

Abstract
Retroviral Gag polyproteins have specific regions, commonly referred to as late assembly (L) domains, which are required for the efficient separation of assembled virions from the host cell. The L domain of HIV-1 is in the C-terminal p6gag domain and contains an essential P(T/S)AP core motif that is widely conserved among lentiviruses. In contrast, the L domains of oncoretroviruses such as Rous sarcoma virus (RSV) have a more N-terminal location and a PPxY core motif. In the present study, we used chimeric Gag constructs to probe for L domain activity, and observed that the unrelated L domains of RSV and HIV-1 both induced the appearance of Gag-ubiquitin conjugates in virus-like particles (VLP). Furthermore, a single-amino acid substitution that abolished the activity of the RSV L domain in VLP release also abrogated its ability to induce Gag ubiquitination. Particularly robust Gag ubiquitination and enhancement of VLP release were observed in the presence of the candidate L domain of Ebola virus, which contains overlapping P(T/S)AP and PPxY motifs. The release defect of a minimal Gag construct could also be corrected through the attachment of a peptide that serves as a physiological docking site for the ubiquitin ligase Nedd4. Furthermore, VLP formation by a full-length Gag polyprotein was sensitive to lactacystin, which depletes the levels of free ubiquitin through inhibition of the proteasome. Our findings suggest that the engagement of the ubiquitin conjugation machinery by L domains plays a crucial role in the release of a diverse group of enveloped viruses.
The efficient separation of assembled HIV-1 virions from the cell surface requires the presence of p6gag, which forms the C-terminal domain of the Gag polyprotein (1). Vigorous HIV-1 budding can be observed in the absence of p6gag, but the assembled particles remain attached to the plasma membrane via a thin stalk and consequently accumulate at the cell surface (1). Within p6gag, an invariant P(T/S)AP motif near the N terminus of the otherwise relatively variable domain is crucial for virus release (1, 2). The P(T/S)AP motif is conserved among the p6gag domains of all known primate lentiviruses. Furthermore, in nonprimate lentiviruses, which lack a p6gag domain, the P(T/S)AP motif is found at the immediate C terminus of the Gag polyprotein. An exception is equine infectious anemia virus, which utilizes a YxxL motif within its unique C-terminal Gag domain for virus release (3).
Gag domains that are required during late stages of virus assembly, collectively referred to as late assembly (L) domains, have also been identified in oncoretroviruses. The L domain of Rous sarcoma virus (RSV) has been mapped to p2bgag, an 11-amino acid peptide in the N-terminal half of the Gag polyprotein (4, 5). Furthermore, the Gag polyproteins of Mason–Pfizer monkey virus and Moloney murine leukemia virus (Mo-MLV) contain L domains in an equivalent location (6, 7). In all three viruses, the core of the L domain contains the polyproline motif PPPY, which is also found in the Gag polyproteins of other oncoretroviruses. Recently, it has become apparent that L domains may also be present in other enveloped viruses, because a portion from the matrix protein of vesicular stomatitis virus (VSV) that included a PPPY motif was able to substitute for the L domain of RSV (8).
The mechanism by which L domains promote virus release remains unknown. L domains constitute autonomous modules that are not dependent on a particular position within Gag and that are transferable between unrelated retroviruses (9), suggesting that they provide docking sites for cellular factors necessary for virus release. The polyproline motif in oncoretroviral L domains matches the PPxY core consensus of WW domain ligands, and in vitro binding assays show that L domains that contain the PPxY motif can interact with specific WW domains from the Yes-kinase-associated protein and from members of the Nedd4 ubiquitin ligase family (10–12). Interestingly, ubiquitin ligases have recently been implicated in the endocytosis of several plasma membrane proteins (13, 14), a process that exhibits certain similarities to virus budding. Specifically, Nedd4 is involved in the internalization of the amiloride-sensitive epithelial Na+ channel (ENaC) through PPxY motifs in its cytoplasmic tails (15). A possible involvement of a ubiquitin ligase in L domain function is suggested by the fact that retroviruses contain ubiquitin (16, 17) and by the observation that 2–5% of the HIV-1 p6gag and of the Mo-MLV p12gag present in virions is monoubiquitinated (17). Although they occupy different positions within the Gag precursor, HIV-1 p6gag and Mo-MLV p12gag both harbor an L domain (1, 7).
We recently reported that the inclusion of the RSV L domain p2bgag in a chimeric minimal Gag construct induced the incorporation of several unidentified protein species into virus-like particles (VLPs) (18). In the present study, we show that the extra bands represent Gag-ubiquitin conjugates. Gag ubiquitination strictly depended on the presence of a functional RSV L domain and was also induced by the unrelated L domain of HIV-1 and by the candidate L domains of human T cell leukemia virus type I (HTLV-I) and Ebola virus. We also show that inhibition of the proteasome, which reduces the level of free ubiquitin (19), interfered with VLP release. Together with the observations described in the accompanying papers (20, 21), these results point to a general role for ubiquitin ligase recruitment in retrovirus release.
Materials and Methods
Plasmids.
The ZWT, ZWT-p6, Δ-ZWT, and Δ-ZWT-p2b Gag constructs have been described previously and are based on the vpu-positive HXBH10 variant of HIV-1HXB2 (18). ZWT encodes a C-terminally truncated HIV-1 Gag precursor that has the NC-p1-p6 region replaced by the GCN4 leucine zipper domain (18). ZWT-p6 has the HIV-1 p6gag coding sequence directly fused to the 3′ end of the GCN4 sequence (18). Δ-ZWT is a variant of ZWT that lacks gag codons 8 through 277, and Δ-ZWT-p2b is a variant of Δ-ZWT that has a synthetic sequence encoding RSV p2bgag attached to the 3′ end of the GCN4 sequence (18). The Y/G p2bgag variant of Δ-ZWT-p2b has p2bgag codon 9 (TAC) replaced by GGC, and the PY/GG p2bgag variant additionally has p2bgag codon 8 (CCG) replaced by GGG. The 2X p2bgag variant of Δ-ZWT-p2b has a synthetic sequence that encodes two tandem copies of p2bgag, followed by a stop codon, fused to the GCN4 sequence. Synthetic sequences coding for the HTLV-I and Ebola virus peptides shown in Table 1 and for the ENaC-derived peptide TAPPPAYATLG were fused to the 3′ end of the GCN4 sequence in Δ-ZWT and are followed by a stop codon. The nucleotide sequences at the junctions are 5′ GGT GAG GAC CCA CAA ATC CCC CCT CCC TAT GTT GAG CCT ACA GCC CCC TGA 3′, 5′ GGT GAG ATC TTG CCT ACT GCT CCT CCT GAA TAT ATG GAG GCC TGA 3′, and 5′ GGT GAG ACA GCA CCC CCT CCC GCG TAC GCT ACC CTG GGG TGA 3′, respectively (GCN4 sequences are in bold). Variant ZWT constructs were obtained from the corresponding Δ-ZWT-p2b constructs by reinserting HIV-1 gag codons 8 through 277. HXBH10/SIVgag, which was used to express the SIVmac239 Gag polyprotein, has been described previously (22). The coding sequence for an octameric ubiquitin precursor with a hemagglutinin (HA) tag at the N terminus of each unit was excised from a previously described expression vector (23) (obtained from Dirk Bohmann, European Molecular Biology Laboratory, Heidelberg, Germany) and transferred into the pBJ5stop expression vector (obtained from Richard Bram, St. Jude Children's Research Hospital, Memphis, TN).
Table 1.
Conservation of PPxY and P(T/S)AP motifs in confirmed and candidate L domains
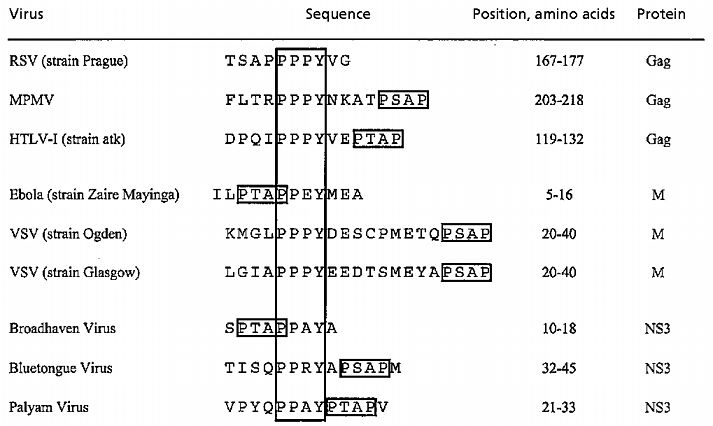 |
MPMV, Mason–Pfizer monkey virus.
Transfection and Analysis of VLP Formation.
HeLa cells (1.4 × 106) or 293T cells (3.5 × 106) were seeded into 80-cm2 tissue culture flasks 24 h before transfection by a calcium phosphate precipitation technique. The cultures were metabolically labeled with [35S]methionine (50 μCi/ml) from 48 to 60 h posttransfection (HeLa cells), or from 24 to 36 h posttransfection (293 T cells). Supernatants were clarified by low-speed centrifugation and passaged through 0.45-μm-pore-size filters. VLP released during the labeling period were spun through 20% sucrose cushions (in PBS) for 2 h at 4°C and 27,000 rpm in a Beckman SW28 rotor. Pelleted VLP were lysed in radioimmunoprecipitation assay buffer and viral proteins were directly analyzed by SDS/PAGE as described (18). For immunoblot analysis, aliquots of the lysed VLP samples were resolved by SDS/PAGE and electroblotted onto Hybond-C Extra membranes (Amersham Pharmacia). The membranes were incubated with a mouse monoclonal anti-HA antibody, followed by a peroxidase-conjugated anti-mouse Ig antibody, and the blots were developed with enhanced chemiluminescence reagents (Amersham Pharmacia) as described (18).
Results
RSV L Domain-Induced Modification of a Minimal Gag Molecule.
We recently observed efficient VLP formation by minimal Gag molecules that harbor only the HIV-1 Gag myristylation signal, the C-terminal domain of HIV-1 CA, a leucine zipper domain, and an L domain (18). Interestingly, the presence of RSV p2bgag in a minimal Gag construct termed Δ-ZWT-p2b led to the incorporation of several unidentified protein species into VLP that migrated more slowly than the expected 16-kDa Gag product (18) (also see Fig. 1, Lane 1). The 11-amino acid p2bgag peptide, which constitutes the L domain of the RSV Gag polyprotein, harbors a PPPPY motif that is crucial for RSV budding (5). As shown in Fig. 1, changing the tyrosine in the PPPPY motif to glycine (Y/G p2bgag) essentially abrogated VLP formation by the Δ-ZWT-p2b construct (compare Lanes 1 and 2). Changing the tyrosine together with the preceding proline residue to glycine (PY/GG p2bgag) had no additional effect (Lane 3). These results confirm that VLP formation by the Δ-ZWT-p2b minimal Gag construct strictly requires the presence of a functional L domain. To determine whether the unidentified protein species in Δ-ZWT-p2b VLP were derived from the 16-kDa minimal Gag molecule, we generated a variant that has two tandem copies of p2bgag at the C terminus (2× p2bgag). As expected, the presence of an additional copy of p2bgag induced a shift in the migration of the minimal Gag product (Fig. 1; compare Lanes 1 and 4). Moreover, the slower migrating bands visible in VLP were shifted to the same degree into higher molecular mass forms, indicating that they represented modified forms of the minimal Gag product.
Figure 1.

An intact RSV L domain rescues VLP formation by a minimal Gag construct and induces the incorporation of modified Gag molecules. HeLa cells were transfected with Δ-ZWT-p2b, which encodes a chimeric minimal Gag molecule with a WT copy of RSV p2bgag at the C terminus (Lane 1), or with the indicated variants of Δ-ZWT-p2b (Lanes 2–4). The Y/G variant (Lane 2) has the single tyrosine in p2bgag changed to glycine, the PY/GG variant (Lane 3) has both the tyrosine and the preceding proline replaced by glycine residues, and the 2× variant (Lane 4) has two tandem copies of p2bgag at the C terminus. After metabolic labeling of the transfected cells with [35S]methionine, VLPs released into the medium were pelleted through sucrose and analyzed by SDS/PAGE. The arrows indicate the positions of protein species that migrated more slowly than the expected minimal Gag molecules. The positions of migration of molecular mass markers (in kilodaltons) are indicated on the right.
Gag Modification Correlates with L Domain Function.
To examine whether the appearance of modified Gag products in VLP was related to L domain function, we made use of the previously described ZWT Gag construct, which does not require a recognizable L domain for the efficient production of VLP (18). The ZWT construct encodes a truncated HIV-1 Gag precursor that retains all of MA and CA but has a leucine zipper domain in place of the C-terminal NC-p1-p6 region (18). Consequently, ZWT lacks known HIV-1 L domain sequences, which are located in p6gag (1). For unknown reasons, the roles of NC and p6gag in assembly and release can both be replaced by the GCN4 leucine zipper domain, and the 40-kDa ZWT molecule produces extracellular VLP with nearly the same efficiency as the wild-type HIV-1 Gag polyprotein (18). To obtain versions that harbor either an intact or a nonfunctional RSV L domain, we attached either WT or Y/G p2bgag to the C terminus of the ZWT Gag molecule. Furthermore, 2× p2bgag was attached to the C terminus of ZWT. The three constructs produced similar amounts of extracellular particles, as judged from the intensities of the principal bands present in VLP, which migrated at the positions expected for the chimeric Gag precursors (Fig. 2A, Left). The presence of WT p2bgag led to the incorporation of several higher molecular mass bands (Lane 1), the electrophoretic mobilities of which were shifted by a second copy of p2bgag (Lane 2), confirming that the extra bands represented modified forms of the chimeric Gag precursors. Remarkably, the Y/G single amino acid substitution in p2bgag completely prevented the appearance of slower migrating forms of Gag, and only the band corresponding to the unmodified Gag precursor remained visible in VLP (Lane 3).
Figure 2.

Induction of Gag ubiquitination by functional L domains. 293T (A) or HeLa (B) cells were transfected with proviral constructs expressing variants of the L domain-independent ZWT Gag molecule that have either WT p2bgag, 2× p2bgag, or Y/G p2bgag (A) or p6gag (B) attached to the C terminus. As controls, a proviral construct unable to express Gag (A) and the unmodified ZWT construct (B) were used. To compare the levels of VLP formation, particulate material released into the medium during metabolic labeling with [35S]methionine was pelleted through sucrose and analyzed directly by SDS/PAGE (Left panels). To detect ubiquitin conjugates, each Gag construct was cotransfected with an expression vector for HA-tagged ubiquitin (HA-Ub), or with the empty vector, and sucrose-purified VLPs were analyzed by immunoblotting with a HA-specific monoclonal antibody (Right panels).
The RSV L Domain-Induced Bands Are Ubiquitin Conjugates.
The most prominent of the extra bands induced by WT p2bgag migrated approximately 7 kDa and 14 kDa more slowly than unmodified Gag. Because of this spacing, we considered the possibility that WT p2bgag induced the attachment of ubiquitin, which has 76 amino acids. To test this possibility, 293T cells were transiently transfected with different versions of ZWT together with a vector that expresses HA-tagged ubiquitin from an octameric precursor, or with an empty control vector. After metabolic labeling, VLPs released into the medium were pelleted through sucrose, and aliquots were directly analyzed by SDS/PAGE and autoradiography to verify that similar amounts of VLPs were recovered in each case (data not shown). In parallel, equivalent aliquots of the pelleted material were subjected to SDS/PAGE and electrotransferred to a nitrocellulose membrane, and ubiquitin conjugates were detected with a HA-specific antibody. When the version of ZWT that harbored WT p2bgag at the C terminus was coexpressed with HA-tagged ubiquitin, the anti-HA antibody revealed a number of distinct ubiquitinated bands that converged into a faint smear toward the top of the gel (Fig. 2A, Right, Lane 1). As expected, these bands were absent when the chimeric Gag construct was cotransfected with the empty vector control (Lane 2). Furthermore, the fastest migrating ubiquitin conjugates detected by immunoblotting mirrored the pattern of bands obtained by metabolic labeling and autoradiography. The pattern of ubiquitin conjugates obtained with the 2× p2bgag version of ZWT was shifted slightly to higher molecular mass forms (Lane 3), confirming that the bands detected by the anti-HA antibody represented conjugates between ubiquitin and the chimeric Gag molecules. Importantly, the presence of Y/G p2bgag at the C terminus of ZWT did not result in Gag ubiquitination (Lane 5).
Taken together with the finding that Y/G p2bgag lacked L domain function in a context where an L domain was required for VLP formation (see Fig. 1), these results indicate that the induction of Gag ubiquitination by p2bgag depends on its ability to act as an L domain.
Unrelated L Domains Induce Ubiquitination.
To examine whether the ability to induce Gag ubiquitination is a common property of retroviral L domains, we used the previously described ZWT-p6 construct (18), which encodes a variant of ZWT that has the 52-amino acid HIV-1 p6gag domain attached at the C terminus. Although HIV-1 p6gag and RSV p2bgag are unrelated at the primary sequence level, they are functionally exchangeable as L domains (9, 18). To facilitate the detection of Gag ubiquitination, the parental ZWT construct and the variant containing p6gag were coexpressed with HA-ubiquitin. As previously reported (18), metabolic labeling of the transfected cells showed that the presence of p6gag had only a small effect on VLP formation (Fig. 2B, Left). However, immunoblotting revealed that p6gag induced the appearance of a ladder of four ubiquitin conjugates in VLP that were intensely stained with the anti-HA antibody, whereas only a single faint band was visible in the absence of p6gag (Fig. 2B, Right). Furthermore, the HA-reactive bands induced by p6gag, which are also faintly visible in the autoradiogram shown in Fig. 2B (Left), migrated as expected for Gag-ubiquitin conjugates. We conclude that the unrelated L domains of RSV and HIV-1 both recruit ubiquitin ligase activity to assembling VLP.
Potent Induction of Ubiquitination and L Domain Activity by a Peptide from Ebola Virus Matrix.
The L domains of RSV, Mason–Pfizer monkey virus, and Mo-MLV all contain an essential PPPY motif (5–7). In contrast, HIV-1 lacks this motif and, like most other lentiviruses, harbors a P(T/S)AP motif that is required for L domain function (1, 2). Interestingly, a P(T/S)AP motif is often found close to a PPPY motif in nonlentiviral Gag polyproteins, for instance, in the known L domain of Mason–Pfizer monkey virus and in the candidate L domain of HTLV-I (Table 1). Also, closely spaced PPPY and P(T/S)AP motif are present near the N terminus of the matrix (M) protein of VSV and are conserved among different VSV strains (Table 1). It was recently shown that an N-terminal portion of VSV M can substitute for the L domain of RSV (8). We also note that the candidate L domain in the Ebola virus M protein (11) harbors entwined P(T/S)AP and PPxY motifs (Table 1). To investigate their potential to provide L domain function, the HTLV-I and Ebola virus sequences shown in Table 1 were fused to the C terminus of the Δ-ZWT minimal Gag construct, which lacks an L domain (18). As previously reported (18), VLP formation by the Δ-ZWT construct is negligible but approaches the level obtained with the full-length HIV-1 Gag polyprotein if HIV-1 p6gag or RSV p2bgag is added to the C terminus of Δ-ZWT (also see Fig. 4). Remarkably, in repeated experiments the Ebola virus peptide consistently boosted VLP formation significantly beyond the level achieved with the RSV L domain, and a more moderate improvement was also seen with the HTLV-I peptide (Fig. 3, Left). Furthermore, Gag-ubiquitin conjugates were visible in each case, and these were most prominent in the presence of the Ebola virus peptide. The coexpression of HA-ubiquitin followed by immunoblotting of VLP samples with anti-HA antibody confirmed that the HTLV-I and Ebola virus peptides, like the RSV L domain, induced the ubiquitination of the chimeric Gag molecules (Fig. 3, Right). Because of their differential electrophoretic mobilities, the four fastest migrating bands visible in lanes 1 through 3 evidently represent Gag-ubiquitin conjugates; however, the higher molecular weight bands could also be VLP-associated cellular contaminants. These data indicate that PPxY and P(T/S)AP motifs together can provide potent L domain activity, particularly the combination found in the Ebola virus matrix protein. Furthermore, the results confirm the relationship between ubiquitin ligase recruitment and L domain function.
Figure 4.

A physiological interaction site for the ubiquitin ligase Nedd4 can act as an L domain. HeLa cells were transfected with proviral constructs expressing the L domain-dependent minimal Gag molecule Δ-ZWT (lane 1), or variants of Δ-ZWT that contain at the C terminus either RSV p2bgag (lane 2) or the sequence TAPPPAYATLG from the α subunit of the human ENaC (lane 3). To compare the levels of VLP formation, particulate material released into the medium during metabolic labeling with [35S]methionine was pelleted through sucrose and analyzed directly by SDS/PAGE. In lane 1, the faint band just below the position of the 14 kDa molecular mass marker corresponds to the parental Δ-ZWT molecule. The arrows indicate the positions of Gag-ubiquitin conjugates.
Figure 3.

Potent L domain activity and ubiquitin ligase recruitment by HTLV-I and Ebola virus peptides with combined PPxY and P(T/S)AP motifs. HeLa cells were transfected with proviral constructs expressing the L domain-dependent minimal Gag molecule Δ-ZWT (Lane C) or variants of Δ-ZWT that contain at the C terminus either RSV p2bgag or the candidate L domains of HTLV-I or Ebola virus listed in Table 1 (Lanes 1–3). Unless an L domain is added, the Δ-ZWT molecule forms negligible amounts of VLP (see Fig. 4). VLPs released during metabolic labeling were pelleted through sucrose and analyzed directly by SDS/PAGE (Left). To confirm the presence of ubiquitin conjugates in VLP, the Gag constructs were cotransfected with an expression vector for HA-tagged ubiquitin, and sucrose-purified VLPs were analyzed by immunoblotting with a HA-specific monoclonal antibody (Right).
A Physiological Interaction Site for the Ubiquitin Ligase Nedd4 Can Substitute for a Viral L Domain.
It has been noted that the PPPY motif in retroviral L domains matches the PPxY core sequence recognized by a subgroup of WW domains (10). Furthermore, it has been shown that the RSV L domain can interact with several of the multiple WW domains present in members of the Nedd4 family of E3 ubiquitin ligases (11, 12). A physiological target of Nedd4 is the amiloride-sensitive ENaC, which harbors PPxY motifs in the cytoplasmic domains of each of its subunits (15, 24). Nedd4 binds these PPxY motifs via its WW domains and mediates the ubiquitin-dependent down-regulation of the ENaC (15, 24). To examine whether a known interaction site for Nedd4 can provide L domain function, we fused the peptide TAPPPAYATLG from the cytoplasmic tail of the human ENaC α subunit to the C terminus of Δ-ZWT. As shown in Fig. 4, the EnaC peptide was about as potent as RSV p2bgag in rescuing VLP formation by the minimal Gag construct.
Inhibition of VLP Formation by the Proteasome Inhibitor Lactacystin.
Inhibition of the proteasome causes an accumulation of polyubiquitinated proteins and a concomitant depletion of unconjugated ubiquitin in the cytosol (19). In an attempt to reduce the levels of free ubiquitin, we treated cells expressing the simian immunodeficiency virus (SIVmac) Gag polyprotein with lactacystin, a highly specific irreversible inhibitor of the proteasome (25). The Gag polyprotein of SIVmac was used because we have observed that VLPs formed by this protein contain particularly high levels of ubiquitin, both free and conjugated to Gag (data not shown). This level of ubiquitin may be related to the fact that in addition to the conserved L domain core motif in p6gag, the SIVmac Gag polyprotein contains a second P(T/S)AP motif at the C terminus of matrix, a position analogous to that of oncoretroviral L domains. Although prolonged treatment with lactacystin inhibited Gag protein synthesis, the levels of cell-associated SIVmac Gag were minimally affected when transfected cells were exposed to 10 μM lactacystin for 2 h and then radiolabeled with [35S]methionine for 4 h in the absence of lactacystin (Fig. 5, Left). However, under these conditions the amount of Gag precursor released in the form of VLP was reduced to about 20% of the amount observed in the absence of lactacystin, as quantified by PhosphorImager analysis (Fig. 5, Right). Also, VLP produced in the absence of lactacystin contained clearly visible bands that migrated at the expected positions of mono- and di-ubiquitinated forms of Gag. However, only the faster migrating band remained faintly visible in VLP produced in the presence of lactacystin, even after overexposure of the autoradiograph shown in Fig. 5. Taken together, these results are consistent with a reduction in the amount of ubiquitin available for conjugation to Gag and support a role for ubiquitin in VLP formation.
Figure 5.

Inhibition of VLP formation by lactacystin. HeLa cells were transfected with the chimeric proviral construct HXBH10/SIVgag, which expresses the full-length Gag polyprotein of SIVmac239 in the absence of a viral protease. The cells were split 48 h posttransfection and kept for 2 h in the presence or absence of 10 μM lactacystin, followed by 4 h of radiolabeling with [35S]methionine in the absence of lactacystin. The cells were then lysed, and cell-associated viral proteins were immunoprecipitated and resolved by SDS/PAGE (Left). VLPs released during the 4-h labeling period were pelleted through sucrose and analyzed directly by SDS/PAGE (Right). The arrows indicate the expected positions of mono- and di-ubiquitinated forms of Gag.
Discussion
The results presented in this study provide evidence that the enhancement of virus release by L domains involves the recruitment of ubiquitin ligase activity. The unrelated L domains of RSV and HIV-1 both induced the conjugation of ubiquitin to Gag constructs, and a single amino acid substitution that inactivated the RSV L domain prevented Gag ubiquitination. The RSV L domain lacks lysine residues to which ubiquitin could be attached, suggesting that it merely serves as a docking site for the ubiquitination machinery. A second line of evidence that ubiquitin plays a role in virus release is the observation that the specific proteasome inhibitor lactacystin inhibited VLP formation by the full-length SIVmac Gag polyprotein. Although we cannot exclude a direct role for the proteasome, our data are consistent with the interpretation that lactacystin inhibited virus release indirectly by depleting the pool of free ubiquitin. In agreement with our findings, proteasome inhibitors have also been found to inhibit the budding of RSV and HIV-1 (20, 21). However, a difference compared with our results and those obtained with RSV (20) is that the inhibitory effect on HIV-1 budding required the presence of an active viral protease (21). Interestingly, the effect of a proteasome inhibitor on RSV budding could be suppressed by the overexpression of ubiquitin and by the fusion of ubiquitin directly to the RSV Gag precursor (20). These observations support the interpretation that proteasome inhibitors interfere with retrovirus budding, at least in part, because they limit the pool of free ubiquitin.
In general, we found that the presence of a functional L domain led to the appearance of only a limited number of prominent Gag-ubiquitin conjugates in VLP, suggesting that L domains do not induce progressive polyubiquitination, which often serves as a signal for protein degradation (26). A small fraction of the p6gag in HIV-1 virions was previously shown to be mono-ubiquitinated, whereas in Mo-MLV ubiquitin was found attached to p12gag (17), which occupies a different position in the Gag precursor. Interestingly, p6gag harbors the L domain of HIV-1, and a PPPY motif in p12gag serves as the L domain for Mo-MLV (1, 7). Although the levels of Gag-ubiquitin conjugates in wild-type HIV-1 and Mo-MLV virions are low (17), it is conceivable that only a small amount of ubiquitinated Gag is needed to attract cellular factors to the site of assembly that are needed during the release step. A role for ubiquitinated Gag in the budding process is supported by the observation that a RSV Gag-ubiquitin chimera was relatively resistant to the inhibitory effects of proteasome inhibitors (20). However, it also remains possible that L domain-facilitated virus release involves the conjugation of ubiquitin to a cellular target at the site of particle assembly, such as to a component of the plasma membrane-associated cytoskeleton.
Based on the finding that a YXXL motif within the L domain of equine infectious anemia virus interacts in vitro with a subunit of the AP-2 complex, it has been suggested that L domains may recruit proteins that are normally involved in endocytosis (27). It is noteworthy in this respect that ubiquitin plays a key role in the down-regulation of a number of plasma membrane-associated proteins by endocytosis (13, 14). While the precise mechanism remains unknown, mono-ubiquitination of a cell surface protein can be sufficient to trigger its rapid internalization (28, 29). Furthermore, in the case of the GH receptor, internalization requires merely the recruitment of the ubiquitin conjugation system and not the conjugation of ubiquitin to the receptor itself (30).
The ubiquitin-dependent down-modulation of the ENaC involves proline-rich tail regions in each of the channel subunits that match the PPxY consensus motif found in oncoretroviral L domains. The PPxY motifs in the ENaC tails interact with the WW domains of Nedd4, which mediates the internalization of the channel (31, 32). Nedd4 belongs to a family of E3 ubiquitin ligases with multiple WW domains, which also includes WWP1, WWP2, and AIP4 (24). Because the present study shows that L domains recruit ubiquitin ligase activity and demonstrates that the Nedd4 interaction site in the ENaC α subunit can substitute for a viral L domain, we considered the possibility that a Nedd4-like ubiquitin ligase is involved in the function of oncoretroviral L domains. However, transient overexpression of wild-type human Nedd4, WWP1, WWP2, or AIP4 in HeLa or 293T cells did not improve RSV L-domain-facilitated VLP formation. Moreover, potentially dominant negative forms of Nedd4 or WWP1 with a cysteine-to-alanine substitution at the active site cysteine in the hect domain did not interfere with the function of the RSV L domain (B.S. and H.G.G., unpublished data).
A peptide from the Ebola virus matrix protein with overlapping P(T/S)AP and PPxY L domain core motifs induced robust Gag ubiquitination and exhibited exceptionally potent L domain activity. In addition to the Ebola virus matrix protein, a SwissProt database search (release 37) with the sequence P(T/S)APPXY identified the gap junction protein connexin45, Gag proteins of rodent intracisternal A-type particles, and the NS3 protein of Broadhaven virus. In connexin45, the P(T/S)APPxY motif is in the putative cytoplasmic domain. Interestingly, connexin45 and other gap junction proteins turn over rapidly, and in the case of connexin43, which also harbors adjacent P(T/S)AP and PPxY motifs in the cytoplasmic domain, the recruitment of a ubiquitin ligase has been implicated in its degradation (33). Intracisternal A-type particles are related to retroviruses but bud into the endoplasmic reticulum rather than at the plasma membrane (34). Broadhaven virus, a member of the Orbivirus genus of Reoviridae, is nonenveloped (35). However, in the case of Bluetongue virus, the best studied of the orbiviruses, it has been shown that viral particles can be released by budding through the plasma membrane, where they transiently acquire a membrane (36). Intriguingly, the nonstructural NS3 protein is required for the release of virus particles by budding (37, 38). Despite low overall sequence homology among the NS3 proteins of different orbiviruses (39), PPxY and P(T/S)AP motifs appear highly conserved (Table 1) and are located in a region that is likely to be cytoplasmic (40). The conservation of these motifs in NS3 raises the possibility that L domains can occur both in structural and in nonstructural viral proteins.
Acknowledgments
We thank Candace Summerford for help with plasmid construction and Dirk Bohmann and Richard Bram for providing reagents. A.C. and G.P. were supported by the Ministero dell'Universita e della Ricerca Scientifica e Tecnologica (MURST), and by grants from ISS-AIDS and CNR Biotechnology. M.A.A. was supported by National Cancer Institute Training Grant T32 CA09141. This work was supported by National Institutes of Health Grants AI29873 and AI28691 (Center for AIDS Research), and by a gift from the G. Harold and Leila Y. Mathers Charitable Foundation.
Abbreviations
- RSV
Rous sarcoma virus
- VLP
virus-like particles
- Mo-MLV
Moloney murine leukemia virus
- VSV
vesicular stomatitis virus
- ENaC
epithelial sodium channel
- HTLV-I
human T cell leukemia virus type 1
- HA
hemagglutinin
- SIVmac
simian immunodeficiency virus from Macaca mulatta
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 12945.
References
- 1.Gottlinger H G, Dorfman T, Sodroski J G, Haseltine W A. Proc Natl Acad Sci USA. 1991;88:3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang M, Orenstein J M, Martin M A, Freed E O. J Virol. 1995;69:6810–6818. doi: 10.1128/jvi.69.11.6810-6818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puffer B A, Parent L J, Wills J W, Montelaro R C. J Virol. 1997;71:6541–6546. doi: 10.1128/jvi.71.9.6541-6546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wills J W, Cameron C E, Wilson C B, Xiang Y, Bennett R P, Leis J. J Virol. 1994;68:6605–6618. doi: 10.1128/jvi.68.10.6605-6618.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiang Y, Cameron C E, Wills J W, Leis J. J Virol. 1996;70:5695–5700. doi: 10.1128/jvi.70.8.5695-5700.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yasuda J, Hunter E. J Virol. 1998;72:4095–4103. doi: 10.1128/jvi.72.5.4095-4103.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan B, Li X, Goff S P. EMBO J. 1999;18:4700–4710. doi: 10.1093/emboj/18.17.4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craven R C, Harty R N, Paragas J, Palese P, Wills J W. J Virol. 1999;73:3359–3365. doi: 10.1128/jvi.73.4.3359-3365.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parent L J, Bennett R P, Craven R C, Nelle T D, Krishna N K, Bowzard J B, Wilson C B, Puffer B A, Montelaro R C, Wills J W. J Virol. 1995;69:5455–5460. doi: 10.1128/jvi.69.9.5455-5460.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garnier L, Wills J W, Verderame M F, Sudol M. Nature (London) 1996;381:744–745. doi: 10.1038/381744a0. [DOI] [PubMed] [Google Scholar]
- 11.Harty R N, Paragas J, Sudol M, Palese P. J Virol. 1999;73:2921–2929. doi: 10.1128/jvi.73.4.2921-2929.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pirozzi G, McConnell S J, Uveges A J, Carter J M, Sparks A B, Kay B K, Fowlkes D M. J Biol Chem. 1997;272:14611–14616. doi: 10.1074/jbc.272.23.14611. [DOI] [PubMed] [Google Scholar]
- 13.Hicke L. Trends Cell Biol. 1999;9:107–112. doi: 10.1016/s0962-8924(98)01491-3. [DOI] [PubMed] [Google Scholar]
- 14.Strous G J, Govers R. J Cell Sci. 1999;112:1417–1423. doi: 10.1242/jcs.112.10.1417. [DOI] [PubMed] [Google Scholar]
- 15.Staub O, Abriel H, Plant P, Ishikawa T, Kanelis V, Saleki R, Horisberger J D, Schild L, Rotin D. Kidney Int. 2000;57:809–815. doi: 10.1046/j.1523-1755.2000.00919.x. [DOI] [PubMed] [Google Scholar]
- 16.Putterman D, Pepinsky R B, Vogt V M. Virology. 1990;176:633–637. doi: 10.1016/0042-6822(90)90035-p. [DOI] [PubMed] [Google Scholar]
- 17.Ott D E, Coren L V, Copeland T D, Kane B P, Johnson D G, Sowder R C, II, Yoshinaka Y, Oroszlan S, Arthur L O, Henderson L E. J Virol. 1998;72:2962–2968. doi: 10.1128/jvi.72.4.2962-2968.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Accola M A, Strack B, Gottlinger H G. J Virol. 2000;74:5395–5402. doi: 10.1128/jvi.74.12.5395-5402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mimnaugh E G, Chen H Y, Davie J R, Celis J E, Neckers L. Biochemistry. 1997;36:14418–14429. doi: 10.1021/bi970998j. [DOI] [PubMed] [Google Scholar]
- 20.Patnaik A, Chau V, Wills J W. Proc Natl Acad Sci USA. 2000;97:13069–13074. doi: 10.1073/pnas.97.24.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schubert U, Ott D E, Chertova E N, Welker R, Tessmer U, Princiotta M, Bennink J R, Kräusslich H-G, Yewdell J W. Proc Natl Acad Sci USA. 2000;97:13057–13062. doi: 10.1073/pnas.97.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh C T, Sodroski J, Gottlinger H G. Nature (London) 1994;372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- 23.Treier M, Staszewski L M, Bohmann D. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- 24.Harvey K F, Kumar S. Trends Cell Biol. 1999;9:166–169. doi: 10.1016/s0962-8924(99)01541-x. [DOI] [PubMed] [Google Scholar]
- 25.Fenteany G, Schreiber S L. J Biol Chem. 1998;273:8545–8548. doi: 10.1074/jbc.273.15.8545. [DOI] [PubMed] [Google Scholar]
- 26.Hochstrasser M. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 27.Puffer B A, Watkins S C, Montelaro R C. J Virol. 1998;72:10218–10221. doi: 10.1128/jvi.72.12.10218-10221.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terrell J, Shih S, Dunn R, Hicke L. Mol Cell. 1998;1:193–202. doi: 10.1016/s1097-2765(00)80020-9. [DOI] [PubMed] [Google Scholar]
- 29.Shih S C, Sloper-Mould K E, Hicke L. EMBO J. 2000;19:187–198. doi: 10.1093/emboj/19.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Govers R, ten Broeke T, van Kerkhof P, Schwartz A L, Strous G J. EMBO J. 1999;18:28–36. doi: 10.1093/emboj/18.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. EMBO J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- 32.Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laing J G, Beyer E C. J Biol Chem. 1995;270:26399–26403. doi: 10.1074/jbc.270.44.26399. [DOI] [PubMed] [Google Scholar]
- 34.Kuff E L, Lueders K K. Adv Cancer Res. 1988;51:183–276. doi: 10.1016/s0065-230x(08)60223-7. [DOI] [PubMed] [Google Scholar]
- 35.Schoehn G, Moss S R, Nuttall P A, Hewat E A. Virology. 1997;235:191–200. doi: 10.1006/viro.1997.8685. [DOI] [PubMed] [Google Scholar]
- 36.Hyatt A D, Eaton B T, Brookes S M. Virology. 1989;173:21–34. doi: 10.1016/0042-6822(89)90218-3. [DOI] [PubMed] [Google Scholar]
- 37.Hyatt A D, Gould A R, Coupar B, Eaton B T. J Gen Virol. 1991;72:2263–2267. doi: 10.1099/0022-1317-72-9-2263. [DOI] [PubMed] [Google Scholar]
- 38.Hyatt A D, Zhao Y, Roy P. Virology. 1993;193:592–603. doi: 10.1006/viro.1993.1167. [DOI] [PubMed] [Google Scholar]
- 39.Moss S R, Jones L D, Nuttall P A. Virology. 1992;187:841–844. doi: 10.1016/0042-6822(92)90491-7. [DOI] [PubMed] [Google Scholar]
- 40.Bansal O B, Stokes A, Bansal A, Bishop D, Roy P. J Virol. 1998;72:3362–3369. doi: 10.1128/jvi.72.4.3362-3369.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]