

Abstract
Retroviruses contain relatively large amounts of ubiquitin, but the significance of this finding has been unknown. Here, we show that drugs that are known to reduce the level of free ubiquitin in the cell dramatically reduced the release of Rous sarcoma virus, an avian retrovirus. This effect was suppressed by overexpressing ubiquitin and also by directly fusing ubiquitin to the C terminus of Gag, the viral protein that directs budding and particle release. The block to budding was found to be at the plasma membrane, and electron microscopy revealed that the reduced level of ubiquitin results in a failure of mature virus particles to separate from each other and from the plasma membrane during budding. These data indicate that ubiquitin is actually part of the budding machinery.
Ubiquitin (Ub) is a 76-aa protein present in cells either as a free molecule or covalently attached to lysines in a wide variety of proteins. Polyubiquitation of short-lived proteins serves as a “tag” for proteolysis mediated by the 26S proteasome (1). However, Ub also has other roles in the cell, including one at the plasma membrane, where monoubiquitination of certain receptor proteins promotes their internalization and down-regulation in a proteasome-independent manner (2–4). The mechanism by which Ub triggers endocytosis of cell surface proteins is currently unclear, but recent work suggests that an endocytic signal within Ub plays a critical role (3, 5).
The plasma membrane is also the site of budding for retroviruses, and, 10 years ago, Volker Vogt and his colleagues (6) showed that avian retroviruses contain unexpectedly large amounts of free Ub, amounting to about 100 molecules per virion. This level is 5-fold higher than that of unconjugated Ub in the cytosol, and packaging appears to be specific because other low molecular weight proteins were not detected in the virions. More recently, similar amounts of free Ub have been found in HIV-1, simian immunodeficiency virus, and murine leukemia virus (7). The mechanism by which Ub is packaged into retrovirions is unknown, but it does not involve the viral glycoproteins (Env) because mutants that lack these still contain Ub (6). In some cases (7), a small amount (about 30%) of the virion-associated Ub has been found to be conjugated to Gag (Fig. 1), the viral protein responsible for particle assembly and budding (8); however, the Ub ligases involved have not been identified, and the significance of Ub for budding has been unknown.
Figure 1.

RSV Gag derivatives used in this study. The wild-type polyprotein is shown at the top and its proteolytic cleavage products are indicated. The domains required for budding are indicated below Gag. The M domain mediates the binding of Gag to the cytoplasmic face of the plasma membrane. The I domains provide the major regions of interaction among the 1,500 molecules that create a virion particle. The L domain is required for the virus–cell separation steps that occur late in the budding pathway. The foreign sequences in Gag-GFP, Gag-Ub, T10C-GFP, and T10C-Ub replace the protease (PR) sequence and the last six residues of the nucleocapsid (NC) sequence.
In contrast to the role of Ub, a great deal is known about the functions of Gag proteins in virus assembly and budding (8). These proteins (Fig. 1) are synthesized on free ribosomes and are subsequently directed to the cytoplasmic face of the plasma membrane by their N-terminal membrane-binding (M) domains. There, approximately 1,500 molecules (9) are packed together into very tight complexes, primarily by means of their interaction (I) domains. The M and I domains lead to the emergence of buds on the surface of the cell, but these are not efficiently released unless the “late” (L) domain is present. Although the amino acid sequences of M, I, and L are not conserved, these domains are functionally equivalent and exchangeable, even between distantly related viruses. The function provided by L domains is also positionally independent (10).
L domains are thought to recruit the cellular machinery needed for virus–cell separation on the plasma membrane. In the case of avian retroviruses, the critical residues of the L domain, PPPPY, are contained within the p2b sequence (Fig. 1) and have been shown to be a ligand for WW domains (11–13). A similar sequence has been found in the p12 sequence of murine leukemia virus (14), the pp16 protein of Mason-Pfizer monkey virus (15), and the matrix protein of rhabdoviruses (16, 17). For HIV-1, the critical residues are PTAP (18, 19), located in the p6 product, and these are possibly involved in binding with an SH3 domain. For equine infectious anemia virus, the critical residues are YPDL in the p9 sequence (20), and these have been shown to bind to and colocalize with adaptor protein (AP)-2, a component of the endocytic machinery on the plasma membrane (21).
Interestingly, there appears to be a correlation between L domains and Ub. In those viruses where Ub has been found to be linked to Gag, the conjugated lysines are in very close proximity to the L domain (7), and ubiquitination of Gag proteins has been found to be L domain-dependent [see accompanying paper by Strack et al. (22)]. Moreover, a search for proteins that bind to the Rous sarcoma virus (RSV) late domain has turned up the WW-containing protein Nedd4, a Ub ligase involved in down regulating sodium channels from the plasma membrane (J. Leis, personal communication).
Our experiments were inspired by the discovery that budding and proteolytic maturation of HIV-1 is reduced when infected cells are treated with a variety of proteasome inhibitors [see accompanying paper by Schubert et al. (23)]. The block appears to be at the plasma membrane and is reminiscent to that of L domain mutants. There are two hypotheses that could explain this defect. One is that misfolded Gag proteins, which would normally be targeted for destruction by the proteasome, interfere with budding as they accumulate in the drug-treated cells. The other possibility is that Ub plays an active role in budding, either as part of Gag or as part of another host protein. In this second hypothesis, there is no role for the proteasome, and the inhibitors merely provide a way to reduce the levels of free Ub in the cell (24).
To test these hypotheses, we made use of RSV, an avian retrovirus. We found that the release of RSV from infected cells is dramatically reduced in response to proteasome inhibitors. This effect can be substantially overcome by overexpressing Ub in trans or as a Gag-Ub fusion. These results argue against the misfolded protein hypothesis and strongly suggest that Ub is a component of the retrovirus budding machinery.
Materials and Methods
Cell Lines and DNAs.
QT6 cells, a line of transformed quail cells (25), were cultured in F10 medium (GIBCO) supplemented with 10% tryptose phosphate broth, 5% FCS, and 1% chicken serum. pRCV8, a plasmid containing an infectious copy of the RSV Prague C genome, has been previously described (26), as has Gag deletion mutant T10C, which lacks the L domain (10, 12). pGag-GFP, pT10C-GFP, pGag-Ub, and pT10C-Ub were constructed by using pEGFP-N2 (CLONTECH). Initially, the RSV gag gene, minus the coding sequences for the last six residues of nucleocapsid and the entire protease, was inserted between the SstI and ApaI sites to create a Gag-GFP fusion (27). Subsequently, the T10C deletion was introduced into pGag-GFP to create pT10C-GFP, and later, the human Ub sequence (minus the C-terminal glycine normally involved in linkage to lysines; refs. 28–30) was inserted in the place of GFP to create Gag-Ub and T10C-Ub.
Proteasome Inhibitors.
MG-132 (also known as zLLL) and lactacystin were obtained from Calbiochem, whereas epoxomicin and boro-MG-132 were kind gifts from Ulrich Schubert (National Institute of Allergy and Infectious Diseases, Bethesda, MD). All of the inhibitors were dissolved in DMSO and used within 2 wk at the final concentrations indicated in the text.
Budding Assay, Metabolic Labeling, and Immunoprecipitation.
RSV-infected QT6 cells were either untreated, pretreated with proteasome inhibitor for 90 min in addition to treatment for 2.5 h during the metabolic labeling period with [35S]methionine, or treated only during the labeling period. Alternatively, QT6 cells were transfected with the indicated DNA constructs by the calcium phosphate transfection method, and, the next day, transfected cells were MG-132-treated as mentioned above. The cells and particles in the growth medium were detergent lysed, and the Gag proteins were immunoprecipitated with polyclonal rabbit serum against whole RSV by using previously described methods (31). Proteins were separated by electrophoresis in SDS/12% polyacrylamide gels, which were then fixed and dried. The labeled proteins were detected by autoradiography using Kodak X-Omat AR5 film at −80°C. The negative impact of MG-132 on budding was quantitated by using a PhosphorImager (Molecular Dynamics).
The levels of free Ub present in MG-132-treated cells were measured by standard immunoblotting techniques using rabbit anti-Ub serum (kindly provided by Caroline E. Shamu, Department of Cell Biology, Harvard Medical School, Boston).
Confocal Microscopy.
QT6 cells were transfected with the indicated pGag-GFP constructs, and, 24 h later, the cells were treated for 3 h with either DMSO (vehicle control) or 80 μM MG-132, and analyzed directly by confocal microscopy (Zeiss).
Electron Microscopy.
RSV-infected QT6 cells on Permanox dishes (Electron Microscopy Sciences, Fort Washington, PA) were treated for 3 h with DMSO (negative control) or 80 μM MG-132 before the analysis. Cells were fixed, dehydrated, embedded, and sectioned for electron microscopy by using standard methods.
Results
Negative Effects of Proteasome Inhibitors on RSV Budding.
To determine whether proteasome inhibitors affect budding of RSV as they do HIV-1 (23), infected cells were treated with carbobenzoxy-l-leucyl-l-leucyl-l-leucinal (MG-132), a reversible peptide aldehyde inhibitor of the proteasome. As assayed by [35S]methionine labeling, we found that the release of RSV from infected cells is dramatically reduced in response to MG-132 (Fig. 2A, lanes 1 and 2). Pretreatment of the cells for 90 min before labeling resulted in a greater defect than adding the drug only during the labeling (Fig. 2B). Budding was reduced to a similar extent by treatments with 20 μM lactacystin (32), 20 μM epoxomicin (33), or 20 μM boro-MG-132 (34), three inhibitors that have greater specificity for the proteasome than MG-132 (not shown). Quantitation of data from multiple experiments reveals that treatment with MG-132 reduces proteolytic cleavage of Gag by approximately 25–50%, a much milder effect than that seen for HIV-1 (23).
Figure 2.

Proteasome inhibitors block RSV budding. (A) RSV-infected QT6 (quail) cells were untreated (lanes 1) or pretreated with 80 μM MG-132 for 90 min before metabolically labeling with [35S]methionine for 2.5 h, also in the presence of the drug (lanes 2). Alternatively, QT6 cells were transfected with pGag-GFP, which expresses a recombinant RSV Gag protein in which C-terminal sequences have been replaced with GFP. The next day, the transfected cells were untreated (lanes 3) or pretreated with 80 μM MG-132 (lanes 4) before labeling as above. The cells and particles in the growth medium were detergent lysed, and the Gag proteins were immunoprecipitated and electrophoresed in SDS/12% polyacrylamide gels before detection by autoradiography. (B) The negative impact of MG132 on budding was quantitated by using a PhosphorImager. Cells were either pretreated with MG-132 for 90 min in addition to treatment during the 2.5-h labeling period, or treated only during the labeling period. The budding efficiency in each culture was calculated as the amount of Gag protein in the medium (CA products) divided by the total in the lysates (Gag precursor and CA products) and media. The effects of MG-132 on budding (% release relative to untreated) were then determined by computing the ratios of budding efficiency in the absence and presence of drug. The averages from two no-pretreatment and three pretreatment experiments are shown .
We also examined the effects of MG-132 on Gag-GFP (Fig. 1). This chimera normally produces particles with the same high efficiency as wild-type Gag (27), even though it has the green fluorescence protein (GFP) inserted in the place of the viral protease (PR). MG-132 inhibited the release of this chimera to the same degree observed for authentic RSV (Fig. 2A, lanes 3 and 4; and Fig. 2B), indicating that none of the other viral products, including Env and PR, is involved.
Restoration of Budding by Ub Overexpression.
When proteasomes are inhibited, the levels of free Ub in mammalian cells are rapidly reduced (23, 24). This is true also in drug-treated QT6 cells, where MG-132 treatment reduced free Ub to 30% of normal within 30 min and to <5% of untreated levels within 90 min, as assayed by immunoblotting (data not shown). With this in mind, we decided to test the effects of overexpressing Ub. If the sole purpose of Gag ubiquitination is to tag misfolded proteins for destruction by proteasomes, then overexpression of Ub in MG-132-treated cells would not restore any degree of budding because there would still be no means for eliminating the interfering Gag molecules. However, if Ub plays an active role in the budding mechanism and the drug-induced block is a result of not having enough free Ub to provide that function, then overexpression should suppress the effects of MG-132. We found that overexpression of Ub has the effect of increasing budding in the presence of MG-132 (Fig. 3). In the absence of MG-132, overexpression of Ub typically resulted in approximately 3-fold increase in free Ub levels, whereas, in the presence of MG-132, an approximately 5-fold increase in free Ub levels was obtained by overexpression of the plasmid (data not shown). The increased levels of Ub did not bring budding back to the level of untreated controls, and this is likely because of the fact that proteasome inhibitors exert a wide variety of “nonspecific” effects on cells, which include a generalized decrease in protein synthesis, while inducing the selective synthesis of heat shock proteins hsp72 and hsp90 (24), and not all of these would be expected to be suppressed by the overexpression of Ub. Nevertheless, our data show that a specific gene product (Ub) can largely restore budding to MG-132-treated cells.
Figure 3.

Overexpression of Ub suppresses the effects of proteasome inhibitors. QT6 cells were cotransfected with 5 μg of (pGag.GFP) and 5 μg of either a human Ub expression plasmid (pUb) or the same plasmid containing the GFP-coding sequence instead (pGFP; negative control). The next day, cells were pretreated with 20 μM MG-132 for 90 min before labeling for 2.5 h with [35S]methionine, also in the presence of 20 μM MG-132. The labeled Gag proteins were collected and analyzed as described in Fig. 2. The graph shows the average of two experiments.
Inhibitor-Resistant Properties of a Gag-Ub Chimera.
It seemed most likely that Ub would provide its budding function after being linked to Gag, rather than to a host protein involved in budding, because monoubiquitinated Gag proteins have been identified (7). If this were the case, then genetic fusion of the Gag- and Ub-coding sequences (Fig. 1) might create a chimera that is capable of directing budding even in the presence of MG-132. Similar direct-fusion approaches have been used successfully in studies of receptor proteins that are down-regulated by Ub (3, 4). However, if Ub provides its budding function as part of a host protein, then the Gag-Ub chimera would still be inhibited by MG-132. We found that Gag-Ub minus the C-terminal glycine normally involved in linkage to Lys (28–30) is indeed much less sensitive to the drug than the control (Fig. 4). Moreover, in the absence of the drug (and in the presence of active proteasomes), this chimera behaves like the wild-type with regard to its stability and budding efficiency despite having Ub attached. These observations further support the hypothesis that Ub is an essential component of the budding machinery. By directly attaching Ub to Gag, the need for a pool of free Ub is bypassed. Based on its role in down-regulating receptor proteins from the plasma membrane, we hypothesize that the purpose of Ub in budding is to divert machinery normally involved in endocytosis to the purpose of separating nascent virus particles from the surface of the cell.
Figure 4.

A Gag-Ub chimera is comparatively insensitive to proteasome inhibitors. QT6 cells were transfected with either pGag-GFP or pGag-Ub (clones 1 and 2). The next day, the cells were pretreated with 80 μM MG-132 for 90 min before labeling with [35S]methionine, also in the presence of drug. (A) Gag proteins from cells that had been labeled for only 5 min were collected to compare the initial rates of protein synthesis of the treated (95 min, total) and untreated cells. (B and C) Identical sets of plates were labeled for 2.5 h to allow budding to proceed, and Gag proteins were collected from the cell lysates and media of the treated (4 h, total) and untreated cultures, as in Fig. 1. (D) PhosphorImager analysis of the budding efficiencies of Gag-GFP and Gag-Ub in the presence of MG-132. Data from three different experiments were used to calculate the averages by two different methods. Method 1 used only the data from the plates labeled for 2.5 h, which was used to calculate the percent of the total Gag protein found in the medium in treated vs. untreated cells. Method 2 used the data from the plates labeled for only 5 min to normalize the signals in the media after 2.5 h of labeling. Both methods show that Gag-Ub is much less sensitive to the effects of MG-132.
Subcellular Localization of Inhibited Gag Proteins.
It is not known when Ub is normally added to Gag; however, membrane-associated Ub ligases have been identified (35). If Ub is required for an event at the plasma membrane during budding, then Gag proteins would be expected to be blocked there in the presence of MG-132. Confocal microscopy of cells transfected with pGag-GFP revealed that Gag proteins do continue to reach the plasma membrane in the presence of the drug (Fig. 5), much like mutants that lack the late domain required for virus–cell separation (10, 12, 18, 20), but unlike mutants that lack the plasma membrane-binding domain (36, 37).
Figure 5.
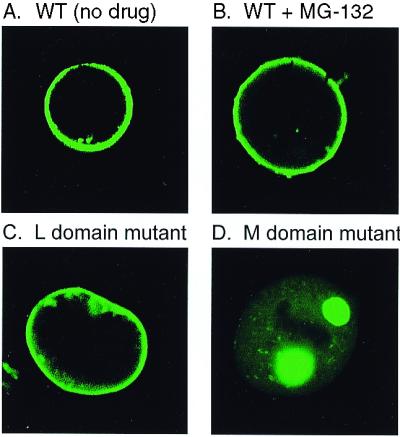
Gag accumulates on the plasma membrane in the presence of MG-132. QT6 cells were transfected with the indicated Gag-GFP constructs, and, 24 h, later the cells were examined by confocal microscopy. (A, C, and D) Untreated cells. (B) Cells treated with 80 μM MG-132 for 3 h.
To get a better look at the surface of MG-132-treated cells, transmission electron microscopy was used to examine RSV-infected cells (Fig. 6). In contrast to untreated cells, which had small groups of particles in random association (Fig. 6A), the surfaces of drug-treated cells were covered with particles over a 5- to 6-fold greater surface area (Fig. 6 B and C). Even more striking, these particles were often found in multilayered crystalline-like arrays, some of which were very large (Fig. 6 D and E). Approximately 50% of the cells scored positive for budding aggregates/crystalline arrays of varying size. In contrast, only 7% of untreated cells scored positive for the budding aggregates/crystalline arrays. The enveloped particles were morphologically mature, indicating that the block does not prevent proteolysis of Gag by the viral protease, and the layers of particles appear to be connected both vertically and horizontally within the arrays. The packing of particles was very tight, and, as a result, the layers often alternated between those in which the virion cores are within the plane of the section and those in which they are setback from the plane. The electron micrographs suggest that Ub is involved at a very late step in the budding pathway, possibly for separation of nascent viral particles from each other and the cell surface.
Figure 6.

Electron microscopy of MG-132-treated RSV-infected cells. (A) DMSO-treated RSV-infected cells, magnified ×27,500. (B—E) 80 μM MG-132-treated RSV-infected cells, magnified ×13,280 (B), ×10,950 (C), ×23,300 (D), and ×12,500 (E).
Corequirement for the L Domain and Ub.
It has been shown that ubiquitination of Gag depends on the presence of the late domain (22). If the sole purpose of the L domain during virus–cell separation is to recruit a specific Ub ligase to modify Gag, then the budding of L domain mutants would be restored by the attachment of Ub to their C termini. To test this hypothesis, we used deletion mutant T10C, which lacks the L domain and does not release particles (Fig. 7, lanes 1). Although this mutant has a large internal deletion, its budding defect is due only to the absence of the L domain. In particular, T10C can be rescued into virus particles by complementation using budding-competent Gag proteins (12, 38), and it becomes budding competent on its own when the L domains of HIV-1 or equine infectious anemia virus are fused to its C terminus (10). However, when Ub was attached to the C terminus of T10C, budding was not restored (Fig. 7, lanes 3). This result indicates that both the late domain and Ub are needed for budding.
Figure 7.

Corequirement for the L domain and Ub. QT6 cells were transfected with the indicated Gag-GFP and Gag-Ub constructs, and, 24 h later, the cells were labeled for 2.5 h with [35S]methionine. No proteasome inhibitors were used in this experiment.
Discussion
The release of budding viruses from their host cells requires the fusion of membranes as the emerging particles “pinch off.” This mixing of lipids in the apposing membranes of the bud is thought to be energetically unfavorable, and like other membrane fusion events, it must be mediated by a specific mechanism. Gag proteins do not possess the machinery required for virus–cell separation, but instead contain very small sequences, L domains, which serve to recruit the fusion machinery needed for efficient budding.
The data shown in this paper illustrate a function of Ub in retrovirus budding. The results from accompanying papers (22, 23) suggest that retroviral L domains recruit Ub ligase activity, which modifies Gag and triggers subsequent downstream events. Because genetic linkage of Ub to an L domain deletion mutant of Gag does not rescue the budding defect, it suggests that this domain has additional role(s) besides recruitment of the ubiquitination machinery (i.e., it might act as a nucleation site for an entire multifunctional complex required for budding).
Effect of Proteasome Inhibitors on Retroviral Protease Activity.
Proteasome inhibitor treatment of RSV-infected avian cells resulted in only a mild reduction in Gag processing by the viral protease. In addition, a protease-negative version of RSV Gag treated with MG-132 was defective for virus release to the same degree as observed for protease-positive Gag. These results imply that the detrimental effects of proteasome inhibitors on virus release are not mediated via a block in viral protease activity. These findings are in contrast to HIV (23) and EIAV (to be presented elsewhere), in which proteasome inhibitors strongly interfere with Gag processing by the viral protease. The above discrepancy is reconciled by the observation that RSV budding is not stimulated by an active viral protease, whereas maximum efficiency of HIV particle production requires viral protease activity (39).
Use of Endocytic Machinery for Retrovirus Budding.
Although very little is known about the host proteins recruited by L domains, three lines of evidence point to the involvement of machinery used for endocytosis. First, the L domain of equine infectious anemia virus has been found to contain YXXL, a motif involved in the endocytosis of a wide variety of proteins on the plasma membrane. This L domain has been shown to interact with the AP-50 subunit of the AP-2 complex in vitro, and it colocalizes with this complex in vivo (20, 21). Second, a search for binding partners of the RSV L domain has revealed Nedd4 (J. Leis, personal communication), a WW-domain-containing Ub ligase that is involved in down-regulating sodium channels from the plasma membrane (35, 40). Interestingly, yeast mutants with defects in the Nedd4 homolog, named Rsp5, show defects in endocytosis (41). The third line of evidence for the involvement of endocytic machinery in budding is our finding that Ub is required at the plasma membrane in a proteasome-independent manner that is reminiscent of Ub's role in down-regulating surface receptors (3, 5). The data presented in the accompanying papers further support the involvement of Ub in budding (22, 23).
The mechanism by which Ub triggers endocytosis is unclear. It does not seem to require polyubiquitination because removal of all of the lysines within Ub does not destroy endocytic activity (2, 3). As demonstrated by receptor-Ub chimeras, the normal linkage of the C terminus of Ub to internal lysines of the target protein is not essential, either. Currently, it is thought that Ub contains an endocytic signal that only comes into play when ubiquitination of a membrane-bound protein occurs. Studies in yeast suggest that this involves two hydrophobic patches on the surface of Ub in which Phe4 and Ile44 are critical residues (3). In mammalian cells, a “di-leucine” motif consisting of Ub residues Leu43 and Ile44 has been shown to be involved (5). Di-leucines promote endocytosis by mediating an interaction with AP-2.
How do retroviruses use machinery normally involved in membrane invagination for the topologically reversed process of budding? This question cannot be answered without further details regarding the budding machinery; however, it is instructive to consider the similarities between virus budding and the formation of multivesicular bodies (MVBs) in yeast. Vesicular budding occurs into the lumen of these large, cytoplasmic structures (for an illustration, see ref. 42). These cell-driven invaginations are topologically identical to retrovirus budding. It will be interesting to learn whether the host machinery involved in MVB formation overlaps with that required for retrovirus budding.
An alternative model for the role of Ub in budding is that ubiquitination of Gag promotes its movement into specialized regions of the plasma membrane, possibly detergent-insoluble membrane rafts (43, 44), where the budding machinery may reside. In support of this “lipid selectivity” model, the IgE receptor undergoes ligand-stimulated ubiquitination, which causes it to move into detergent-resistant domains of the plasma membrane upon stimulation (45).
Sites of Budding on the Plasma Membrane.
In RSV, proteasome inhibitors result in the production of particles that are tightly packed, often in large clusters, on the surface of the cell. Close examination of these arrays reveals thin, possibly membranous, connectors that hold the group of particles together. These clusters have important implications for retrovirus budding. They strongly suggest that budding occurs from many closely arranged, fixed sites on the membrane, each of which is capable of generating many particles. The production of a new particle at one of these sites can begin even before the previous particle has been released. Thus, when Ub levels are limiting, particles in a group emerging from the cell surface fail to fully separate their membranes from one another (horizontally) and from the next group of particles forming below (vertically). Whether these budding factories are unique entities created by Gag proteins or preexisting locations to which Gag proteins are directed remains to be determined. However, if the sites of budding are fixed in position on the membrane, then it may be possible to isolate and characterize them.
L Domains and Particle Size.
The data presented here may provide insight to the previously described influence of L domains on the size of HIV-1 particles. Our laboratory has shown that HIV-1 Gag mutants lacking the L-domain-containing p6 sequence produce particles that are extremely large as assayed by sucrose gradient sedimentation (46). Normal size was restored by replacing p6 with the p2b sequence of RSV or the p9 sequence of equine infectious anemia virus (47). It will be interesting to see whether the large HIV-1 particles produced in the absence of all L domains are actually clusters of many normal-sized particles that failed to separate from one another. If so, then it is likely that the role of Ub in virus separation is not unique to RSV but is also common to HIV.
Significance of Free Ub.
The simplest interpretation of our data is that budding requires ubiquitination of the RSV Gag protein. At first glance, this may seem to be in conflict with the fact that Ub-conjugated forms of Gag were not observed in RSV (6). However, even in those viruses where conjugated forms of Gag have been found, the majority of Ub is still free (7). These observations may be an indication that the mechanism of budding includes a host-encoded, deubiquitinating enzyme, some of which is incorporated into the particles. Alternatively, it is possible that the viral protease itself removes all or a large portion of the Ub sequence from Gag. It is also worth considering the possibility that only a few conjugated Gag proteins (too few to have been seen in previous experiments with RSV) are required for the formation of each bud, as is the case for L domains (12). Moreover, it may be that the Gag-Ub molecules required for budding are ones that remain tethered to other host proteins and are not actually packaged into the particle, serving only to recruit the appropriate cellular machinery. This would not explain why free Ub is concentrated in particles relative to the cytoplasm, however. Further investigations are clearly warranted.
In conclusion, our data and those of the accompanying papers (22, 23) suggest that one of the host proteins recruited by the L domain is a Ub ligase, which modifies Gag in a manner necessary for the poorly understood process of retrovirus budding. Because other enveloped viruses (e.g., rhabdoviruses) have been shown to contain late domain activities (16, 17), these observations may have broad significance for virology as a whole.
Acknowledgments
We thank Rebecca C. Craven and Richard J. Courtney for their thoughtful comments and suggestions on the manuscript. We are deeply indebted to Roland Meyers for his superb skills in electron microscopy. We also thank Brad Bowzard, Tina Cairns, Eric Callahan, Josh Loomis, and Carol Wilson for their help and suggestions. This work was supported by a grant from the National Institutes of Health awarded to J.W.W. (CA47482).
Abbreviations
- Ub
ubiquitin
- RSV
Rous sarcoma virus
- L domain
late domain
- GFP
green fluorescent protein
- AP
adaptor protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 12945.
References
- 1.Ciechanover A, Orian A, Schwartz A L. BioEssays. 2000;22:442–451. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 2.Hicke L. FASEB J. 1997;11:1215–1226. doi: 10.1096/fasebj.11.14.9409540. [DOI] [PubMed] [Google Scholar]
- 3.Shih S C, Sloper-Mould K E, Hicke L. EMBO J. 2000;19:187–198. doi: 10.1093/emboj/19.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Terrel J, Shih S, Dunn R, Hicke L. Mol Cell. 1998;1:193–202. doi: 10.1016/s1097-2765(00)80020-9. [DOI] [PubMed] [Google Scholar]
- 5.Nakatsu F, Sakuma M, Matsuo Y, Arase H, Yamasaki H, Nakamura N, Saito T, Ohno H. J Biol Chem. 2000;275:26213–26219. doi: 10.1074/jbc.M907720199. [DOI] [PubMed] [Google Scholar]
- 6.Putterman D, Pepinsky R B, Vogt V M. Virology. 1990;176:633–637. doi: 10.1016/0042-6822(90)90035-p. [DOI] [PubMed] [Google Scholar]
- 7.Ott D E, Coren L V, Copeland T D, Kane B P, Johnson D G, Sowder R C, Yoshinaka Y, Oroszlan S, Arthur L O, Henderson L E. Proc Natl Acad Sci USA. 1998;72:2962–2968. doi: 10.1128/jvi.72.4.2962-2968.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swanstrom R, Wills J W. In: Retroviruses. Weiss R, Teich N, Varmus H, Coffin J M, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1997. pp. 263–334. [Google Scholar]
- 9.Vogt V, Simon M N. J Virol. 1999;73:7050–7055. doi: 10.1128/jvi.73.8.7050-7055.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parent L J, Bennett R P, Craven R C, Nelle T D, Krishna N K, Bowzard J B, Wilson C B, Puffer B A, Montelaro R C, Wills J W. J Virol. 1995;69:5455–5460. doi: 10.1128/jvi.69.9.5455-5460.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garnier L, Wills J W, Verderame M F, Sudol M. Nature (London) 1996;381:744–745. doi: 10.1038/381744a0. [DOI] [PubMed] [Google Scholar]
- 12.Wills J W, Cameron C E, Wilson C B, Xiang Y, Bennett R P, Leis J. J Virol. 1994;68:6605–6618. doi: 10.1128/jvi.68.10.6605-6618.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiang Y, Cameron C E, Wills J W, Leis J. J Virol. 1996;70:5695–5700. doi: 10.1128/jvi.70.8.5695-5700.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan B, Li X, Goff S. EMBO J. 1999;18:4700–4710. doi: 10.1093/emboj/18.17.4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yasuda J, Hunter E. J Virol. 1998;72:4095–4103. doi: 10.1128/jvi.72.5.4095-4103.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Craven R C, Harty R N, Paragas J, Palese P, Wills J W. J Virol. 1999;73:3359–3365. doi: 10.1128/jvi.73.4.3359-3365.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harty R N, Paragas J, Sudol M, Palese P. J Virol. 1999;73:2921–2929. doi: 10.1128/jvi.73.4.2921-2929.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Göttlinger H G, Dorfman T, Sodroski J G, Haseltine W A. Proc Natl Acad Sci USA. 1991;88:3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang M, Orenstein J M, Martin M A, Freed E O. J Virol. 1995;69:6810–6818. doi: 10.1128/jvi.69.11.6810-6818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puffer B A, Parent L J, Wills J W, Montelaro R C. J Virol. 1997;71:6541–6546. doi: 10.1128/jvi.71.9.6541-6546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puffer B A, Watkins S C, Montelaro R C. J Virol. 1998;72:10218–10221. doi: 10.1128/jvi.72.12.10218-10221.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strack B, Calistri A, Accola M A, Palú G, Göttlinger H G. Proc Natl Acad Sci USA. 2000;97:13063–13068. doi: 10.1073/pnas.97.24.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schubert U, Ott D, Chertova E N, Welker R, Tessmer U, Princiotta M, Bennink J R, Kräusslich H-G, Yewdell J W. Proc Natl Acad Sci USA. 2000;97:13057–13062. doi: 10.1073/pnas.97.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mimnaugh E G, Chen H Y, Davie J R, Celis J E, Neckers L. Biochemistry. 1997;36:14418–14429. doi: 10.1021/bi970998j. [DOI] [PubMed] [Google Scholar]
- 25.Moscovici C, Moscovici M G, Jimenez H, Lai M M C, Hayman M J, Vogt P K. Cell. 1977;11:95–103. doi: 10.1016/0092-8674(77)90320-8. [DOI] [PubMed] [Google Scholar]
- 26.Craven R C, Leure-duPree A E, Erdie C R, Wilson C B, Wills J W. J Virol. 1993;67:6246–6252. doi: 10.1128/jvi.67.10.6246-6252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowzard J B, Visalli R J, Wilson C B, Callahan E M, Courtney R J, Wills J W. J Virol. 2000;74:8692–8699. doi: 10.1128/jvi.74.18.8692-8699.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodgins R R W, Ellison K S, Ellison M J. J Biol Chem. 1992;267:8807–8812. [PubMed] [Google Scholar]
- 29.Pickart C M, Kasperek E M, Beal R, Kim A. J Biol Chem. 1994;269:7115–7123. [PubMed] [Google Scholar]
- 30.Wilkinson K D, Tashayev V L, O'Connor L B, Larsen C N, Kasparek E, Pickart C M. Biochemistry. 1995;34:14535–14546. doi: 10.1021/bi00044a032. [DOI] [PubMed] [Google Scholar]
- 31.Weldon R A, Jr, Erdie C R, Oliver M G, Wills J W. J Virol. 1990;64:4169–4179. doi: 10.1128/jvi.64.9.4169-4179.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee D H, Goldberg A L. J Biol Chem. 1996;271:27280–27284. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- 33.Meng L, Mohan R, Kwok B H, Elofsson M, Sin N, Crews C M. Proc Natl Acad Sci USA. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCormack T, Baumeister W, Grenier L, Moomaw C, Plamondon L, Pramanik B, Slaughter C, Soucy F, Stein R, Zuhl F, Dick L. J Biol Chem. 1997;272:26103–26109. doi: 10.1074/jbc.272.42.26103. [DOI] [PubMed] [Google Scholar]
- 35.Plant P J, Yeger H, Staub O, Howard P, Rotin D. J Biol Chem. 1997;272:32329–32336. doi: 10.1074/jbc.272.51.32329. [DOI] [PubMed] [Google Scholar]
- 36.Sandefur S, Varthakavi V, Spearman P. J Virol. 1998;72:2723–2732. doi: 10.1128/jvi.72.4.2723-2732.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wills J W, Craven R C, Weldon R A, Jr, Nelle T D, Erdie C R. J Virol. 1991;65:3804–3812. doi: 10.1128/jvi.65.7.3804-3812.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennett R P, Wills J W. J Virol. 1999;73:2045–2051. doi: 10.1128/jvi.73.3.2045-2051.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaplan A H, Manchester M, Swanstrom R. J Virol. 1994;68:6782–6786. doi: 10.1128/jvi.68.10.6782-6786.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Staub O, Abriel H, Plant P, Ishikawa T, Kanelis V, Saleki R, Horisberger J D, Schild L, Rotin D. Kidney Int. 2000;57:809–815. doi: 10.1046/j.1523-1755.2000.00919.x. [DOI] [PubMed] [Google Scholar]
- 41.Zoladek T, Tobiasz A, Vaduva G, Boguta M, Martin N C, Hopper A K. Genetics. 1997;145:595–603. doi: 10.1093/genetics/145.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Odorizzi G, Babst M, Emr S D. Cell. 1998;95:847–858. doi: 10.1016/s0092-8674(00)81707-9. [DOI] [PubMed] [Google Scholar]
- 43.Brown D A, London E. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 44.Rietveld A, Simons K. Biochim Biophys Acta. 1998;1376:467–479. doi: 10.1016/s0304-4157(98)00019-7. [DOI] [PubMed] [Google Scholar]
- 45.Field K A, Holowka D, Baird B. Proc Natl Acad Sci USA. 1995;92:9201–9205. doi: 10.1073/pnas.92.20.9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garnier L, Ratner L, Rovinski B, Cao S-X, Wills J W. J Virol. 1998;72:4667–4677. doi: 10.1128/jvi.72.6.4667-4677.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garnier L, Parent L J, Rovinski B, Cao S X, Wills J W. J Virol. 1999;73:2309–2320. doi: 10.1128/jvi.73.3.2309-2320.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]