google Scholar]
- 6.Giles GI. The redox regulation of thiol dependent signaling pathways in cancer. Curr Pharm Des. 2006;12:4427–4443. doi: 10.2174/138161206779010549. [DOI] [PubMed] [Google Scholar]
- 7.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 8.Hirose K, Longo DL, Oppenheim JJ, Matsushima K. Overexpression of mitochondrial manganese superoxide dismutase promotes the survival of tumor cells exposed to interleukin-1, tumor necrosis factor, selected anticancer drugs, and ionizing radiation. FASEB J. 1993;7:361–368. doi: 10.1096/fasebj.7.2.8440412. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Rosen DG, Zhou Y, Feng L, Yang G, Liu J, Huang P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J Biol Chem. 2005;280:39485–39492. doi: 10.1074/jbc.M503296200. [DOI] [PubMed] [Google Scholar]
- 10.Kakolyris S, Giatromanolaki A, Koukourakis M, Powis G, Souglakos J, Sivridis E, Georgoulias V, Gatter KC, Harris AL. Thioredoxin expression is associated with lymph node status and prognosis in early operable non-small cell lung cancer. Clin Cancer Res. 2001;7:3087–3091. [PubMed] [Google Scholar]
- 11.Kim HJ, Chae HZ, Kim YJ, Kim YH, Hwangs TS, Park EM, Park YM. Preferential elevation of Prx I and Trx expression in lung cancer cells following hypoxia and in human lung cancer tissues. Cell Biol Toxicol. 2003;19:285–298. doi: 10.1023/b:cbto.0000004952.07979.3d. [DOI] [PubMed] [Google Scholar]
- 12.Kim SJ, Miyoshi Y, Taguchi T, Tamaki Y, Nakamura H, Yodoi J, Kato K, Noguchi S. High thioredoxin expression is associated with resistance to docetaxel in primary breast cancer. Clin Cancer Res. 2005;11:8425–8430. doi: 10.1158/1078-0432.CCR-05-0449. [DOI] [PubMed] [Google Scholar]
- 13.Kinnula VL, Paakko P, Soini Y. Antioxidant enzymes and redox regulating thiol proteins in malignancies of human lung. FEBS Lett. 2004;569:1–6. doi: 10.1016/j.febslet.2004.05.045. [DOI] [PubMed] [Google Scholar]
- 14.Lee HC, Kim DW, Jung KY, Park IC, Park MJ, Kim MS, Woo SH, Rhee CH, Yoo H, Lee SH, Hong SI. Increased expression of antioxidant enzymes in radioresistant variant from U251 human glioblastoma cell line. Int J Mol Med. 2004;13:883–887. [PubMed] [Google Scholar]
- 15.Lillig CH, Holmgren A. Thioredoxin and related molecules—from biology to health and disease. Antioxid Redox Signal. 2007;9:25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 16.Miranda-Vizuete A, Damdimopoulos AE, Spyrou G. The mitochondrial thioredoxin system. Antioxid Redox Signal. 2000;2:801–810. doi: 10.1089/ars.2000.2.4-801. [DOI] [PubMed] [Google Scholar]
- 17.Mirkovic N, Voehringer DW, Story MD, McConkey DJ, McDonnell TJ, Meyn RE. Resistance to radiation-induced apoptosis in Bcl-2-expressing cells is reversed by depleting cellular thiols. Oncogene. 1997;15:1461–1470. doi: 10.1038/sj.onc.1201310. [DOI] [PubMed] [Google Scholar]
- 18.Park JH, Kim YS, Lee HL, Shim JY, Lee KS, Oh YJ, Shin SS, Choi YH, Park KJ, Park RW, Hwang SC. Expression of peroxiredoxin and thioredoxin in human lung cancer and paired normal lung. Respirology. 2006;11:269–275. doi: 10.1111/j.1440-1843.2006.00849.x. [DOI] [PubMed] [Google Scholar]
- 19.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Update. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Powis G, Mustacich D, Coon A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic Biol Med. 2000;29:312–322. doi: 10.1016/s0891-5849(00)00313-0. [DOI] [PubMed] [Google Scholar]
- 21.Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park JB, Rhim JS, Harris CC. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- 22.Reddy PG, Bhuyan DK, Bhuyan KC. Lens-specific regulation of the thioredoxin-1 gene, but not thioredoxin-2, upon in vivo photochemical oxidative stress in the Emory mouse. Biochem Biophys Res Commun. 1999;265:345–349. doi: 10.1006/bbrc.1999.1691. [DOI] [PubMed] [Google Scholar]
- 23.Rhee SG, Kang SW, Chang TS, Jeong W, Kim K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life. 2001;52:35–41. doi: 10.1080/15216540252774748. [DOI] [PubMed] [Google Scholar]
- 24.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soini Y, Kahlos K, Napankangas U, Kaarteenaho-Wiik R, Saily M, Koistinen P, Paaakko P, Holmgren A, Kinnula VL. Widespread expression of thioredoxin and thioredoxin reductase in non-small cell lung carcinoma. Clin Cancer Res. 2001;7:1750–1757. [PubMed] [Google Scholar]
- 26.Sun J, Chen Y, Li M, Ge Z. Role of antioxidant enzymes on ionizing radiation resistance. Free Radic Biol Med. 1998;24:586–593. doi: 10.1016/s0891-5849(97)00291-8. [DOI] [PubMed] [Google Scholar]
- 27.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 28.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Kobayashi M, Sakurada K, Imamura M, Moriuchi T, Hosokawa M. Possible roles of an adult T-cell leukemia (ATL)-derived factor/thioredoxin in the drug resistance of ATL to adriamycin. Blood. 1997;89:2480–2487. [PubMed] [Google Scholar]
- 30.Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vasquez–Vivar J, Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med. 2003;34:1359–1368. doi: 10.1016/s0891-5849(03)00142-4. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101:4098–4104. doi: 10.1182/blood-2002-08-2512. [DOI] [PubMed] [Google Scholar]