

Summary
Nucleotide excision repair (NER) is a vital cellular defense system against carcinogen-DNA adducts, which, if not repaired, can initiate cancer development. The structural features of bulky DNA lesions that account for differences in NER efficiencies in mammalian cells are not well understood. In vivo, the predominant DNA adduct derived from metabolically activated benzo[a]pyrene (BP), a prominent environmental carcinogen, is the 10S (+)-trans-anti-[BP]-N2-dG adduct (G*), which resides in the B-DNA minor groove 5′-oriented along the modified strand. We have compared the structural distortions in double-stranded DNA, imposed by this adduct, in the different sequence contexts 5′-…CGG*C…, 5′-…CG*GC…, 5′-…CIG*C… (“I”= 2′-deoxyinosine), and 5′-…CG*C…. Based on electrophoretic mobilities, all duplexes manifest moderate bends except the 5′-…CGG*C…duplex, which exhibits an anomalous, slow mobility attributed to a pronounced flexible kink at the site of the lesion. This kink, resulting from steric hindrance between the 5′-flanking guanine amino group and the BP aromatic rings, both positioned in the minor groove, is abolished in the 5′-…CIG*C…duplex (the 2′-deoxyinosine group, I, lacks this amino group). In contrast, the sequence-isomeric 5′-… CG*GC…duplex exhibits only a moderate bend, but displays a remarkably increased opening rate at the 5′-flanking base pair of G*, indicating a significant destabilization of Watson-Crick hydrogen bonding. The NER dual incision product yields were compared for these different sequences embedded in otherwise identical 135-mer duplexes in cell-free human HeLa extracts. The yields of excision products varied by a factor of as much as ∼4 in the order 5′-…CG*GC… >5′…CGG*C…≥5′…CIG*C… ≥5′-…CG*C…. Overall, destabilized Watson-Crick hydrogen bonding, manifested in the 5′-…CG*GC…duplex, elicits the most significant NER response, while the flexible kink displayed in the sequence-isomeric 5′-…CGG*C…duplex represents a less significant signal in this series of substrates. These results demonstrate that the identical lesion can be repaired with markedly variable efficiencies in different local sequence contexts that differentially alter the structural features of the DNA duplex around the lesion site.
Keywords: Nucleotide excision repair susceptibility, benzo[a]pyrenyl-guanine lesion, structure-function relationship, flexible DNA bend, sequence-dependence, dual incision efficiencies
The global genome nucleotide excision repair (NER) mechanism is a repair pathway in eukaryotes that removes bulky DNA adducts in vivo.1-3 The eukaryotic NER system consists of a set of ∼30 distinct subunits4 that collaborate in the excision of lesion-containing oligonucleotides, 24-32 nucleotide-long, from the damaged strand. One of the key and vital features of the NER machinery is its ability to recognize and excise an astounding variety of bulky DNA lesions. The protein XPC, tightly associated with another protein, HR23B, in vivo5 and in vitro,6-9 plays an initial role in recognizing DNA lesions in cellular environments.7, 10-16 The XPC/HR23B complex initiates the recruitment of other NER factors that are essential for ultimately stimulating the dual incisions of the damaged strand.4, 12, 13, 17 The first among these other factors is the multi-protein transcription factor TFIIH.18-20 The helicases XPB and XPD, components of TFIIH, cause the unwinding of a 20-25 nucleotide patch around the site of the lesion in an ATP-dependent manner.11, 14 The factor XPA then binds to this complex, presumably acting as a wedge to stabilize this “bubble”-like structure, and XPC/HR23B is released. Another factor, RPA, a single-strand binding protein, further stabilizes this nucleoprotein complex. Subsequently the structure-specific endonucleases XPG and XPF/ERCC1 bind to the complex and incise the damaged strand on the 3′- and 5′-sides of the lesion, respectively, thus releasing the 24-32 nucleotide-long fragments that are the hallmark of successful mammalian NER incision activity.11, 13, 18, 21, 22
The rate of repair of chemically and conformationally different lesions by the mammalian NER apparatus varies over several orders of magnitude.2, 21, 23 The structures of the DNA lesions that elicit efficient or inefficient NER repair have been a subject of considerable interest over the years.1-3, 21, 24 It has been suggested that the NER factors do not recognize the lesion itself, but the local distortions in the DNA that are associated with the lesions.7, 13, 18, 23, 25 Further insights into these issues is provided by the X-ray crystallographic structure of a truncated form of Rad4/Rad23 (the S. cerevisiae homologues of XPC and HR23B, respectively) in a complex with an oligonucleotide containing a cyclobutane pyrimidine dimer (CPD) lesion.26 One of the three β-hairpin domains of Rad4 is inserted into the DNA helix, thus separating the CPD lesion from the unmodified strand. The CPD lesion is positioned in a disordered region of the crystal where it has no visible contacts with the protein.26 In contrast, the two thymines in the complementary strand that were positioned opposite the CPD in the duplex, interact with Rad4 amino acid residues. As discussed by Schärer,24, 27 this structure indicates that the interaction of the unmodified complementary strand with the protein plays an important role in lesion recognition, which had been suggested by the experiments of Buterin et al.28 The structure points to the necessity of local strand separation that should be facilitated by lesions causing significant local thermodynamic destabilization of duplex DNA. In turn, this local destabilization is affected by multiple factors such as the interactions of the lesions themselves with the local DNA residues that can cause the weakening of local hydrogen bonding and base stacking interactions. Thus, multiple structural deviations, or a multipartite mechanism, may be involved in the recognition of DNA lesions.29, 30 Other structural perturbations that lead to the recognition of DNA lesions have been considered. These include the recognition of flipped out bases from the complementary strand,28 DNA flexibility around the lesion site,31 and oscillatory motions of the unmodified complementary strand.3, 32, 33 All of these phenomena involve lesion-induced deviations from the normal B-DNA duplex structural parameters.34 Many of these parameters are coupled,35, 36 and the various deviations are therefore not mutually exclusive. Our interests have been directed towards an understanding of the multipartite changes in the DNA structural parameters and how they affect the recognition and subsequent dual incisions that characterize both prokaryotic29, 30, 37-39 and eukaryotic NER.29
In order to elucidate the relationships between NER efficiencies and the conformations of bulky DNA adducts and the DNA distortions they cause, we have been studying lesions derived from the binding of metabolites of polycyclic aromatic hydrocarbons (PAH) to DNA.40 Adducts derived from the binding of the diol epoxide reactive intermediate 7r,8t-dihydroxy-t9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene (anti-BPDE), a metabolite of the cancer-causing compound benzo[a]pyrene,41, 42 are excellent substrates for probing the features of DNA lesions that are recognized by the NER apparatus. The most abundant stable adduct derived from the reaction of BPDE with DNA in mammalian cells43, 44 is the 10S (+)-trans-anti-[BP]-N2-dG adduct (Figure 1a). However, the reactions of BPDE with DNA also yield other, stereoisomeric adducts to dG and dA, with varying conformations and differences in structural perturbations of double-stranded DNA.45 The susceptibilities of the different PAH-DNA adducts to NER depend on adduct stereochemistry46, 47 and PAH topology.48 The [BP]-DNA adducts, unless removed by DNA repair mechanisms,49 are highly mutagenic.50, 51
Figure 1.
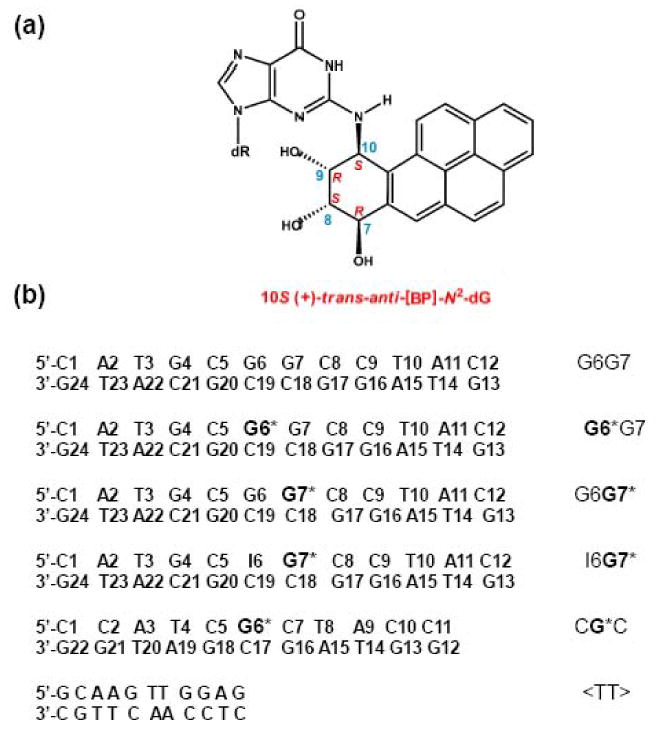
Structure and stereochemical properties of the 10S (+)-trans-anti-[BP]-N2-dG adduct (top). The base sequence contexts of the different duplexes studied and their abbreviations are defined here where G* = 10S (+)-trans-anti-[BP]-N2-dG. The NMR properties52, 58, 70 and the gel electrophoresis results (Figure 2) were obtained with the duplexes shown. The NER studies were conducted with the same duplexes embedded in 135-mer duplexes that otherwise had the same base sequence (Supporting Information).
We have shown recently by NMR methods that the identical 10S (+)-trans-anti-[BP]-N2-dG adduct (G*, Figure 1a) positioned either at G6 or at G7 in the G6G7 duplex (Figure 1b) exhibits remarkably different, base sequence-dependent structural characteristics.52 The G6G7 dinucleotide sequence was initially selected for study because it constitutes a mutational hotspot in E. coli53, 54 and in mammalian cells.50 The bulky aromatic residue in G* adopts a minor groove adduct conformation in both the G6G7* and the G6*G7 12-mer duplexes. However, the characteristics of these two sequence-isomeric duplexes differ in significant aspects. Specifically, in the G6*G7 case, the duplex exhibits a moderate bend, and the flanking C5:G20 base pair is destabilized at ambient temperatures, while all other base pairs flanking the G6*:C19 base pairs remain intact.52 In contrast, the G6G7* 12-mer duplex manifests a pronounced flexible bend or kink at the site of the lesion, but all base pairs flanking the G7*:C18 pair are intact.52 Since these duplexes are identical except for the positions of the modified guanine residues, we wished to determine whether the NER dual incision efficiencies are affected by these physical differences.
The relative NER efficiencies were determined by incubating the G6*G7 and the G6G7* 12-mer duplexes (Figure 1b) embedded in otherwise identical 135-mer duplex sequences, in cell-free extracts from human HeLa cells, as described previously.46, 47 The yields of dual incision products were also investigated using the G6G7* duplex analog I6G7*. In the latter, G6 in the G6G7* duplex was replaced by inosine (I6)‡ and is of interest because the I6G7* duplex exhibits a moderate bend, rather than the pronounced kink/bend exhibited by the G6G7* duplex. Finally, the new NER results reported here are compared to the dual incision yields obtained with the CPD and 6-4<TT> photodimers,7, 13, 22, 55-57 and the same minor groove 10S (+)-trans-anti-[BP]-N2-dG adduct58 in the CG*C sequence context (Figure 1b) reported earlier.46, 47
An unusual flexible kink in the G6G7* duplex
The impact of the 10S (+)-trans-anti-[BP]-N2-dG adduct on the three-dimensional characteristics of the modified oligonucleotide duplexes can be assessed by comparing the electrophoretic mobilities of the BP-modified and unmodified duplexes in native (non-denaturing) polyacrylamide gels.59-62 The G6*G7, G6G7* and I6G7* duplexes (Figure 1b) exhibit extraordinary differences in electrophoretic mobilities (Figure 2). The mobility of the 12-mer G6*G7 duplex is about 10% lower than that of the unmodified G6G7 duplex (Figure 2, lanes 3 and 4, respectively). These results are comparable to the mobility of the CG*C duplex (∼ 5% lower mobility) and with G* in other sequence contexts.59-62 Remarkably, the mobility of the G6G7* duplex is lowered by 42% (Figure 2, lanes 2 and 4, respectively). This result indicates that the lesion at G7* induces an usually strong bend 59 or kink52 in the G6G7* sequence context. Interestingly, the anomalous slow electrophoretic mobility of the G6G7* duplex is abolished by substituting the guanine base G6 by inosine. The mobility of the I6G7* duplex is identical to the mobility of the G6*G7 duplex, suggesting that the overall three-dimensional shapes of the G6*G7 and I6G7* duplexes are similar. Since inosine is identical to guanine except that it lacks the latter's exocyclic amino group, these results indicate that the exocyclic amino group of guanine at G6 plays a dominant role in causing the prominent kink in the G6G7* duplex.52, 63
Figure 2.
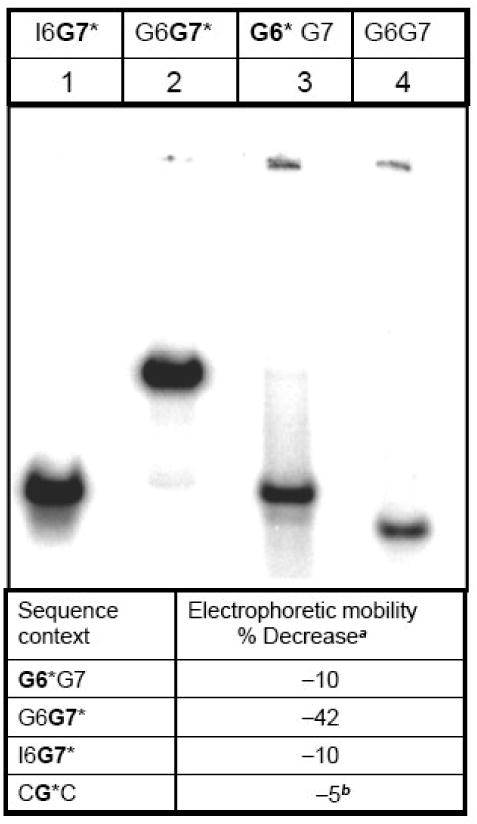
Relative electrophoretic mobilities of unmodified and the 10S (+)-trans-anti-[BP]-N2-dG adduct-modified oligonucleotide duplexes defined in Figure 1 with their natural complementary strands in native 8% polyacrylamide gels at room temperature. The faint bands at the top of the gel represent the wells. Table: % decrease in electrophoretic mobilitya: (Mobility of the modified duplex – mobility of the unmodified duplex) × 100 / mobility of the unmodified duplex. bData from Xu et al.66
The difference between rigid, directed bends and flexible, hinge-like joints can be established by examining the relationships between the electrophoretic mobility patterns of self-ligation products and the phasing of the bends with respect to the helical repeat64 (10.3-10.6 base pairs65 per helical turn), as discussed by Hagerman.64 These techniques were previously employed to demonstrate that the prominent kink induced by the 10S (+)-trans-anti-[BP]-N2-dG adduct in the G6G7* duplex is highly flexible.66 In contrast, for the same 10S (+)-trans-anti-[BP]-N2-dG adduct in the G6*G7 and I6G7* duplexes, the bending is moderate (Figure 2) and the bends are rigid rather than flexible, as shown earlier in our laboratory.66 Furthermore, when G* is flanked by cytosine on both sides as in the CG*C sequence context, the bend is also rigid, as shown by phasing methods60, 62 In the case of the stereoisomerically different 10R (+)-cis-anti-[BP]-N2-dG adduct in the CG*C duplex, which adopts a base-displaced/intercalated conformation (Figure S1, Supporting Information),67 the extent of bending is smaller than in the case of the (+)-trans-adduct in the same sequence context.68
Relative NER Efficiencies
The relative efficiencies of dual incisions of the modified strand of G6*G7, G6G7*, I6G7* and CG*C 135-mer duplexes containing identical single 10S (+)-trans-anti-[BP]-N2-dG adducts were compared in cell-free HeLa cell extracts (details in Supporting Information). Briefly, the NER activities were assessed using denaturing gel electrophoresis to resolve the characteristic 24-32 oligonucleotide dual incision products. Phosphorimager methods were then employed to evaluate the yields of these products (radioactivity in the 24-32 band region divided by the total radioactivity in the same lane). These yields varied in different cell extracts (2-6%), and this variability was accounted for by including experiments with a standard stereoisomeric 10R (+)-cis-anti-[BP]-N2-dG adduct in the CG*C sequence context, in each set of experiments. Thus, in experiments conducted with different cell extract preparations, the dual incision efficiencies for the 10S (+)-trans adduct in different sequence contexts were normalized with respect to the 10R (+)-cis positive control standard. The 10R (+)-cis-adduct, with its base-displaced/intercalated conformation 67, is removed ∼5 times more efficiently than the minor groove 10S (+)-trans-adduct 58 in the same CG*C sequence context in HeLa cell extracts, and by a factor of ∼2.5 by a set of reconstituted and purified NER factors.47 We compared the time dependence of NER dual incision efficiencies of the 10S (+)-trans-anti-[BP]-N2-dG adduct in the CG*C duplex with those characterizing the extensively studied cis-syn cyclobutane pyrimidine dimer (CPD) and the 6-4 UV photodimer under identical conditions.13, 22, 55, 57, 69 Typical densitometry traces are shown (Figure 3a) and were used for the quantitative evaluation of relative NER efficiencies. The time dependence is linear up to 40 minute incubation time (Figure 3b). The relative rates decreased in the order (6-4 <TT>) ∼ 10R (+)-cis-anti-[BP]-N2-dG > 10S (+)-trans-anti-[BP]-N2-dG > CPD <TT>. The [6-4 <TT>]/[CPD <TT>] ratio of dual incision rates is ∼ 19. This compares with ratios of ≥ 10 also found in human cell extracts in vitro57 and in vivo in UVB/UVA-irradiated human skin,56 and a ratio of ∼ 8 observed in Cho cell extracts.22
Figure 3.
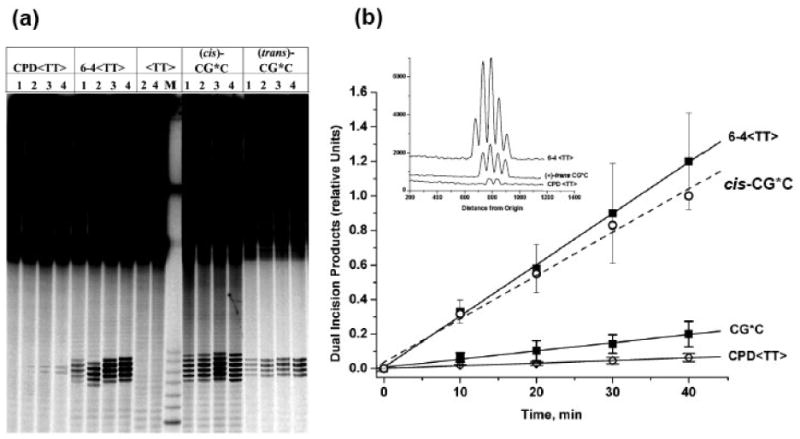
(a) Comparison of nucleotide excision repair results in HeLa cell extracts for 135-mer duplexes containing the UV photodimer CPD <TT> or 6-4 <TT> duplexes, the 10S (+)-trans-anti-[BP]-N2-dG (CG*C), or the stereoisomeric (+)-cis adducts in the same CG*C sequence context (cis-CG*C). The (+)-cis-duplex was used as a reference standard in each set of experiments to adjust for variabilities in the NER activities of cell extracts prepared at different times (see text for details). The overall radioactivity levels in each lane were comparable, to allow for a visual inspection of the differences in dual incision efficiencies. Lanes 1, 2, 3, and 4 represent incubation times of 10, 20, 30, and 40 minutes respectively. The lane marked <TT> represents parallel incubations with unmodified <TT> duplexes, while lane M contains unmodified oligonucleotide markers 32, 30, 28, 26, and 24 nucleotides in length. In this experiment, the maximum extent of NER incisions (observed in the case of the 10R (+)-cis-anti-[BP]-N2-dG adduct in the cis-CG*C duplex at 40 min), was 4.6%. (b) Time dependence of formation of dual incision products. The average values and standard deviations are based on five independent experiments in the case of the two UV photodimers, and eight experiments in the case of the 10S (+)-trans-anti-[BP]-N2-dG adduct in the CG*C sequence. The straight lines are the least square fits to the data points, and the relative values of the slopes and the standard errors are summarized in Table S1, Supporting Information. In all these independent experiments, the data points for the the CPD <TT>, 6-4 <TT>, and CG*C (10S (+)-trans-anti-[BP]-N2-dG adduct) were normalized to the 40 min value of the 10R (+)-cis-anti-[BP]-N2-dG adduct-containing CG*C duplex (open circles) obtained in the same experiment. The inset in Figure 3b shows the densitometric tracings of the 40 min lanes shown in the gel (panel (a)).
Sequence-context dependent differential excision
The excision efficiencies of the CG*C, G6*G7, and G6G7* sequences embedded in otherwise identical 135-mer duplexes are compared in Figure 4a. The same 10S (+)-trans-anti-[BP]-N2-dG adduct embedded in different sequence contexts is excised with different efficiencies in the order G6*G7 > G6G7* > CG*C (Figure 4). Both XPC, a critical component of the global genome NER apparatus, and XPA, are essential to the NER activities shown. In XPC-deficient, or XPA-deficient cell lines, the NER dual incision efficiencies are reduced by 90% or more, but are restored in mixtures of cell extracts from XPA and XPC cells, as expected if an NER mechanism is operative (Figure S2, Supporting Information).
Figure 4.

Comparison of dual incisions elicited by the 10S (+)-trans-anti-[BP]-N2-dG adduct in CG*C, G6G7* (abbreviated as G7*), and G6*G7 (abbreviated as G7*) 135-mer duplexes in HeLa cell extracts after an incubation time of 40 min. Control samples: G6G7, unmodified duplex without or after treatment with cell extracts: CG*C, G6G7*, and G6*G7: untreated controls. The overall radioactivity levels in each lane were comparable, to allow for a visual inspection of the differences in dual incision efficiencies. Densitometry tracings of the lanes in panel comparing dual incision efficiencies of the same 10S (+)-trans-anti-[BP]-N2-dG adduct embedded in the different sequence contexts are shown in Figure S4, Supporting Information. The relative NER efficiency of the (+)-cis-anti-[BP]-N2-dG adduct used as a standard (See text) is also shown.
In order to compare the initial rates of excision of these sequences, and to take into account possible variations obtained with different cell extracts, we examined the time course of incisions in the CG*C, G6*G7, G6G7* and I6G7* 135-mer duplexes. A representative example of a gel is shown in Figure 5a. Results from multiple experiments with different cell extracts and substrate samples are summarized in Figure 5b, together with the standard deviations of each set of time-dependent data points. The key feature of the findings (Table S1, Supporting Information) is that the NER efficiencies of the identical 10S (+)-trans-anti-[BP]-N2-dG lesions are markedly dependent on the bases flanking the lesion, spanning a factor of as much as ∼4 (G6*G7 vs. CG*C duplexes). Particularly remarkable are the differences between the sequence isomers G6*G7 and G6G7*, the G6*G7/G6G7* ratios of dual incision efficiencies being 2.4 ± 0.3.
Figure 5.
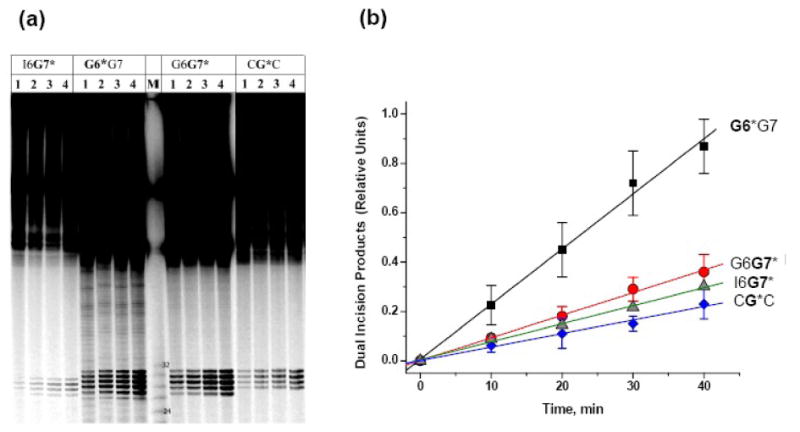
(a) Denaturing gel showing the appearance of dual excisions elicited by the 10S (+)-trans-anti-[BP]-N2-dG adducts in the I6G7*, G6*G7, G6G7* and CG*C duplexes as a function of incubation time in HeLa cell extracts. Lanes 1, 2, 3, and 4 represent incubation times of 10, 20, 30, and 40 min, respectively. Representative densitometry tracings, adjusted for the total radioactivity in each lane to compensate for loading factors and differences in the radioactivities of the samples (the concentration of the I6G7* was lower than in the case of the other samples). (b) Time course of dual incision product formation elicited by the 10S (+)-trans-anti-[BP]-N2-dG adduct in the G6*G7, G6G7*, I6G7* and CG*C duplexes. The experimental points are averages of seven independent experiments, and the error bars represent the standard deviations. The error bars for the I6G7* data points are omitted for clarity, but they do overlap with the error bars of the G6G7* and CG*C data points. The straight lines are the least square fits to the data points, and the relative values of the slopes and the standard error are summarized in Table S1, Supporting Information. In all these experiments, the data points were normalized to unity based on the 40 min value of the 10R (+)-cis-anti-[BP]-N2-dG in the CG*C duplex obtained in each individual experiment with a given cell extract (not shown). The lanes for the I6G7* duplex are uniformly lighter (panel a, left) than in the case of the other sequences; this was due to the lower quantities of I6G7* sample available. However, this difference is not reflected in panel b, since these data represent ratios of [dual excision products]/[total DNA substrates]) (see text).
Differences in conformations dictated by base sequence context
The NMR solution structures of the 10S (+)-trans-anti-[BP]-N2-dG adduct embedded in the G6*G7,52 G6G7*,52 I6G7*,70 and CG*C58 duplexes have been investigated and the results show that there are subtle sequence-dependent conformational differences among double-stranded DNA duplexes. In all cases the bulky BP aromatic ring system is positioned in the minor groove, pointing in the 5′-direction relative to the modified guanine base. However, only the G6*G7 duplex manifests a remarkable destabilization of the base pair flanking the lesion on its 5′-side in a temperature range much lower than the global melting temperature of the modified duplex. Specifically, the overall thermal stabilities of the G6*G7 and G6G7* 12-mer duplexes (Tm = 55 ± 1 °C and 57 ± 1 °C, respectively) are not very different from one another.52 But the temperature-dependence of the line widths and chemical shifts of the G20 imino proton NMR resonances in the G6*G7 duplex suggest that the C5:G20 base pair is destabilized within the temperature interval of 18-30 °C. Furthermore, analysis of the Molecular Dynamics (MD) – refined structure based on the NMR data shows that the G6G7* duplex shows a unique local untwisting accompanied by an enlarged Roll52 (Roll is the angle between two neighboring base pair planes and produces bending into the major or minor groove.71 See also Figure S5, Supporting information). Enlarged Roll coupled with untwisting is known to be largely responsible for DNA bending or kink formation35, 36, 72 There are also changes in local base-base stacking interactions, but without a strong impact on Watson-Crick pairing except for a modest perturbation at the lesion site itself.63 The properties of the I6G7* duplex were also studied by NMR methods.70 Multiple conformations were found, although two major conformers with the BP residues positioned in the minor groove and oriented towards the 5′-direction of the modified strand could be identified.70 The CG*C and G6G7* duplexes exhibit only a single conformer and all neighboring Watson-Crick base pairs remain intact.52, 58
Extensive structural analyses,63 utilizing MD simulation methods, of the 10S (+)-trans-anti-[BP]-N2-dG adduct in the duplexes investigated here have provided further molecular insights into the origins of the experimentally observed sequence-dependent structural properties.63 The critical elements that produce differences in the structural perturbations are the variable steric hindrance effects from the guanine amino groups that are positioned in the vicinity of the bulky aromatic BP ring system. Both compete for accommodation in the duplex minor groove. For the G6*G7 duplex, steric hindrance from the guanine amino group of G7 causes the episodic rupturing of the Watson-Crick hydrogen bonds of the C5:G20 base pair 5′-flanking the lesion. The G7 guanine amino group crowds the BP benzylic ring on the 3′-side of the lesion, causing the BP moiety to rotate about the linkage site; as a result, the BP distal aromatic ring system episodically intrudes into the hydrogen-bonded region of the C5:G20 base pair, flanking the lesion on its 5′-side (Figure S6, Supporting Information). Furthermore, in the G6G7* duplex, the anomalously slow electrophoretic mobility associated with the flexible kink, manifested in our MD simulation,63 as a dynamic untwisting and enlarged Roll at the C5:G20/G6:C19 base pair step, is caused by steric hindrance between the G6 guanine amino group and the BP aromatic ring system (Figure S6, Supporting Information). This role of the G6 guanine amino group was also substantiated by MD studies of the dynamics of the I6G7* duplex63 which does not exhibit untwisting or enlarged Roll. In contrast, in the CG*C sequence, the amino groups of the guanines G18 and G16 in the C5:G18 and C7:G16 base pairs, respectively, flanking the lesion, tend to place the BP moiety in a more confined region of the minor groove, thus causing the bending to be rigid.
Structural DNA distortions/destabilizations recognized by the human NER apparatus
The highest NER efficiency in this series of minor groove-aligned, stereochemically identical 10S (+)-trans-anti-[BP]-N2-dG adduct in the different sequence contexts considered is observed in the G6*G7 duplex; in this case, the efficiency is ∼2.4 times greater than in the sequence-isomeric G6G7* duplex and ∼4.1 times greater than in the CG*C sequence context (Figure 6 and Table S1, Supporting Information). In terms of structural distortions, only the G6G7* duplex displays a significant destabilization and episodic denaturation of Watson-Crick hydrogen bonding at the 5′-side base pair flanking the lesion modification site; this is correlated with the highest NER efficiency in this series of BP-modified duplexes. In contrast, only the G6G7* duplex has the hallmarks of a flexible kink or bend,66 manifested in the anomalous electrophoretic mobility observed in Figure 2 (lane 2); this is correlated with the second highest repair efficiency in this series of substrates. Therefore, in this set of minor-groove aligned sequence-isomeric BP-modified guanine lesions, the weakening of Watson-Crick base pairing seems to be a more effective factor for eliciting mammalian NER than a flexible, sharply bent or kinked duplex. The I6G7* duplex lacks this flexible kink, but its repair efficiency is similar to that of the sharply bent G6G7* duplex substrate with the flexible kink. However, replacing G6 by I6 changes other, local interactions that could affect NER, since the conformation of the I6G7* duplex is conformationally heterogeneous and displays at least two different minor groove conformations.70 By contrast the G6G7* duplex has only one predominant conformation.52 The conformational heterogeneity of I6G7* is attributed to a weaker I6:C19 base pair, with only two hydrogen bonds flanking the lesion on the 5′-side. Furthermore, MD studies63 show that this I6:C19 base pair has one of the two hydrogen bonds episodically ruptured, which could account for its moderate repair susceptibility.63 The least structurally distorted CG*C duplex, without disrupted Watson-Crick hydrogen bonding or flexible kink, exhibits the lowest susceptibility to NER in this series. The repair in this case seems to be related to its enlarged minor groove,47 and modestly perturbed Watson-Crick hydrogen bonding at the lesion site;63 these two features are common to all of the substrates we considered here. These structure/function correlations are summarized in Figure 6 and Table S1, Supporting Information.
Figure 6.
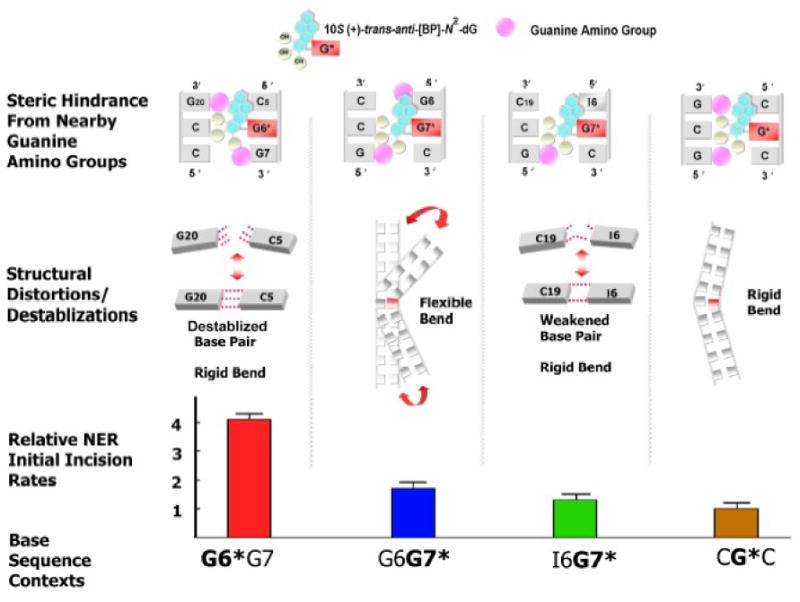
Summary and comparisons of structural effects on relative NER efficiencies. Differential steric hindrance by nearby guanine amino groups governs sequence-dependent distortions and duplex destabilization63,70 that influence the DNA sequence-governed NER dual incision efficiencies. In the series of minor groove lesion-containing duplexes studied here, the strongest signal that elicits the NER dual incision response is a disrupted Watson-Crick base pair in the G6*G7 duplex. The flexible kink in G6G7* appears to be less important since the bends are more rigid in I6G7* and the other two duplexes studied (see text). Minor groove enlargement47,63 together with modest perturbation of Watson-Crick base pairing at the lesion site63 is common to all DNA sequences examined here.
The DNA duplex with the stereoisomerically different 10R (+)-cis-anti-[BP]-N2-dG adduct, with base-displaced/intercalated conformation is excised with similar efficiencies as the 6-4 <TT> dimer, and about ∼5 times better than the stereoisomeric 10S (+)-trans-anti-[BP]-N2-dG adduct in the CG*C sequence context.46, 47 The large distortions associated with this conformation, namely, the prominently flipped out base pair,28 with completely ruptured Watson-Crick hydrogen bonding as well as the enlarged minor groove,47 can provide the significant recognition signals in the family of base-displaced/intercalated [BP]-N2-dG adducts; this also fits well with the hierarchy of NER susceptibilities for the cases investigated here.
Conclusions
Analyses of the structural properties of the series of minor groove 10S (+)-trans-anti-[BP]-N2-dG adducts positioned in different sequence contexts of double-stranded DNA by NMR methods, electrophoretic mobilities and MD simulations,63 have provided new insights into the structural distortions/destabilizations that elicit and modulate the efficiencies of the initial steps in NER (Figure 6). In the case of the [BP]-N2-dG adduct studied in this work, the episodic denaturation of Watson-Crick hydrogen bonding in the base pair flanking the lesion on the 5′-side provides a stronger recognition signal than a flexible kink/bend. The origins of these structural differences stem from steric hindrance between the exocyclic amino groups of unmodified guanines in the vicinity of the bulky [BP]-N2-dG adducts in the minor groove. The impact of these exocyclic amino groups on the physical properties of the duplexes depends on the positions of the unmodified guanines in the duplexes. Our results are consistent with the finding that in this family of bulky lesions, differences in the local structural distortions and destabilizations caused by the adducts are correlated with local helix opening patterns caused by the XPC/HR23B binding and their differential dual incision efficiencies.47 Depending on the sequence context, the lesion-containing duplexes manifest different structural distortions/destabilizations63. In turn, strand separation, base flipping, and β-hairpin insertion by the Rad426 or XPC recognition factors may depend on the local base sequence context. Future detailed studies of the structural properties of different PAH-derived lesions with different conformations in selected base sequence contexts, should provide further clues to the mechanisms underlying the recognition and excision of bulky DNA lesions by the mammalian NER apparatus, and their mutagenic and tumorigenic properties.
Acknowledgments
This work was supported by NIH Grant CA-099194 (N.E.G.) and by Grant CA-28038 (S.B.). Partial support for computational infrastructure and systems management was also provided by CA75449 (S.B.). Support for this work to D.J.P. was provided by CA-046533. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Abbreviations
- BP
benzo[a]pyrene
- (+)-anti-BPDE
(benzo[a]pyrene diol epoxide):(+)-(7R,8S,9S,10R)-7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene
- dG
2′-deoxyguanosine
- NER
nucleotide excision repair
- CPD
cyclobutane pyrimidine dimer
Footnotes
Formally, inosine is the nucleoside, while hypoxanthine is the correct name for the corresponding base. For simplicity we utilize the term inosine only
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reardon JT, Sancar A. Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol. 2005;79:183–235. doi: 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- 2.Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106:253–76. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 3.Maillard O, Camenisch U, Blagoev KB, Naegeli H. Versatile protection from mutagenic DNA lesions conferred by bipartite recognition in nucleotide excision repair. Mutation Res. 2008;658:271–286. doi: 10.1016/j.mrrev.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–68. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 5.van der Spek PJ, Eker A, Rademakers S, Visser C, Sugasawa K, Masutani C, Hanaoka F, Bootsma D, Hoeijmakers JH. XPC and human homologs of RAD23: intracellular localization and relationship to other nucleotide excision repair complexes. Nucleic Acids Res. 1996;24:2551–9. doi: 10.1093/nar/24.13.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugasawa K, Ng JM, Masutani C, Maekawa T, Uchida A, van der Spek PJ, Eker AP, Rademakers S, Visser C, Aboussekhra A, Wood RD, Hanaoka F, Bootsma D, Hoeijmakers JH. Two human homologs of Rad23 are functionally interchangeable in complex formation and stimulation of XPC repair activity. Mol Cell Biol. 1997;17:6924–31. doi: 10.1128/mcb.17.12.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugasawa K, Shimizu Y, Iwai S, Hanaoka F. A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair (Amst) 2002;1:95–107. doi: 10.1016/s1568-7864(01)00008-8. [DOI] [PubMed] [Google Scholar]
- 8.Kusumoto R, Masutani C, Sugasawa K, Iwai S, Araki M, Uchida A, Mizukoshi T, Hanaoka F. Diversity of the damage recognition step in the global genomic nucleotide excision repair in vitro. Mutat Res. 2001;485:219–27. doi: 10.1016/s0921-8777(00)00082-3. [DOI] [PubMed] [Google Scholar]
- 9.Araki M, Masutani C, Takemura M, Uchida A, Sugasawa K, Kondoh J, Ohkuma Y, Hanaoka F. Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J Biol Chem. 2001;276:18665–72. doi: 10.1074/jbc.M100855200. [DOI] [PubMed] [Google Scholar]
- 10.Batty D, Rapic'-Otrin V, Levine AS, Wood RD. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J Mol Biol. 2000;300:275–90. doi: 10.1006/jmbi.2000.3857. [DOI] [PubMed] [Google Scholar]
- 11.Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 2003;22:5293–303. doi: 10.1093/emboj/cdg489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, Hanaoka F, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2:223–32. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 13.Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15:507–21. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tapias A, Auriol J, Forget D, Enzlin JH, Scharer OD, Coin F, Coulombe B, Egly JM. Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J Biol Chem. 2004;279:19074–83. doi: 10.1074/jbc.M312611200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thoma BS, Vasquez KM. Critical DNA damage recognition functions of XPC-hHR23B and XPA-RPA in nucleotide excision repair. Mol Carcinog. 2003;38:1–13. doi: 10.1002/mc.10143. [DOI] [PubMed] [Google Scholar]
- 16.Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, van Zeeland AA, Mullenders LH. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–24. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 17.Wakasugi M, Sancar A. Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc Natl Acad Sci U S A. 1998;95:6669–74. doi: 10.1073/pnas.95.12.6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997;16:6559–73. doi: 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uchida A, Sugasawa K, Masutani C, Dohmae N, Araki M, Yokoi M, Ohkuma Y, Hanaoka F. The carboxy-terminal domain of the XPC protein plays a crucial role in nucleotide excision repair through interactions with transcription factor IIH. DNA Repair (Amst) 2002;1:449–61. doi: 10.1016/s1568-7864(02)00031-9. [DOI] [PubMed] [Google Scholar]
- 20.Yokoi M, Masutani C, Maekawa T, Sugasawa K, Ohkuma Y, Hanaoka F. The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J Biol Chem. 2000;275:9870–5. doi: 10.1074/jbc.275.13.9870. [DOI] [PubMed] [Google Scholar]
- 21.Gunz D, Hess MT, Naegeli H. Recognition of DNA adducts by human nucleotide excision repair. Evidence for a thermodynamic probing mechanism. J Biol Chem. 1996;271:25089–98. doi: 10.1074/jbc.271.41.25089. [DOI] [PubMed] [Google Scholar]
- 22.Reardon JT, Sancar A. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 2003;17:2539–51. doi: 10.1101/gad.1131003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood RD. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie. 1999;81:39–44. doi: 10.1016/s0300-9084(99)80036-4. [DOI] [PubMed] [Google Scholar]
- 24.Scharer O. A molecular basis for damage recognition in eukaryotic nucleotide excision repair. ChemBioChem. 2008;9:21–23. doi: 10.1002/cbic.200700619. [DOI] [PubMed] [Google Scholar]
- 25.Fujiwara Y, Masutani C, Mizukoshi T, Kondo J, Hanaoka F, Iwai S. Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J Biol Chem. 1999;274:20027–33. doi: 10.1074/jbc.274.28.20027. [DOI] [PubMed] [Google Scholar]
- 26.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–5. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 27.Scharer OD. Achieving broad substrate specificity in damage recognition by binding accessible nondamaged DNA. Mol Cell. 2007;28:184–6. doi: 10.1016/j.molcel.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Buterin T, Meyer C, Giese B, Naegeli H. DNA quality control by conformational readout on the undamaged strand of the double helix. Chem Biol. 2005;12:913–22. doi: 10.1016/j.chembiol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Geacintov NE, Naegeli H, Patel DJ, Broyde S. Structural aspects of polycyclic aromatic carcinogen-damaged DNA and its recognition by NER proteins. In: Siede W, Kow YW, Doetsch PW, editors. DNA Damage and Recognition. Taylor and Francis; London: 2006. pp. 263–296. [Google Scholar]
- 30.Geacintov NE, Broyde S, Buterin T, Naegeli H, Wu M, Yan S, Patel DJ. Thermodynamic and structural factors in the removal of bulky DNA adducts by the nucleotide excision repair machinery. Biopolymers. 2002;65:202–10. doi: 10.1002/bip.10239. [DOI] [PubMed] [Google Scholar]
- 31.Isaacs RJ, Spielmann HP. A model for initial DNA lesion recognition by NER and MMR based on local conformational flexibility. DNA Repair (Amst) 2004;3:455–64. doi: 10.1016/j.dnarep.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Blagoev KB, Alexandrov BS, Goodwin EH, Bishop AR. Ultra-violet light induced changes in DNA dynamics may enhance TT-dimer recognition. DNA Repair (Amst) 2006;5:863–7. doi: 10.1016/j.dnarep.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Maillard O, Camenisch U, Clement FC, Blagoev KB, Naegeli H. DNA repair triggered by sensors of helical dynamics. Trends Biochem Sci. 2007;32:494–9. doi: 10.1016/j.tibs.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–21. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorin AA, Zhurkin VB, Olson WK. B-DNA twisting correlates with base-pair morphology. J Mol Biol. 1995;247:34–48. doi: 10.1006/jmbi.1994.0120. [DOI] [PubMed] [Google Scholar]
- 36.Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci U S A. 1998;95:11163–8. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai Y, Patel DJ, Geacintov NE, Broyde S. Dynamics of a benzo[a]pyrene-derived guanine DNA lesion in TGT and CGC sequence contexts: enhanced mobility in TGT explains conformational heterogeneity, flexible bending, and greater susceptibility to nucleotide excision repair. J Mol Biol. 2007;374:292–305. doi: 10.1016/j.jmb.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruan Q, Liu T, Kolbanovskiy A, Liu Y, Ren J, Skorvaga M, Zou Y, Lader J, Malkani B, Amin S, Van Houten B, Geacintov NE. Sequence context- and temperature-dependent nucleotide excision repair of a benzo[a]pyrene diol epoxide-guanine DNA adduct catalyzed by thermophilic UvrABC proteins. Biochemistry. 2007;46:7006–15. doi: 10.1021/bi700294k. [DOI] [PubMed] [Google Scholar]
- 39.Zou Y, Liu TM, Geacintov NE, Van Houten B. Interaction of the UvrABC nuclease system with a DNA duplex containing a single stereoisomer of dG-(+)- or dG-(-)-anti-BPDE. Biochemistry. 1995;34:13582–93. doi: 10.1021/bi00041a038. [DOI] [PubMed] [Google Scholar]
- 40.Naegeli H, Geacintov NE. In: Mechanisms of repair of PAH-induced DNA damage The Carcinogenic Effects of Polycyclic Aromatic Hydrocarbons. Luch A, editor. London: World Scientific Press, The Imperial College of London; 2005. pp. 211–258. [Google Scholar]
- 41.Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res. 1982;42:4875–917. [PubMed] [Google Scholar]
- 42.Luch A. Nature and nurture - lessons from chemical carcinogenesis. Nat Rev Cancer. 2005;5:113–25. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- 43.Koreeda M, Moore PD, Wislocki PG, Levin W, Yagi H, Jerina DM. Binding of benzo[a]pyrene 7,8-diol-9,10-epoxides to DNA, RNA, and protein of mouse skin occurs with high stereoselectivity. Science. 1978;199:778–81. doi: 10.1126/science.622566. [DOI] [PubMed] [Google Scholar]
- 44.Weinstein IB, Jeffrey AM, Jennette KW, Blobstein SH, Harvey RG, Harris C, Autrup H, Kasai H, Nakanishi K. Benzo[a]pyrene diol epoxides as intermediates in nucleic acid binding in vitro and in vivo. Science. 1976;193:592–5. doi: 10.1126/science.959820. [DOI] [PubMed] [Google Scholar]
- 45.Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ. NMR solution structures of stereoisometric covalent polycyclic aromatic carcinogen-DNA adduct: principles, patterns, and diversity. Chem Res Toxicol. 1997;10:111–46. doi: 10.1021/tx9601418. [DOI] [PubMed] [Google Scholar]
- 46.Hess MT, Gunz D, Luneva N, Geacintov NE, Naegeli H. Base pair conformation-dependent excision of benzo[a]pyrene diol epoxide-guanine adducts by human nucleotide excision repair enzymes. Mol Cell Biol. 1997;17:7069–76. doi: 10.1128/mcb.17.12.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mocquet V, Kropachev K, Kolbanovskiy M, Kolbanovskiy A, Tapias A, Cai Y, Broyde S, Geacintov NE, Egly JM. The human DNA repair factor XPC-HR23B distinguishes stereoisomeric benzo[a]pyrenyl-DNA lesions. EMBO J. 2007;26:2923–32. doi: 10.1038/sj.emboj.7601730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buterin T, Hess MT, Luneva N, Geacintov NE, Amin S, Kroth H, Seidel A, Naegeli H. Lack of enzymatic repair of fjord polycyclic aromatic hydrocarbon-DNA adducts in ras codon 61 mutational hotspots. Cancer Res. 2000;60:1849–1856. [PubMed] [Google Scholar]
- 49.Wei D, Maher VM, McCormick JJ. Site-specific rates of excision repair of benzo[a]pyrene diol epoxide adducts in the hypoxanthine phosphoribosyltransferase gene of human fibroblasts: correlation with mutation spectra. Proc Natl Acad Sci U S A. 1995;92:2204–8. doi: 10.1073/pnas.92.6.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandes A, Liu T, Amin S, Geacintov NE, Grollman AP, Moriya M. Mutagenic potential of stereoisomeric bay region (+)- and (−)-cis-anti-benzo[a]pyrene diol epoxide-N2-2′-deoxyguanosine adducts in Escherichia coli and simian kidney cells. Biochemistry. 1998;37:10164–72. doi: 10.1021/bi980401f. [DOI] [PubMed] [Google Scholar]
- 51.Seo KY, Nagalingam A, Tiffany M, Loechler EL. Mutagenesis studies with four stereoisomeric N2-dG benzo[a]pyrene adducts in the identical 5′-CGC sequence used in NMR studies: G->T mutations dominate in each case. Mutagenesis. 2005;20:441–8. doi: 10.1093/mutage/gei061. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez FA, Cai Y, Lin C, Tang Y, Kolbanovskiy A, Amin S, Patel DJ, Broyde S, Geacintov NE. Exocyclic amino groups of flanking guanines govern sequence-dependent adduct conformations and local structural distortions for minor groove-aligned benzo[a]pyrenyl-guanine lesions in a GG mutation hotspot context. Nucleic Acids Res. 2007;35:1555–68. doi: 10.1093/nar/gkm022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez H, Loechler EL. Mutational specificity of the (+)-anti-diol epoxide of benzo[a]pyrene in a supF gene of an Escherichia coli plasmid: DNA sequence context influences hotspots, mutagenic specificity and the extent of SOS enhancement of mutagenesis. Carcinogenesis. 1993;14:373–83. doi: 10.1093/carcin/14.3.373. [DOI] [PubMed] [Google Scholar]
- 54.Jelinsky SA, Liu T, Geacintov NE, Loechler EL. The major, N2-Gua adduct of the (+)-anti-benzo[a]pyrene diol epoxide is capable of inducing G–>A and G–>C, in addition to G–>T, mutations. Biochemistry. 1995;34:13545–53. doi: 10.1021/bi00041a034. [DOI] [PubMed] [Google Scholar]
- 55.Banerjee SK, Christensen RB, Lawrence CW, LeClerc JE. Frequency and spectrum of mutations produced by a single cis-syn thymine-thymine cyclobutane dimer in a single-stranded vector. Proc Natl Acad Sci U S A. 1988;85:8141–5. doi: 10.1073/pnas.85.21.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mouret S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci U S A. 2006;103:13765–70. doi: 10.1073/pnas.0604213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szymkowski DE, Lawrence CW, Wood RD. Repair by human cell extracts of single (6-4) and cyclobutane thymine-thymine photoproducts in DNA. Proc Natl Acad Sci U S A. 1993;90:9823–7. doi: 10.1073/pnas.90.21.9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cosman M, de los Santos C, Fiala R, Hingerty BE, Singh SB, Ibanez V, Margulis LA, Live D, Geacintov NE, Broyde S, et al. Solution conformation of the major adduct between the carcinogen (+)-anti-benzo[a]pyrene diol epoxide and DNA. Proc Natl Acad Sci U S A. 1992;89:1914–8. doi: 10.1073/pnas.89.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu T, Xu J, Tsao H, Li B, Xu R, Yang C, Amin S, Moriya M, Geacintov NE. Base sequence-dependent bends in site-specific benzo[a]pyrene diol epoxide-modified oligonucleotide duplexes. Chem Res Toxicol. 1996;9:255–61. doi: 10.1021/tx9501086. [DOI] [PubMed] [Google Scholar]
- 60.Tsao H, Mao B, Zhuang P, Xu R, Amin S, Geacintov NE. Sequence dependence and characteristics of bends induced by site- specific polynuclear aromatic carcinogen-deoxyguanosine lesions in oligonucleotides. Biochemistry. 1998;37:4993–5000. doi: 10.1021/bi980291c. [DOI] [PubMed] [Google Scholar]
- 61.Xu R, Mao B, Amin S, Geacintov NE. Bending and circularization of site-specific and stereoisomeric carcinogen-DNA adducts. Biochemistry. 1998;37:769–78. doi: 10.1021/bi971785x. [DOI] [PubMed] [Google Scholar]
- 62.Xu R, Mao B, Xu J, Li B, Birke S, Swenberg CE, Geacintov NE. Stereochemistry-dependent bending in oligonucleotide duplexes induced by site-specific covalent benzo[a]pyrene diol epoxide-guanine lesions. Nucleic Acids Res. 1995;23:2314–9. doi: 10.1093/nar/23.12.2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cai Y, Patel DJ, Geacintov NE, Broyde S. Differential nucleotide excision repair susceptibility of bulky DNA adducts in different sequence contexts: Hierarchies of recognition signals. J Mol Biol. 2009;385:30–44. doi: 10.1016/j.jmb.2008.09.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hagerman PJ. Sequence dependence of the curvature of DNA: a test of the phasing hypothesis. Biochemistry. 1985;24:7033–7. doi: 10.1021/bi00346a001. [DOI] [PubMed] [Google Scholar]
- 65.Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag New York, Inc.; New York, NY: 1984. p. 241. [Google Scholar]
- 66.Xu J. PhD Thesis, Chemistry Department. New York University; 1999. Sequence-dependence of carcinogen-induced DNA bending. [Google Scholar]
- 67.Cosman M, de los Santos C, Fiala R, Hingerty BE, Ibanez V, Luna E, Harvey R, Geacintov NE, Broyde S, Patel DJ. Solution conformation of the (+)-cis-anti-[BP]dG adduct in a DNA duplex: intercalation of the covalently attached benzo[a]pyrenyl ring into the helix and displacement of the modified deoxyguanosine. Biochemistry. 1993;32:4145–55. doi: 10.1021/bi00067a001. [DOI] [PubMed] [Google Scholar]
- 68.Suh M, Jankowiak R, Ariese F, Mao B, Geacintov NE, Small GJ. Flanking base effects on the structural conformation of the (+)-trans-anti-benzo[a]pyrene diolepoxide adduct to N2-dG in sequence-defined oligonucleotides. Carcinogenesis. 1994;15:2891–8. doi: 10.1093/carcin/15.12.2891. [DOI] [PubMed] [Google Scholar]
- 69.Mouret S, Charveron M, Favier A, Cadet J, Douki T. Differential repair of UVB-induced cyclobutane pyrimidine dimers in cultured human skin cells and whole human skin. DNA Repair (Amst) 2008;7:704–12. doi: 10.1016/j.dnarep.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 70.Rodriguez FA. Ph.D. Thesis, Chemistry Department. New York University; 2007. Nuclear magnetic resonance solution structure of covalent polycyclic aromatic carcinogen-DNA adducts: Influence of base sequence context and carcinogen topology. [Google Scholar]
- 71.Bloomfield VA, Crothers DM, Tinoco I. Nucleic acids: Structures, properties, and functions. University science books; New York: 2000. [Google Scholar]
- 72.Dickerson RE. DNA bending: the prevalence of kinkiness and the virtues of normality. Nucleic Acids Res. 1998;26:1906–26. doi: 10.1093/nar/26.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]