

Abstract
Although it is a widely studied psychiatric syndrome, major depressive disorder remains a poorly understood illness, especially with regard to the disconnect between treatment initiation and the delayed onset of clinical improvement. We have recently validated chronic social defeat stress in mice as a model in which a depression-like phenotype is reversed by chronic, but not acute, antidepressant administration. Here, we use chromatin immunoprecipitation (ChIP)-chip assays—ChIP followed by genome wide promoter array analyses—to study the effects of chronic defeat stress on chromatin regulation in the mouse nucleus accumbens (NAc), a key brain reward region implicated in depression. Our results demonstrate that chronic defeat stress causes widespread and long-lasting changes in gene regulation, including alterations in repressive histone methylation and in phospho-CREB (cAMP response element-binding protein) binding, in the NAc. We then show similarities and differences in this regulation to that observed in another mouse model of depression, prolonged adult social isolation. In the social defeat model, we observed further that many of the stress-induced changes in gene expression are reversed by chronic imipramine treatment, and that resilient mice—those resistant to the deleterious effects of defeat stress—show patterns of chromatin regulation in the NAc that overlap dramatically with those seen with imipramine treatment. These findings provide new insight into the molecular basis of depression-like symptoms and the mechanisms by which antidepressants exert their delayed clinical efficacy. They also raise the novel idea that certain individuals resistant to stress may naturally mount antidepressant-like adaptations in response to chronic stress.
Introduction
Depression, a common debilitating illness, is a heterogeneous syndrome comprised of multiple subtypes that remain poorly defined (Krishnan and Nestler, 2008). Although many brain regions are implicated in depression, our laboratory has focused on the nucleus accumbens (NAc) as one substrate for depressive-like behavior in rodent models. Cellular and molecular manipulations within this brain region exert dramatic effects in animal models of depression (Willner, 1983; Zacharko and Anisman, 1991; Willner et al., 1992; Nestler and Carlezon, 2006), and there is increasing evidence for abnormalities within the NAc in human depression (Tremblay et al., 2005; Krishnan et al., 2007, 2008).
We have validated a chronic social defeat stress paradigm in mice, which causes depressive-like symptoms that are reversed by chronic, but not acute, administration of antidepressant medications (Berton et al., 2006; Tsankova et al., 2006; Krishnan et al., 2007). Interestingly, inbred mice exhibit two distinct responses to chronic defeat stress: a susceptible response characterized by depressive symptoms as well as an unsusceptible response where the animals appear “resilient,” avoiding most deleterious effects of defeat stress. This model can be used to study naturally occurring variations in vulnerability to chronic stress. Contrasting with defeat stress, a type of “active stress,” we have demonstrated that prolonged social isolation of adult mice, a form of “passive stress,” induces depression-like behavior, although with partly distinct symptoms seen under these two conditions (Wallace et al., 2009). Chronic defeat stress and prolonged social isolation could model subtypes of human depression or, alternatively, show phenotypic differences resulting from the differential severity of the stressors involved (Antonijevic, 2008).
We set out to characterize long-lasting changes in gene transcription within the NAc in these two depression models. We used chromatin immunoprecipitation (ChIP)-chip assays that provide a genome-wide view of chromatin regulation at gene promoters (Renthal et al., 2009). We analyzed dimethylation of Lys9 and Lys27 in histone H3 (dimethylK9/K27-H3), well characterized repressive modifications associated with reduced gene expression (Felsenfeld and Groudine, 2003; Kouzarides, 2007). We also analyzed binding of the transcriptionally active form of cAMP response element binding protein (CREB), phospho-CREB, since increased CREB activity within the NAc induces depression-like behavior in several models (Pliakas et al., 2001; Barrot et al., 2002; Newton et al., 2002; Carlezon et al., 2005; Blendy, 2006). Using the social defeat model, we also used the dimethylK9/K27-H3 and phospho-CREB markers of transcriptional regulation to analyze changes in chromatin structure associated with antidepressant treatment as well as with the susceptible versus resilient phenotypes.
These approaches make it possible for the first time to analyze the stable epigenetic changes that occur in brain in depressive-like states and in response to treatment. Our findings indicate that imipramine treatment and resiliency share a common set of regulated genes, supporting the notion that certain individuals are naturally resistant to stressors in the environment (Charney, 2004; Charney and Manji, 2004; Yehuda, 2004; Hoge et al., 2007), in part by mounting active responses that overlap with those induced by antidepressant treatment.
Materials and Methods
Social defeat and imipramine treatment.
Male C57BL/6ByJ (hereafter, C57) mice, 7–9 weeks old, were obtained from Jackson Laboratory and housed on a 12 h light/dark cycle with access to food and water ad libitum. Male CD1 retired breeder mice (9–13 months old) were obtained from Charles River Laboratories. Animals were permitted to acclimate to the facility for 1 week before experimentation. Mice were subjected to chronic social defeat exactly as described previously (Berton et al., 2006; Krishnan et al., 2007). Briefly, CD1 mice were screened for aggressive behavior after intrusion into their home cages. Those mice exhibiting aggression were used to defeat the C57 mice. The C57 mice were defeated for 10 min per day and then housed for the remainder of the day with the aggressor but separated by a Plexiglas screen. This process was repeated daily for 10 consecutive days with a different CD1 aggressor each day to minimize interaggressor variability. Control animals were housed in divided cages with another C57 mouse. On day 11, defeated animals were subjected to the social interaction test and sorted into either susceptible or unsusceptible (resilient) phenotypes based on interaction scores as described (Krishnan et al., 2007).
Mice were then divided into treatment groups. For each group (control, susceptible, unsusceptible/resilient), one-half of the animals received daily intraperitoneal injections of imipramine (20 mg/kg) for 28 d. The other half in each group received vehicle. Previous work has shown that this imipramine treatment paradigm completely reverses the social interaction deficit seen in susceptible animals, which otherwise persists unabated in vehicle-treated susceptible animals (Berton et al., 2006; Tsankova et al., 2006). All mice were killed 24 h after the last imipramine or vehicle injection, and NAc dissections were obtained by punch dissection as described previously (Kumar et al., 2005). We did not analyze tissue from the nondefeated control mice and resilient mice treated with imipramine since no behavioral consequences of imipramine have been seen under these conditions (Berton et al., 2006; Tsankova et al., 2006).
Social isolation.
Male C57 mice, 7–9 weeks old, were housed on a 12 h light/dark cycle with access to food and water ad libitum. Animals were permitted to acclimate to the facility for 1 week before experimentation. Test animals were housed individually for 8 weeks, whereas control animals were group housed for the same period of time. This paradigm has been shown to induce a range of depression- and anxiety-like symptoms (Wallace et al., 2009). After the 8 week treatment period, NAc samples were obtained as above.
Chromatin immunoprecipitation and microarray analysis.
ChIP was performed for dimethylK9/K27-H3 and for phospho-CREB as described previously (Kumar et al., 2005). Briefly, for each ChIP, bilateral 14 gauge NAc punches were pooled from approximately eight mice, the tissue was lightly fixed to cross-link DNA with associated proteins, and the material was sheared and immunoprecipitated with an antibody that recognizes dimethylK9/K27-H3 or phospho-CREB, and then repaired and amplified (Sikder et al., 2006). Resulting immunoprecipitated DNA and total (input) genomic DNA were labeled with Cy5 and Cy3 fluorescent dyes, respectively. Independent, duplicate samples were then hybridized to NimbleGen mouse promoter arrays that span ∼20,000 genes. The resulting array data were subjected to rigorous statistical analysis (see supplemental Materials and Methods, available at www.jneurosci.org).
Briefly, data were initially preprocessed to log2 ratios and scaled using Tukey's Biweight Method to prevent inclusion of outliers in the final data set. Using median normalization and the floor method, a final data set was created for analysis. Significant alterations between treatment and control conditions were determined by first averaging the mean signal intensity for the mark of interest at each probe across the replicated samples. Since the gene array is tiled, a moving average that took into account three adjacent probes on the chip was used to stabilize the signal, and the p value for each probe was calculated using the ChIPOTle algorithm. False discovery rates for H3 methylation were determined by fitting the p values with a β-uniform mixture model. This method provides an acceptable false discovery rate that is in line with our previous work, as well as the work of others (Renthal et al., 2009). By only taking those probes with p values <0.001 (i.e., >3.1 SDs from the mean), false discovery rates are calculated at 0.18 and 0.16 for repressive histone methylation in social defeat and social isolation, respectively. We have previously shown that this method of data analysis allows us to estimate true false positive rates. (Renthal et al., 2009). Moreover, the overlap rate for the independent data sets in these experiments is in line with other published work on false discovery rates (Pyne et al., 2006), further alleviating concerns with regard to false discovery of genes. Overlap between the replicates was also calculated as a quality control measure. On average, the duplicates in each case showed a 70–80% similarity with Pearson's correlations of 0.6–0.9.
Promoter plots and genome-wide spatial binding patterns.
We used mpeak software (Zheng et al., 2007) to derive the signal peaks and overall spatial pattern of the locations of dimethylK9/K27-H3 and phospho-CREB binding relative to transcriptional starting sites. The empirical densities of the peak location were plotted and compared between social defeat (or isolation) condition and control condition. The empirical densities were estimated using R statistical software with all default settings (adjust = 1). Then, the bandwidth was determined by R density function automatically according to the number of enrichment probes, identified from the mpeak package. The bandwidths for H3 methylation and phospho-CREB binding are 271 and 623, respectively, in social defeat. The bandwidths for H3 methylation and phospho-CREB binding are 315 and 649 respectively, in social isolation.
Heat maps.
The heat maps included in Figures 1–4 depict the signal strength averaged across probes for each gene. Genes listed in the Tables are those with >1.1-fold change in social defeat or isolation that were also present as significantly changed in DNA expression arrays form our previous work (Berton et al., 2006; Krishnan et al., 2008; Wallace et al., 2009). The genes were rearranged using the Spearman hierarchical clustering method (Genespring).
Figure 1.
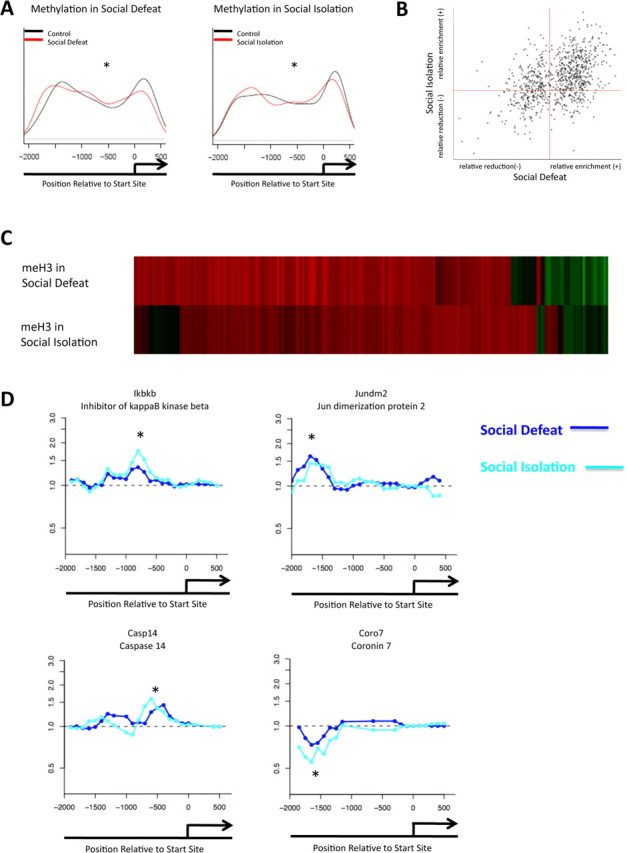
Similar regulation of H3 methylation in two mouse models of depression. A, Promoter plots show significant alterations in repressive dimethylK9/K27-H3 methylation in the NAc between control animals and those subjected to chronic social defeat or chronic social isolation. B, Positive correlation is seen between the two models for H3 methylation (p < 0.0001). C, The top heat map illustrates the fold change of H3 methylation in social defeat, compared with the fold change of H3 methylation of the same genes in the social isolation model. D, Patterns of H3 methylation at representative gene promoters. Light blue is social isolation; dark blue is social defeat. *p < 0.05.
Figure 2.
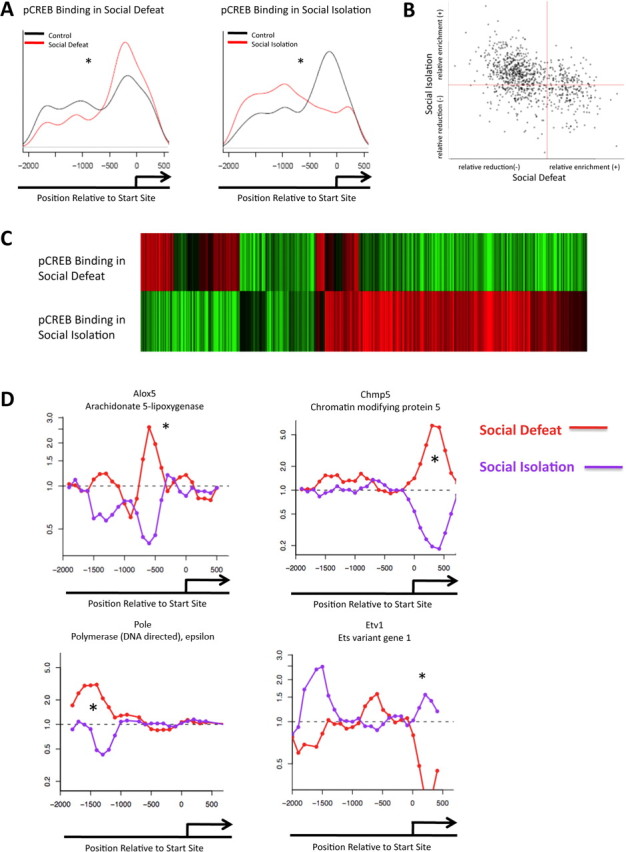
Opposite regulation of phospho-CREB binding in two mouse models of depression. A, Promoter plots show significant alterations in phospho-CREB binding in the NAc between control animals and those subjected to chronic social defeat or chronic social isolation. B, Negative correlation is seen between the two models for phospho-CREB binding (p < 0.0001). C, The top heat map illustrates the fold change of phospho-CREB binding in social defeat, compared with the fold change of phospho-CREB binding of the same genes in the social isolation model. D, Patterns of phospho-CREB binding at representative gene promoters. Red is social isolation; purple is social defeat. *p < 0.05.
Figure 3.

Reversal of H3 methylation and phospho-CREB binding patterns by imipramine treatment. A, Heat maps of genes whose repressive dimethylK9/K27-H3 methylation status seen in the NAc after chronic social defeat stress is reversed by chronic imipramine treatment. The top panel shows the state of the genes 4 weeks after social defeat (with vehicle injections), whereas the bottom shows the same genes after 4 weeks of imipramine injections. The bottom panel is represented as change from the −imipramine condition. C illustrates a similar reversal of phospho-CREB binding. Data are presented in the same manner as in A. B, D, Promoter plots for representative genes. B shows genes for H3 methylation, whereas D shows genes for phospho-CREB binding. For H3 methylation, dark blue is for control animals given vehicle, light blue for defeated animals given vehicle, and dark green for defeated animals given imipramine. For phospho-CREB binding, red is for control animals given vehicle, purple for defeated animals given vehicle, and orange for defeated animals given imipramine. *p < 0.05.
Figure 4.
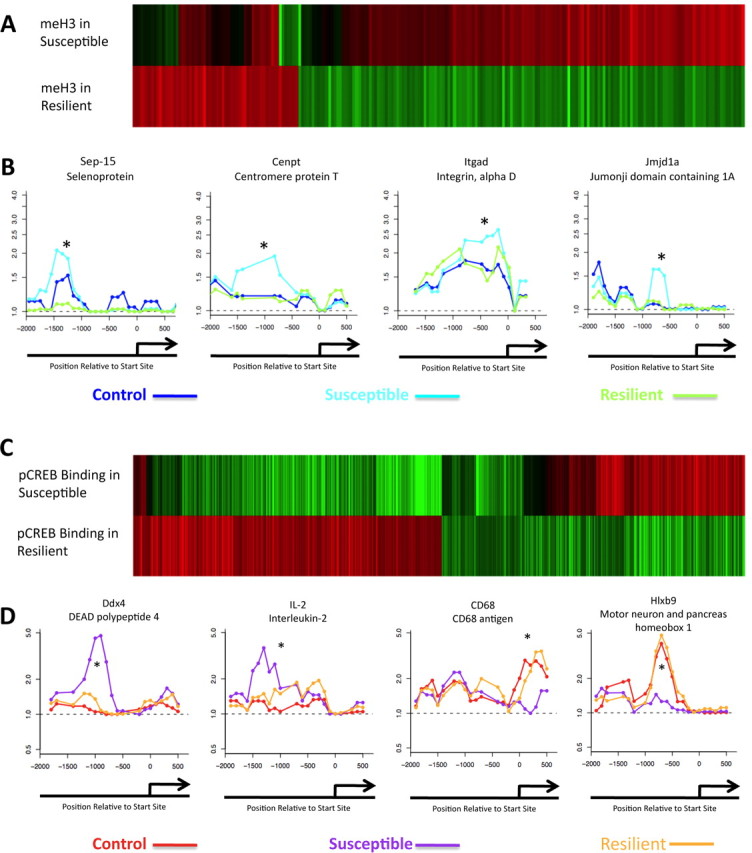
Differential patterns of H3 methylation and phospho-CREB binding in resiliency. A, Heat maps of genes whose repressive dimethylK9/K27-H3 methylation status in the NAc is mostly opposite in susceptible versus resilient animals 4 weeks after chronic social defeat. The top heat map depicts the state of the gene in susceptible animals compared with nondefeated controls, whereas the bottom shows the same genes in resilient animals represented as change from the susceptible condition. C illustrates a similar differential effect for phospho-CREB binding in susceptible versus resilient animals. Data are presented in the same manner as in A. B, D, Promoter plots for representative genes. B shows genes for H3 methylation, whereas D shows genes for phospho-CREB binding. For methylation, dark blue is for control animals given vehicle, light blue for vulnerable animals given vehicle, and dark green for resilient animals given vehicle. For phospho-CREB binding, red is for control animals given vehicle, purple for vulnerable animals given vehicle, and orange for resilient animals given vehicle. *p < 0.05.
Results
Repressive histone methylation is regulated in two mouse models of depression: evidence of similarly regulated genes
As a first step toward characterizing epigenetic regulation associated with depression, we mapped the entire genome of mice that were subjected to one of two recently validated models of chronic stress, chronic social defeat (Berton et al., 2006; Tsankova et al., 2006; Krishnan et al., 2007) or prolonged adult social isolation (Wallace et al., 2009), for levels of dimethylK9/K27-H3 in the NAc. This dimethylation mark has been shown to be a highly reliable indicator of gene repression in many systems (Felsenfeld and Groudine, 2003; Kouzarides, 2007). For this first set of social defeat analyses, only animals that exhibited a susceptible/defeated phenotype—as measured by a social avoidance scale (Berton et al., 2006)—on day 11, the day after 10 d of chronic defeat, were used. Mice were then analyzed 28 d later, a time when the avoidance phenotype remains stable. Control animals received nonaggressive social interactions as described previously (Berton et al., 2006). Socially isolated animals were subjected to isolated housing for 8 weeks compared with group-housed controls.
In both social defeat and social isolation animals, compared with their respective controls, we observed a significant difference (p < 0.001) in the relative levels of dimethylK9/K27-H3 in regions of gene promoters immediately upstream to their start sites (−2000–0 bp), as evidenced by the curves in Figure 1A. Note that these data are averaged across the entire genome, which lends credence to the idea that there are significant and far-reaching changes that occur in the NAc at a genomic level in response to either form of chronic stress.
Heat maps were generated from these data using Genespring software (Agilent Technologies) to visually represent the enrichment or attenuation of dimethylK9/K27-H3 at individual gene promoters (Fig. 1C). For socially defeated mice, a total of 1285 genes showed increased H3 methylation (supplemental Table S1, available at www.jneurosci.org as supplemental material), whereas 799 exhibited decreased methylation (supplemental Table S2, available at www.jneurosci.org as supplemental material). In the social isolation model, 1448 genes showed increased methylation (supplemental Table S3, available at www.jneurosci.org as supplemental material), and 615 displayed decreased methylation (supplemental Table S4, available at www.jneurosci.org as supplemental material). We identified 436 genes—∼20% of the total (supplemental Table S5, available at www.jneurosci.org as supplemental material)—that displayed alterations in H3 methylation (increased or decreased) (p < 0.001) in response to chronic social defeat and social isolation. Two clear results were apparent from these analyses. First, the vast majority of these regulated genes exhibited an increase in dimethylK9/K27-H3, which would be expected to correspond to a decrease in gene transcription. Moreover, the majority of genes (∼75%) were regulated in a similar manner in social defeat and in social isolation (i.e., those genes that showed increased H3 methylation in social defeat also showed increased H3 methylation in social isolation). Of these, 265 genes showed increased H3 methylation in both models (supplemental Table S6, available at www.jneurosci.org as supplemental material), whereas 60 genes showed a decrease (supplemental Table S7, available at www.jneurosci.org as supplemental material) (p < 0.001). Figure 1B depicts this information graphically. The positive correlation (p < 0.0001) between enrichment and attenuation of dimethylK9/K27-H3 binding between the two depression models suggests that social defeat and social isolation involve many similar gene regulatory events in terms of this repressive mark despite the very different type of stress involved. This is consistent with the fact that the two models induce some similar behavioral abnormalities such as depressive symptoms (e.g., anhedonia) and anxiety-like behavior (Krishnan et al., 2007; Wallace et al., 2009).
Genes that showed interesting patterns of methylated H3 regulation in these depression models were studied by Ingenuity molecular pathway analysis. Full lists of regulated genes with fold changes in methylation status are provided in supplemental Tables S1–S7, available at www.jneurosci.org as supplemental material. Interestingly, many of the genes that display increased H3 methylation in social defeat and social isolation are those involved in either inflammatory or cell death pathways (Table 1A). For example, inhibitor of κ-B kinase β (encoded by Ikbkb) usually allows for increased nuclear factor κB (NF-κB) signaling by causing degradation of inhibitor of κ-B (IKB), which normally constitutes a molecular brake on the system. Increased H3 methylation at the Ikbkb promoter (Fig. 1D) may contribute to a decreased ability to activate this pathway, as reduced Ikbkb expression would be associated with higher levels of IKB (Schmid and Birbach, 2008). Another gene of interest is Sep-15, which encodes a selenoprotein. Its primary function is to serve as an antioxidant in cells, thereby removing toxic intermediates (Chen and Berry, 2003; Ashrafi et al., 2007). This gene is dysregulated in several neurodegenerative disorders such as Alzheimer's disease. Increased H3 methylation at this gene promoter may contribute to a decreased ability of NAc neurons to deal with oxidative stress after exposure to chronic social defeat or social isolation stress.
Table 1.
Genes demonstrating similar dimethylK9/K27-H3 states in both stress models
| Genes | Gene activity | Reference |
|---|---|---|
| A | ||
| Aktip/Ft1 | General regulator of apoptosis via PKB/Akt pathway | Remy and Michnick, 2004 |
| Casp14 | Expressed during ischemic neuronal cell death | Krajewska et al., 2004 |
| CD84 | Enhances Ifn-γ secretion during inflammatory responses | Yan et al., 2007 |
| Fcrlb | Involved in cell-mediated cytotoxicity, inflammation, and hypersensitivity | Daëron, 1997 |
| Gstm4 | Reduces oxidative stress damage by inactivating reactive intermediates | Hayes et al., 2005 |
| Ikbkb | Activity allows for activation NfκB-dependent pathways | Schmid and Birbach, 2008 |
| IL-16 | Marker of microglial activation in inflammatory states | Guo et al., 2004 |
| Il1rl1 | Involved in induction and signaling in TNFα, IL-2, IL-6, IL-12, and IL-18 pathways | Subramaniam et al., 2004 |
| IL8rα | Increased expression following inflammatory stimuli (LPS) | Lee et al., 2002 |
| NEDD9 | Implicated in NFκB signaling through Tax | Singh et al., 2007 |
| Sep-15 | Possesses anti-oxidant properties; dysregulation is implicated in neurodegenerative disorders | Chen and Berry, 2003;Ashrafi et al., 2007 |
| SLAM7 | Involved in NK cytotoxity and B-cell activation | Calpe et al., 2008 |
| SLAM9 | Unknown function; homology suggests similar function to SLAM7 | Calpe et al., 2008 |
| B | ||
| Fgd5 | RhoGEF predicted to be involved in actin remodeling via Rac-Pac pathways | Rossman et al., 2005;Buchsbaum, 2007 |
| Sorbs3/vinexin | Involved in cell–cell adhesion and actin dynamics | Kioka et al., 1999 |
| C | ||
| Jdp2/Jundm2 | Binding to c-jun causes transcriptional repression via binding to AP-1 sites; binding to ATF-2 causes transcriptional repression via effects on HDAC3 | Eitoku et al., 2008 |
| Mcm8 | Required for eukaryotic genome replication | Maiorano et al., 2006 |
| Spic | Transcription factor similar to Pu0.1 involved in B-cell development; other transcriptional functions unclear | Bemark et al., 1999 |
| TFIIF | Part of the required core of transcriptional initiation machinery | Thomas and Chiang, 2006 |
A, Genes implicated in cellular inflammation, death, or redox reactions; B, genes implicated in cellular plasticity; C, genes implicated in gene regulation. LPS, Lipopolysaccharide.
Also prominent among gene promoters in which H3 methylation is regulated similarly after social defeat and social isolation are those involved in the regulation of gene transcription (Table 1C). One example is Jun dimerization protein 2 (Jundm2/Jdp2) (Fig. 1D). The binding of this protein to its partner c-Jun allows the complex to bind to AP-1 responsive genes and mediate transcriptional repression. Moreover, Jdp2 causes additional repressive effects on transcription via binding to ATF-2 (activating transcription factor 2) (Eitoku et al., 2008). Consequently, a decrease in the availability of the Jdp2 gene for transcription would result in “de-repression” of AP-1 and ATF-2 regulated genes.
Opposite regulation of phospho-CREB binding in two mouse models of depression
In addition to studying H3 methylation, we analyzed levels of phospho-CREB binding genome-wide in the same extracts of NAc of mice subjected to chronic social defeat or social isolation stress. This analysis was of interest given the fact that these two depression models are associated with opposite changes in phospho-CREB levels in the NAc, despite the fact that some common depressive symptoms are seen. Thus, active forms of stress like social defeat are associated with increased levels of phospho-CREB in the NAc, which has been shown to induce depression-like behaviors (Pliakas et al., 2001; Barrot et al., 2002; Newton et al., 2002; Carlezon et al., 2005; Blendy, 2006), whereas prolonged social isolation decreases phospho-CREB levels in this brain region (Wallace et al., 2009). ChIP-chip has previously been used to characterize CREB binding to gene promoters in brain in response to electroconvulsive seizures (Tanis et al., 2008), but not yet applied to depression models.
In contrast to the similar genome-wide regulation of H3 methylation observed in social defeat and social isolation, phospho-CREB analysis revealed strikingly different results. Furthermore, whereas changes in H3 methylation were confined primarily to the transcription start site of the genes, phospho-CREB binding was seen throughout the promoter region as far as 2000 bp upstream. As with the repressive methylation mark, phospho-CREB binding was significantly altered when averaged across all genes in the region immediately upstream to their start sites. Whereas social defeat increased phospho-CREB binding close to the start site and decreased it further upstream, social isolation decreased phospho-CREB binding around the region of the start site and increased binding further upstream. These data (shown in Fig. 2A) are averaged across the entire genome and provide a global view of the widespread epigenetic changes that occur in the NAc in these two depression models. Figure 2B illustrates the negative correlation between the social defeat and social isolation paradigms in terms of phospho-CREB binding as opposed to the positive correlation found for dimethylK9/K27-H3 binding (see Fig. 1B).
Genes that showed altered levels of phospho-CREB binding after chronic social defeat are listed in supplemental Tables S8 and S9, available at www.jneurosci.org as supplemental material, whereas those that showed altered levels of phospho-CREB binding after chronic social isolation are listed in supplemental Tables S10 and S11, available at www.jneurosci.org as supplemental material (p < 0.0001). Heat maps were generated to visually represent the enrichment or attenuation of phospho-CREB binding at these individual gene promoters (Fig. 2C). In total, 473 genes (supplemental Table S12, available at www.jneurosci.org as supplemental material) exhibited altered phospho-CREB binding in both the social defeat and social isolation model (p < 0.001). Although a larger number of genes exhibited changes in one model or the other, we focused on those that were altered in both. Confirming the negative trend seen globally (Fig. 2B), we found that 346 genes (∼73%) of these genes exhibited opposite changes in phospho-CREB binding in social defeat versus social isolation. Figure 2D illustrates representative examples of these genes.
Molecular pathway analysis indicated that similar gene families showed altered phospho-CREB binding after chronic stress as found for altered dimethylK9/K27-H3 binding, including those involved in inflammation, cell death, redox state, or gene regulation (Table 2). For instance, Ndufa3 and Ndufa5 are subunits of NADH [nicotinamide adenine dinucleotide (reduced form)]:ubiquinone oxireductase (Loeffen et al., 1998) and Gstm4 and Gstm5 are capable of reducing oxidative stress by consuming reactive intermediates (Hayes et al., 2005). All four show altered phospho-CREB binding. This corresponds with the regulation of H3 methylation at the Gstm4 and Sep-15 gene promoters. Additionally, genes such as Edar, the product of which is capable of activating the NF-κB pathway, exhibit altered phospho-CREB binding (Kumar et al., 2001). As these changes are in the opposite direction in social defeat versus social isolation, CREB regulation of these inflammatory pathways presumably proceeds differently in these two models.
Table 2.
Genes demonstrating opposite pCREB binding in both stress models
| Genes | Gene activity | Reference | |
|---|---|---|---|
| A | |||
| ⇓ In defeat | ⇑ in isolation | ||
| Alox5/5-LO | Catalyzes the first step in leukotriene biosynthesis | Murphy and Gijón, 2007 | |
| Etv/ERM | Involved in PKC signaling to CD44 and ICAMs | Larsson, 2006 | |
| Ndufa3 | Subunit of NADH:ubiquinone oxireductase | Loeffen et al., 1998 | |
| Ndufb5 | Subunit of NADH:ubiquinone oxireductase | Loeffen et al., 1998 | |
| Sep-15 | Possesses anti-oxidant properties; dysregulation is implicated in neurodegenerative disorders | Chen and Berry, 2003;Ashrafi et al., 2007 | |
| Thap1 | Zinc finger protein; pro-apoptotic factor; also implicated in interactions with chromatin remodeling enzymes | Bessière et al., 2008 | |
| ⇓ In isolation | ⇑ in defeat | ||
| CD5l | Unknown; inflammatory by similarity | ||
| Edar | Receptor capable of activating NfκB, JNK, and caspase independent death pathways | Kumar et al., 2001 | |
| Gstm4 | Reduces oxidative stress damage by inactivating reactive intermediates | Hayes et al., 2005 | |
| Gstm5 | Reduces oxidative stress damage by inactivating reactive intermediates | Hayes et al., 2005 | |
| IL-2 | Pro-inflammatory cytokine implicated in depression via activation of degradation pathways for W and 5-HT | Müller and Schwarz, 2007 | |
| Klrb1b | Inhibitor of NK cell-mediated cytotoxicity | Calpe et al., 2008;Lanier et al., 1994 | |
| B | |||
| ⇓ In defeat | ⇑ in isolation | ||
| Ank3 | Involved in linking spectrin/actin network to the cytoplasmic part of integral membrane proteins; involved in neuronal sodium channel inactivation | Shirahata et al., 2006 | |
| Ankra2 | Identified as a binding partner for class II HDACs | Wang et al., 2005 | |
| Arhgef9 | Critically involved in neuronal gephyrin clustering and actin dynamics | Harvey et al., 2004 | |
| Dctn3 | Smallest subunit of dynactin; involved in dynein transport, spindle formation, chromosomal organization, protein transport | Karki et al., 1998 | |
| Dync1li2 | Subunit of dynein; may play roles similar to that for dynactin | Karki et al., 1998 | |
| Etv5/ERM | Involved in PKC signaling to allow microfilament rearrangement | Larsson, 2006 | |
| Kptn | Actin binding protein involved with cytoskeletal rearrangements in platelets | Bearer and Abraham, 1999 | |
| Lasp1 | Actin binding protein involved in cystoskeletal remodeling and extension of cellular outgrowths; contains SH3 domain | Schreiber et al., 1998 | |
| C | |||
| ⇓ In defeat | ⇑ in isolation | ||
| Ankra2 | Identified as a binding partner for class II HDACs | Wang et al., 2005 | |
| Cxxc1 | Binds to unmethylated CpG regions of DNA; acts as a gene activator | Voo et al., 2000 | |
| Ddx28 | RNA helicase; involved in transcription and translation | Abdelhaleem, 2005 | |
| Ddx42 | RNA helicase; involved in transcription and translation | Abdelhaleem, 2005 | |
| E4f1 | Transcription factor regulated by E1A | Le Cam et al., 2004 | |
| EII | Elongation factor of RNA polymerase II | Armache et al., 2005 | |
| FoxN3 | Member of fork head/ winged transcription factors | Katoh and Katoh, 2004 | |
| Gabpb2 | Transcription factor; capable of binding to purine rich regions of genome | Muller and Schwarz, 2007;Le Cam et al., 2004 | |
| Gtf2f2 | General transcription factor; binds to RNA pol II | Armache et al., 2005 | |
| Gtf2h3 | Integral member of the TFIIH transcription factor involved in NER | Armache et al., 2005 | |
| Lmo4 | Transcription factor with GC rich regions; required for hematopoeisis | Ferreira et al., 2005 | |
| Med31 | Part of the mediator complex; involved in the transcription of almost all RNA pol II dependent genes | Guglielmi et al., 2007 | |
| Nolc1 | Involved in nucleolar organization and genes transcribed by RNA pol I | Tsai et al., 2008 | |
| Pola1 | DNA polymerase | Loeb and Monnat, 2008 | |
| Pole | DNA polymerase | Loeb and Monnat, 2008 | |
| Rnaseh2a | Ribonuclease; implicated in neuroinflammatory disorders | Rigby et al., 2008 | |
| Smad4 | Involved in TGF-β signaling via binding to DNA along with SMAD1 or 2 | Ross and Hill, 2008 | |
| Sox12 | Group C Sry-HMG related transcription factor | Dy et al., 2008 | |
| Sox17 | Sry-HMG related transcription factor important in early development | Jang and Sharkis, 2007 | |
| Sox4 | Group C Sry-HMG related transcription factor | Dy et al., 2008 | |
| Sp1 | Transcription factor capable of binding to GC rich domains | Li et al., 2004 | |
| Tsen 34 | One of two subunits important for tRNA splicing and pre-mRNA 3′-end formation | Paushkin et al., 2004 | |
| ⇓ In isolation | ⇑ in defeat | ||
| Ints7 | Subunit of the integrator complex which associates with RNA pol II | Nakagawa et al., 2008 | |
| Mcm3 | Required for eukaryotic genome replication | Maiorano et al., 2006 | |
| Msx2 | Transcriptional repressor of cells derived from the neural crest | Ramos and Robert, 2005 | |
| Tmem34 | Possible tumor suppressor gene | Akaishi et al., 2007 | |
A, Genes implicated in cellular inflammation, death, or redox reactions; B, genes implicated in cellular plasticity; C, genes implicated in gene regulation. regulation. Up arrows indicate increased pCREB binding. Down arrows indicate decreased pCREB binding. HDAC, Histone deacetylase; HMG, high mobility group; NK, natural killer; NER, nucleotide excision repair; JNK, c-Jun N-terminal kinase.
Several other genes that are differentially regulated via phospho-CREB after chronic social defeat and chronic social isolation regulate the neuronal cytoskeleton (Table 2B). Several encode proteins that are involved in the reorganization of actin (Ank3, Arghef9, Kptn, and Lasp1). The products of some of these genes function in cell–cell signaling as well. The product of Ank3 regulates sodium channel inactivation (Shirahata et al., 2006), whereas that of Lasp3 accumulates in the cell membrane and contains an SH3 domain that operates in signal transduction pathways (Schreiber et al., 1998).
Last, numerous genes that show altered phospho-CREB binding after social defeat or social isolation encode proteins that themselves control gene activation and expression (Table 2C). The product of Cxxc1 is capable of binding to unmethylated regions of DNA and serving as a gene activator (Voo et al., 2000). Several genes (EII, Gtf2f2, Gtf2h3, and Med31) that display altered phospho-CREB binding encode proteins that are part of the general transcription machinery (Armache et al., 2005; Guglielmi et al., 2007). This suggests that CREB in the NAc acts as a large scale genomic “switch,” which orchestrates a variety of transcriptional changes in response to environmental perturbations.
Imipramine treatment reverses the histone methylation and phospho-CREB changes induced by social defeat
Having established changes induced by chronic social defeat in dimethylK9/K27-H3 and phospho-CREB binding in the NAc, we were next interested in determining the effect of chronic imipramine treatment, which is known to reverse the behavioral deficits associated with this model (Berton et al., 2006; Tsankova et al., 2006). In this experiment, we built on the data shown in Figures 1 and 2, which show chromatin changes in chronically defeated mice 28 d after the last defeat; these animals received vehicle injections. Animals were now analyzed that displayed equivalent levels of social defeat on day 11 but were treated with imipramine for this 28 d period to reverse the behavioral deficits.
Figure 3A shows that, consistent with the behavioral reversal, a large portion of H3 methylation changes induced by social defeat are reversed by chronic imipramine treatment. The top heatmap in Figure 3A shows the stable H3 methylation changes apparent in the NAc 4 weeks after chronic social defeat in the absence of imipramine treatment; the bottom heat map shows the relative H3 methylation states for these same genes after 4 weeks of imipramine treatment. Note that data in the +imipramine map are presented as change compared with the −imipramine state; that is, an equal intensity change in the opposite direction from the −imipramine condition corresponds to a return to baseline methylation status seen in nondefeated controls. Seven hundred and eighty-five total genes were reversed in their methylation pattern after imipramine treatment. Lists of genes in which imipramine reversed decreases or increases in H3 methylation induced by chronic social defeat are shown in supplemental Tables S13 and S14, available at www.jneurosci.org as supplemental material. Together, these data demonstrate that imipramine significantly reversed ∼38% of all of the H3 methylation changes seen after chronic defeat stress. Among the genes normalized by imipramine are several involved in inflammation, redox state, and gene regulation (e.g., Sep-15, CD84, FcrlB, Ikbkb, Il16, and Spi-C). Although we did not study the chromatin effects of imipramine in the social isolation paradigm, it should be noted that each of the genes mentioned was commonly regulated after social defeat and social isolation.
We next analyzed phospho-CREB binding to gene promoters in the NAc 4 weeks after chronic social defeat stress and the effects of chronic imipramine on these adaptations. Heat maps are shown in Figure 3C. The top heat map depicts those genes that exhibited stable alterations in phospho-CREB binding 4 weeks after social defeat; the bottom heat map reveals how those genes were influenced by chronic imipramine treatment. As for the H3 methylation data, the +imipramine heat map shows data compared with the −imipramine heat map, such that a bar of equal intensity but opposite color corresponds to a return to baseline (nondefeated control) levels of phospho-CREB binding.
Analysis of these data revealed that chronic imipramine treatment reversed ∼29% of the genes that showed stable alterations in phospho-CREB binding after chronic social defeat. In this experiment, 350 (∼49%) of these genes showed increased phospho-CREB binding after social defeat that was reversed by imipramine, whereas 280 (∼19%) displayed decreased phospho-CREB binding that was reversed by imipramine. These imipramine-corrected genes were again involved in the same 3 molecular pathways described above (e.g., Acbd4, EII, Prkcsh, Thoc1, Gstm5, Il2, and Mcm3). Full gene lists are provided in supplemental Tables S15 and S16, available at www.jneurosci.org as supplemental material.
Susceptible and resilient mice exhibit differential regulation of histone methylation and phospho-CREB binding
An important feature of the social defeat model is that not all animals subjected to the stress exhibit the characteristic defeat phenotype. Such unsusceptible or resilient animals can be distinguished from susceptible animals based on a measure of social avoidance (Krishnan et al., 2007). These groups, so defined, also show numerous other differences in behavioral and physiological parameters, with the resilient animals exhibiting values closer to those of control animals that are not defeated. This difference led us to examine levels of dimethylK9/K27-H3 and phospho-CREB binding in the NAc in these two distinct populations of mice. Animals were examined 4 weeks after the chronic (10 d) defeat procedure when the susceptible and resilient phenotypes remain stable.
In general, resilient mice displayed patterns of H3 methylation in the NAc that more closely approximated those exhibited by control mice, with susceptible mice being significantly different from both control and resilient animals. Five hundred and forty-six total genes showed such differential levels of H3 methylation in resilient mice compared with susceptible animals. 124 genes showed increased H3 methylation in resiliency that was not seen in susceptible animals, whereas 422 showed increased methylation in susceptible animals, but a significant similarity to baseline (control) status in resilient animals (Fig. 4A; supplemental Tables S17 and S18, available at www.jneurosci.org as supplemental material). The data are shown in Figure 4A such that the top heat map represents susceptible animals compared with controls, with the bottom heat map reflecting resilient animals compared with susceptible animals. Thus, oppositely colored bars of equal intensity suggest that resiliency is associated with normalization of these genes to baseline status, possibly by preventing the modifications in the first place. Among these differentially regulated genes are several involved in inflammation, redox state, and gene regulation (e.g., Sep-15, Cenpt, Cox6c, Itgad, and Spi-C) (Fig. 4B). Importantly, the resilient state was also associated with significant changes in levels of H3 methylation at other gene promoters compared with nondefeated control animals. Some of these genes that showed modified dimethylK9/K27-H3 levels in resilient animals were the same that showed opposite changes in susceptible animals, whereas others were regulated uniquely in the resilient state. A total of 2122 genes showed altered H3 methylation in the resilient state as different from the control state. These findings support the view that resiliency is an active process, associated with chromatin modifications of its own, and not just the absence of modifications seen in defeated animals.
Analogous results were obtained for phospho-CREB binding. In this experiment, 247 genes (Fig. 4C; supplemental Table S19, available at www.jneurosci.org as supplemental material) showed decreased phospho-CREB binding in susceptible mice in which such binding was normalized to control levels in resilient animals, whereas 267 genes (supplemental Table S20, available at www.jneurosci.org as supplemental material) showed increased phospho-CREB binding in susceptible mice, with normal (i.e., control) levels of phospho-CREB binding seen in resilient animals. Again, the top heat map in Figure 4C reflects susceptible animals relative to control, and the bottom heat map reflects resilient animals compared with susceptible animals, such that equal intensity but oppositely colored bars show control levels of phospho-CREB binding in resilient animals. Thus, ∼24% of the genes that showed significant changes in phospho-CREB binding in susceptible and resilient animals exhibited differential regulation under these two conditions. These regulated genes were again involved in the same three molecular pathways stated above (e.g., Etfb, Il2, Mcm3, Ndufb9, Thoc1) (Fig. 4D).
Resiliency and imipramine treatment show similar patterns of chromatin regulation
The observation that imipramine and resiliency each offset a large proportion of the chromatin changes that occur in the social defeat paradigm raised the novel possibility that they do so by modulating similar pathways. Indeed, we found appreciable overlap in H3 methylation patterns under these two conditions. For example, 124 genes showed decreased dimethylK9/K27-H3 in the NAc of susceptible mice that was not observed in resilient mice. One hundred ninety-one genes showed decreased dimethylK9/K27-H3 in the NAc of susceptible mice that was reversed with chronic imipramine treatment. There are 51 overlapping genes in these two datasets. Regulation in the opposite direction (i.e., increased H3 methylation in susceptible mice, but normal H3 methylation in imipramine treatment or in resiliency) shows an overlap of 275 genes (∼31% of the imipramine genes; ∼40% of the resiliency genes) (Fig. 5A). Many of these genes have been previously mentioned, including Cenpt, Cox6c, Dctn3, and Spi-C. Another overlapping gene not mentioned previously is Jmjd1a, a histone demethylase involved in gene regulation.
Figure 5.
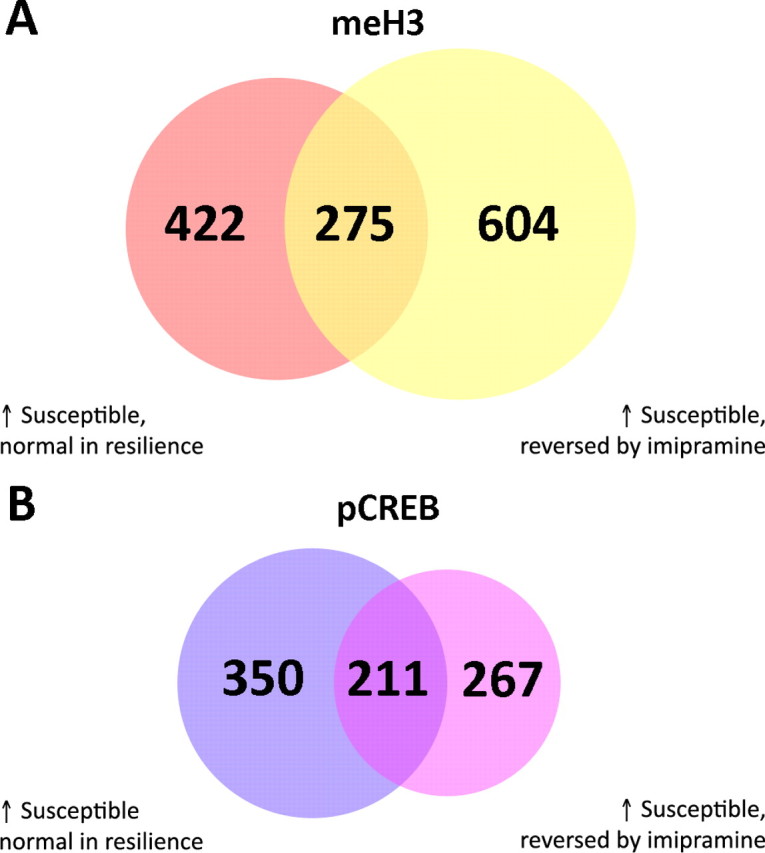
Overlap of chromatin changes in resiliency and after imipramine treatment. A, Venn diagram of genes whose H3 methylation status is regulated in the NAc of susceptible animals after chronic social defeat and reversed by imipramine treatment and not seen in resiliency. B, Venn diagram of genes whose phospho-CREB binding is regulated in the NAc of vulnerable animals after chronic social defeat and reversed by imipramine treatment and not seen in resiliency.
Levels of phospho-CREB binding likewise showed similarities between resiliency and imipramine treatment. Of genes that showed decreased phospho-CREB binding in the NAc of susceptible animals, with no changes in either resiliency (247) or after imipramine treatment (280), there are 140 overlapping genes. Of those that showed increased phospho-CREB binding in the NAc of susceptible mice, and no change in resiliency (561) or after imipramine treatment (478), 211 are overlapping (Fig. 5B). In correspondence with earlier findings, many of these genes are involved in inflammation, redox state, or gene regulation (e.g., Cul4b, Ddx4, Hist1h2bc, IL2, Ndufb9, and Zfp535). Full gene lists are provided in supplemental Tables S21–S24, available at www.jneurosci.org as supplemental material.
Discussion
Recent work has demonstrated the utility of using ChIP-chip analyses of the NAc to study psychiatric phenomena such as drug addiction (Renthal et al., 2009). Our current study extends these techniques to another common psychiatric illness, major depressive disorder, by using two animal models of depression: chronic social defeat stress (Berton et al., 2006; Tsankova et al., 2006; Krishnan et al., 2007) and prolonged social isolation of adult mice (Wallace et al., 2009).
We show that levels of dimethylK9/K27-H3, a marker of gene repression, are highly increased in both the social defeat and social isolation models. Despite the fact that one model involves an active form of stress and the other a type of passive stress, we have shown some common depressive-like symptoms (e.g., anhedonia-like symptoms) and anxiety-related behavior in the two paradigms (Krishnan et al., 2007; Wallace et al., 2009). The increased H3 methylation observed would reduce the ability of the affected genes to be transcribed either at baseline or in response to other stimuli, and the consequences of such increased methylation may be far-reaching. Indeed, a key advantage of this ChIP-chip analysis is that it provides a novel look at genes that are largely repressed or silenced.
Among the regulated genes are several involved in inflammatory pathways and regulation of redox state. One possibility is that the repression of such genes may be protective (e.g., by limiting the consequences of the initial activation of these biochemical pathways), but that this comes at the cost of decreased ability to activate the pathways later in the face of further challenges. Sep-15, which shows differential H3 methylation between stressed and control animals, has been shown to have antioxidant properties, and its dysregulation is implicated in neurodegenerative disorders, such as Alzheimer's disease (Chen and Berry, 2003; Ashrafi et al., 2007). Activity of the Na+/K+ ATPase, as well as activity of enzymes in the mitochondrial respiratory chain, are also disrupted in animal models of stress (Rezin et al., 2009). This corresponds with our findings in that we see regulation of Cox6c and Cox8c, subunits of cytochrome c oxidase. Likewise, the repression of genes involved in NF-κB signaling is interesting in that we have recently demonstrated a prodepressive effect of inhibiting the NF-κB pathway in the NAc (LaPlant et al., 2009). This is in contrast to the effect of NF-κB in hippocampus, where activation of the pathway decreases neurogenesis and depressive-like phenotypes (Pace et al., 2006; Koo and Duman, 2008). This work adds to an evolving literature that has implicated a range of inflammatory, cell stress, and cytokine pathways in depression-related phenomena. Chronic inflammation, through cytokine release, induces depression-like behavior in animals, and treatment of hepatitis C with interferon α induces depression in a large subset of patients (Dantzer et al., 2008). Conversely, behavioral stress, as shown here, activates cytokine signaling in the brain, which may contribute to a range of stress-related behavioral abnormalities (Shintani et al., 1995; Merali et al., 1997; Maier et al., 1999; Anisman et al., 2008).
Another class of genes highly regulated in common in the social defeat and social isolation models are those involved in transcriptional regulation. Jdp2 shows greatly induced H3 methylation in stressed animals. The product of this gene binds to c-Jun, with the resulting complex inducing transcriptional repression by binding to AP-1 sites and preventing the binding of active AP-1 transcription factor complexes. Therefore, repression of Jdp2 would lead to decreased repression of c-Jun and hence to an increase in AP-1 mediated transcription. Moreover, Jdp2 protein binds to ATF-2 and causes transcriptional repression via HDAC3 (Eitoku et al., 2008), and we have recently demonstrated antidepressant-like effects of ATF-2 when overexpressed in the NAc (Green et al., 2008), making this protein an attractive target for future investigations.
For phospho-CREB, our results demonstrate that, unlike H3 methylation state, binding of phospho-CREB to gene promoters is differentially regulated in the social defeat and social isolation models of depression. It is interesting, however, that the same general categories of genes showed altered levels of phospho-CREB binding as found for H3 methylation. Additionally, prominent among the genes that displayed opposite regulation of phospho-CREB binding in the two stress models are those related to actin remodeling. It is well documented that hippocampal volume is decreased in depressed patients (Egger et al., 2008), and that there is hippocampal dendritic atrophy in chronic stress paradigms (Magariños et al., 1996). Identification of stress-induced alterations in several genes involved in actin remodeling in the NAc raises the novel possibility that related morphological changes may occur in NAc neurons. Our data suggest that CREB activity may function as an overall switch that coordinates such regulation in the context of chronic stress.
The clinical efficacy of antidepressants, all of which alter monoaminergic neurotransmission, is well documented; however, it is unclear how chronic antidepressant administration changes the behavioral state of the organism. Our analyses provide new insight into the long-term effects of antidepressant treatments on the brain. Specifically, our data demonstrate that chronic imipramine treatment dramatically reverses many of the effects of chronic social defeat stress on H3 methylation and on phospho-CREB binding in the NAc. Although such actions would not be expected to account for the complete behavioral effects elicited by antidepressant treatment, they provide a beginning framework and suggest several novel molecular pathways to pursue. Among the genes that show reversal of stress regulation by imipramine is Chrh2, which encodes the corticotropin releasing hormone receptor, which has been highly implicated in stress responses (Nemeroff and Vale, 2005).
One of the most intriguing aspects of the social defeat paradigm is its consistent ability to generate animals that have undergone equivalent levels of defeat stress but do not exhibit a depression-like phenotype (Krishnan et al., 2007). Consistent with previous studies, in which we demonstrated that such resilient animals display some unique changes in mRNA expression in the NAc compared with susceptible animals (Krishnan et al., 2007), we now show that resiliency is likewise associated with a set of unique chromatin modifications in the NAc with respect to H3 methylation and phospho-CREB binding, further supporting the notion that resilience is an active process and not simply the absence of changes that occur in vulnerable animals. The identification of such “resilience genes,” therefore, defines a novel drug discovery pathway whereby new antidepressant treatments can be developed to promote resiliency. Given that susceptible and resilient responses are seen among inbred mice raised in identical environmental settings, the factors responsible for these differential adaptations to chronic defeat stress remain unknown. One possibility is that preexisting epigenetic modifications contribute to these distinct responses (Krishnan and Nestler, 2008). Regardless, the deleterious adaptations in susceptible animals may involve “allostatic overload.” Stress in the short-term normally elicits adaptive responses that are beneficial to the animal; in a more chronic context, the system may be overtaxed in some individuals, resulting in dysregulation of multiple neural processes (McEwen, 2008).
One of the most striking findings of the present study is the significant overlap in the chromatin changes in the NAc associated both with chronic imipramine treatment and with resiliency. This observation supports the highly novel view that resilient individuals are less vulnerable to the deleterious effects of chronic stress in part by mounting naturally some of the same changes in gene expression that occur in the NAc. Such overlapping genes provide a rich array of possible mediators of resiliency and antidepressant responses. However, since less than half of all patients treated with available antidepressants show a complete response, the genes that show chromatin regulation in resilient animals and not after antidepressant treatment may provide an even better set of novel targets for the development of new agents with a broader range of clinical efficacy.
The experiments undertaken in this study examined adult male mice only. Previous work has shown that there are important effects of both gender and age that should be considered going forward. Females are more susceptible to depression and more likely exhibit atypical symptoms of depression (Gorman, 2006). Furthermore, a better understanding of childhood and geriatric depression, and improved treatments, are sorely needed (Lenze et al., 2008; Tsapakis et al., 2008). It would also be interesting in future studies to examine individual differences in chromatin mechanisms in susceptible and resilient mice, something not yet feasible because of the current need to pool tissue from multiple animals for ChIP analysis.
The results of the present study illustrate the power of epigenetic investigations in psychiatric illness. ChIP-chip techniques provide novel insight into the detailed transcriptional mechanisms underlying the regulation of previously known gene targets in depression models. They also reveal many previously unknown gene targets and thereby provide a rational framework for the development of new treatment agents. Ultimately, a detailed step by step manipulation of individual genes, or sets of genes, identified by these methods will be needed to validate them as bona fide mediators of depression or antidepressant action. In the meantime, this work enables a more complete appreciation of the molecular changes induced in the brain that underlie vulnerability or resistance to depression and its effective treatment.
Footnotes
This work was supported by grants from the National Institute on Mental Health and Astra Zeneca. A.K. was supported by the National Alliance for Research on Schizophrenia and Depression Young Investigator Award in 2004.
The authors declare no competing financial interests.
References
- Abdelhaleem M. RNA helicases: regulators of differentiation. Clin Biochem. 2005;38:499–503. doi: 10.1016/j.clinbiochem.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Akaishi J, Onda M, Okamoto J, Miyamoto S, Nagahama M, Ito K, Yoshida A, Shimizu K. Down-regulation of an inhibitor of cell growth, transmembrane protein 34 (TMEM34), in anaplastic thyroid cancer. J Cancer Res Clin Oncol. 2007;133:213–218. doi: 10.1007/s00432-006-0159-8. [DOI] [PubMed] [Google Scholar]
- Anisman H, Merali Z, Hayley S. Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: comorbidity between depression and neurodegenerative disorders. Prog Neurobiol. 2008;85:1–74. doi: 10.1016/j.pneurobio.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Antonijevic I. HPA axis and sleep: identifying subtypes of major depression. Stress. 2008;11:15–27. doi: 10.1080/10253890701378967. [DOI] [PubMed] [Google Scholar]
- Armache KJ, Kettenberger H, Cramer P. The dynamic machinery of mRNA elongation. Curr Opin Struct Biol. 2005;15:197–203. doi: 10.1016/j.sbi.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Ashrafi MR, Shams S, Nouri M, Mohseni M, Shabanian R, Yekaninejad MS, Chegini N, Khodadad A, Safaralizadeh R. A probable causative factor for an old problem: selenium and glutathione peroxidase appear to play important roles in epilepsy pathogenesis. Epilepsia. 2007;48:1750–1755. doi: 10.1111/j.1528-1167.2007.01143.x. [DOI] [PubMed] [Google Scholar]
- Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL, Abraham MT. 2E4 (kaptin): a novel actin-associated protein from human blood platelets found in lamellipodia and the tips of the stereocilia of the inner ear. Eur J Cell Biol. 1999;78:117–126. doi: 10.1016/S0171-9335(99)80013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemark M, Mårtensson A, Liberg D, Leanderson T. Spi-C, a novel Ets protein that is temporally regulated during B lymphocyte development. J Biol Chem. 1999;274:10259–10267. doi: 10.1074/jbc.274.15.10259. [DOI] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bessière D, Lacroix C, Campagne S, Ecochard V, Guillet V, Mourey L, Lopez F, Czaplicki J, Demange P, Milon A, Girard JP, Gervais V. Structure-function analysis of the THAP zinc finger of THAP1, a large C2CH DNA-binding module linked to Rb/E2F pathways. J Biol Chem. 2008;283:4352–4363. doi: 10.1074/jbc.M707537200. [DOI] [PubMed] [Google Scholar]
- Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006;59:1144–1150. doi: 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Buchsbaum RJ. Rho activation at a glance. J Cell Sci. 2007;120:1149–1152. doi: 10.1242/jcs.03428. [DOI] [PubMed] [Google Scholar]
- Calpe S, Wang N, Romero X, Berger SB, Lanyi A, Engel P, Terhorst C. The SLAM and SAP gene families control innate and adaptive immune responses. Adv Immunol. 2008;97:177–250. doi: 10.1016/S0065-2776(08)00004-7. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Charney DS. Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am J Psychiatry. 2004;161:195–216. doi: 10.1176/appi.ajp.161.2.195. [DOI] [PubMed] [Google Scholar]
- Charney DS, Manji HK. Life stress, genes, and depression: multiple pathways lead to increased risk and new opportunities for intervention. Sci STKE. 2004;2004:re5. doi: 10.1126/stke.2252004re5. [DOI] [PubMed] [Google Scholar]
- Chen J, Berry MJ. Selenium and selenoproteins in the brain and brain diseases. J Neurochem. 2003;86:1–12. doi: 10.1046/j.1471-4159.2003.01854.x. [DOI] [PubMed] [Google Scholar]
- Daëron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy P, Penzo-Méndez A, Wang H, Pedraza CE, Macklin WB, Lefebvre V. The three SoxC proteins–Sox4, Sox11 and Sox12–exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res. 2008;36:3101–3117. doi: 10.1093/nar/gkn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger K, Schocke M, Weiss E, Auffinger S, Esterhammer R, Goebel G, Walch T, Mechtcheriakov S, Marksteiner J. Pattern of brain atrophy in elderly patients with depression revealed by voxel-based morphometry. Psychiatry Res. 2008;164:237–244. doi: 10.1016/j.pscychresns.2007.12.018. [DOI] [PubMed] [Google Scholar]
- Eitoku M, Sato L, Senda T, Horikoshi M. Histone chaperones: 30 years from isolation to elucidation of the mechanisms of nucleosome assembly and disassembly. Cell Mol Life Sci. 2008;65:414–444. doi: 10.1007/s00018-007-7305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- Ferreira R, Ohneda K, Yamamoto M, Philipsen S. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol Cell Biol. 2005;25:1215–1227. doi: 10.1128/MCB.25.4.1215-1227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman JM. Gender differences in depression and response to psychotropic medication. Gend Med. 2006;3:93–109. doi: 10.1016/s1550-8579(06)80199-3. [DOI] [PubMed] [Google Scholar]
- Green TA, Alibhai IN, Unterberg S, Neve RL, Ghose S, Tamminga CA, Nestler EJ. Induction of activating transcription factors (ATFs) ATF2, ATF3, and ATF4 in the nucleus accumbens and their regulation of emotional behavior. J Neurosci. 2008;28:2025–2032. doi: 10.1523/JNEUROSCI.5273-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmi B, Soutourina J, Esnault C, Werner M. TFIIS elongation factor and Mediator act in conjunction during transcription initiation in vivo. Proc Natl Acad Sci U S A. 2007;104:16062–16067. doi: 10.1073/pnas.0704534104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo LH, Mittelbronn M, Brabeck C, Mueller CA, Schluesener HJ. Expression of interleukin-16 by microglial cells in inflammatory, autoimmune, and degenerative lesions of the rat brain. J Neuroimmunol. 2004;146:39–45. doi: 10.1016/j.jneuroim.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Harvey K, Duguid IC, Alldred MJ, Beatty SE, Ward H, Keep NH, Lingenfelter SE, Pearce BR, Lundgren J, Owen MJ, Smart TG, Lüscher B, Rees MI, Harvey RJ. The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neurosci. 2004;24:5816–5826. doi: 10.1523/JNEUROSCI.1184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- Hoge EA, Austin ED, Pollack MH. Resilience: research evidence and conceptual considerations for posttraumatic stress disorder. Depress Anxiety. 2007;24:139–152. doi: 10.1002/da.20175. [DOI] [PubMed] [Google Scholar]
- Jang YY, Sharkis SJ. Fetal to adult stem cell transition: knocking Sox17 off. Cell. 2007;130:403–404. doi: 10.1016/j.cell.2007.07.027. [DOI] [PubMed] [Google Scholar]
- Karki S, LaMonte B, Holzbaur EL. Characterization of the p22 subunit of dynactin reveals the localization of cytoplasmic dynein and dynactin to the midbody of dividing cells. J Cell Biol. 1998;142:1023–1034. doi: 10.1083/jcb.142.4.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M, Katoh M. Human FOX gene family (Review) Int J Oncol. 2004;25:1495–1500. [PubMed] [Google Scholar]
- Kioka N, Sakata S, Kawauchi T, Amachi T, Akiyama SK, Okazaki K, Yaen C, Yamada KM, Aota S. Vinexin: a novel vinculin-binding protein with multiple SH3 domains enhances actin cytoskeletal organization. J Cell Biol. 1999;144:59–69. doi: 10.1083/jcb.144.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A. 2008;105:751–756. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Rosenthal RE, Mikolajczyk J, Stennicke HR, Wiesenthal T, Mai J, Naito M, Salvesen GS, Reed JC, Fiskum G, Krajewski S. Early processing of Bid and caspase-6, -8, -10, -14 in the canine brain during cardiac arrest and resuscitation. Exp Neurol. 2004;189:261–279. doi: 10.1016/j.expneurol.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Mazei-Robison M, Iñiguez SD, Ables JL, Vialou V, Berton O, Ghose S, Covington HE, 3rd, Wiley MD, Henderson RP, Neve RL, Eisch AJ, Tamminga CA, Russo SJ, Bolaños CA, Nestler EJ. Akt signaling within the ventral tegmental area regulates cellular and behavioral responses to emotional stimuli. Biol Psychiatry. 2008;64:691–700. doi: 10.1016/j.biopsych.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Eby MT, Sinha S, Jasmin A, Chaudhary PM. The ectodermal dysplasia receptor activates the nuclear factor-kappaB, JNK, and cell death pathways and binds to ectodysplasin A. J Biol Chem. 2001;276:2668–2677. doi: 10.1074/jbc.M008356200. [DOI] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Lanier LL, Chang C, Phillips JH. Human NKR-P1A. A disulfide-linked homodimer of the C-type lectin superfamily expressed by a subset of NK and T lymphocytes. J Immunol. 1994;153:2417–2428. [PubMed] [Google Scholar]
- LaPlant Q, Chakravarty S, Vialou V, Mukherjee S, Koo JW, Kalahasti G, Bradbury KR, Taylor SV, Maze I, Kumar A, Graham A, Birnbaum SG, Krishnan V, Truong HT, Neve RL, Nestler EJ, Russo SJ. Role of nuclear factor kappaB in ovarian hormone-mediated stress hypersensitivity in female mice. Biol Psychiatry. 2009;65:874–880. doi: 10.1016/j.biopsych.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson C. Protein kinase C and the regulation of the actin cytoskeleton. Cell Signal. 2006;18:276–284. doi: 10.1016/j.cellsig.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Le Cam L, Lacroix M, Ciemerych MA, Sardet C, Sicinski P. The E4F protein is required for mitotic progression during embryonic cell cycles. Mol Cell Biol. 2004;24:6467–6475. doi: 10.1128/MCB.24.14.6467-6475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Nagai A, Kim SU. Cytokines, chemokines, and cytokine receptors in human microglia. J Neurosci Res. 2002;69:94–103. doi: 10.1002/jnr.10253. [DOI] [PubMed] [Google Scholar]
- Lenze EJ, Sheffrin M, Driscoll HC, Mulsant BH, Pollock BG, Dew MA, Lotrich F, Devlin B, Bies R, Reynolds CF., 3rd Incomplete response in late-life depression: getting to remission. Dialogues Clin Neurosci. 2008;10:419–430. doi: 10.31887/DCNS.2008.10.4/jlenze. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, He S, Sun JM, Davie JR. Gene regulation by Sp1 and Sp3. Biochem Cell Biol. 2004;82:460–471. doi: 10.1139/o04-045. [DOI] [PubMed] [Google Scholar]
- Loeb LA, Monnat RJ., Jr DNA polymerases and human disease. Nat Rev Genet. 2008;9:594–604. doi: 10.1038/nrg2345. [DOI] [PubMed] [Google Scholar]
- Loeffen JL, Triepels RH, van den Heuvel LP, Schuelke M, Buskens CA, Smeets RJ, Trijbels JM, Smeitink JA. cDNA of eight nuclear encoded subunits of NADH:ubiquinone oxidoreductase: human complex I cDNA characterization completed. Biochem Biophys Res Commun. 1998;253:415–422. doi: 10.1006/bbrc.1998.9786. [DOI] [PubMed] [Google Scholar]
- Magariños AM, McEwen BS, Flügge G, Fuchs E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci. 1996;16:3534–3540. doi: 10.1523/JNEUROSCI.16-10-03534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF, Nguyen KT, Deak T, Milligan ED, Watkins LR. Stress, learned helplessness, and brain interleukin-1 beta. Adv Exp Med Biol. 1999;461:235–249. doi: 10.1007/978-0-585-37970-8_13. [DOI] [PubMed] [Google Scholar]
- Maiorano D, Lutzmann M, Méchali M. MCM proteins and DNA replication. Curr Opin Cell Biol. 2006;18:130–136. doi: 10.1016/j.ceb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Central effects of stress hormones in health and disease: Understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol. 2008;583:174–185. doi: 10.1016/j.ejphar.2007.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merali Z, Lacosta S, Anisman H. Effects of interleukin-1beta and mild stress on alterations of norepinephrine, dopamine and serotonin neurotransmission: a regional microdialysis study. Brain Res. 1997;761:225–235. doi: 10.1016/s0006-8993(97)00312-0. [DOI] [PubMed] [Google Scholar]
- Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12:988–1000. doi: 10.1038/sj.mp.4002006. [DOI] [PubMed] [Google Scholar]
- Murphy RC, Gijón MA. Biosynthesis and metabolism of leukotrienes. Biochem J. 2007;405:379–395. doi: 10.1042/BJ20070289. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Tategu M, Yamauchi R, Sasaki K, Sekimachi S, Yoshida K. Transcriptional regulation of an evolutionary conserved intergenic region of CDT2-INTS7. PLoS ONE. 2008;3:e1484. doi: 10.1371/journal.pone.0001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff CB, Vale WW. The neurobiology of depression: inroads to treatment and new drug discovery. J Clin Psychiatry. 2005;66(Suppl 7):5–13. [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Newton SS, Thome J, Wallace TL, Shirayama Y, Schlesinger L, Sakai N, Chen J, Neve R, Nestler EJ, Duman RS. Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J Neurosci. 2002;22:10883–10890. doi: 10.1523/JNEUROSCI.22-24-10883.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, Heim CM. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry. 2006;163:1630–1633. doi: 10.1176/ajp.2006.163.9.1630. [DOI] [PubMed] [Google Scholar]
- Paushkin SV, Patel M, Furia BS, Peltz SW, Trotta CR. Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3′ end formation. Cell. 2004;117:311–321. doi: 10.1016/s0092-8674(04)00342-3. [DOI] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA., Jr Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci. 2001;21:7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne S, Futcher B, Skiena S. Meta-analysis based on control of false discovery rate: combining yeast ChIP-chip datasets. Bioinformatics. 2006;22:2516–2522. doi: 10.1093/bioinformatics/btl439. [DOI] [PubMed] [Google Scholar]
- Ramos C, Robert B. msh/Msx gene family in neural development. Trends Genet. 2005;21:624–632. doi: 10.1016/j.tig.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Remy I, Michnick SW. Regulation of apoptosis by the Ft1 protein, a new modulator of protein kinase B/Akt. Mol Cell Biol. 2004;24:1493–1504. doi: 10.1128/MCB.24.4.1493-1504.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE, III, Maze I, Sikder D, Robison AJ, LaPlant Q, Dietz DM, Russo SJ, Vialou V, Chakravarty S, Kodadek TJ, Stack A, Kabbaj M, Nestler EJ. Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron. 2009;62:335–348. doi: 10.1016/j.neuron.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezin GT, Amboni G, Zugno AI, Quevedo J, Streck EL. Mitochondrial dysfunction and psychiatric disorders. Neurochem Res. 2009;34:1021–1029. doi: 10.1007/s11064-008-9865-8. [DOI] [PubMed] [Google Scholar]
- Rigby RE, Leitch A, Jackson AP. Nucleic acid-mediated inflammatory diseases. Bioessays. 2008;30:833–842. doi: 10.1002/bies.20808. [DOI] [PubMed] [Google Scholar]
- Ross S, Hill CS. How the Smads regulate transcription. Int J Biochem Cell Biol. 2008;40:383–408. doi: 10.1016/j.biocel.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)–a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev. 2008;19:157–165. doi: 10.1016/j.cytogfr.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Moog-Lutz C, Régnier CH, Chenard MP, Boeuf H, Vonesch JL, Tomasetto C, Rio MC. Lasp-1, a novel type of actin-binding protein accumulating in cell membrane extensions. Mol Med. 1998;4:675–687. [PMC free article] [PubMed] [Google Scholar]
- Shintani F, Nakaki T, Kanba S, Sato K, Yagi G, Shiozawa M, Aiso S, Kato R, Asai M. Involvement of interleukin-1 in immobilization stress-induced increase in plasma adrenocorticotropic hormone and in release of hypothalamic monoamines in the rat. J Neurosci. 1995;15:1961–1970. doi: 10.1523/JNEUROSCI.15-03-01961.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahata E, Iwasaki H, Takagi M, Lin C, Bennett V, Okamura Y, Hayasaka K. Ankyrin-G regulates inactivation gating of the neuronal sodium channel, Nav1.6. J Neurophysiol. 2006;96:1347–1357. doi: 10.1152/jn.01264.2005. [DOI] [PubMed] [Google Scholar]
- Sikder D, Johnston SA, Kodadek T. Widespread, but non-identical, association of proteasomal 19 and 20 S proteins with yeast chromatin. J Biol Chem. 2006;281:27346–27355. doi: 10.1074/jbc.M604706200. [DOI] [PubMed] [Google Scholar]
- Singh M, Cowell L, Seo S, O'Neill G, Golemis E. Molecular basis for HEF1/NEDD9/Cas-L action as a multifunctional co-ordinator of invasion, apoptosis and cell cycle. Cell Biochem Biophys. 2007;48:54–72. doi: 10.1007/s12013-007-0036-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Stansberg C, Cunningham C. The interleukin 1 receptor family. Dev Comp Immunol. 2004;28:415–428. doi: 10.1016/j.dci.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Tanis KQ, Duman RS, Newton SS. CREB binding and activity in brain: regional specificity and induction by electroconvulsive seizure. Biol Psychiatry. 2008;63:710–720. doi: 10.1016/j.biopsych.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- Tremblay LK, Naranjo CA, Graham SJ, Herrmann N, Mayberg HS, Hevenor S, Busto UE. Functional neuroanatomical substrates of altered reward processing in major depressive disorder revealed by a dopaminergic probe. Arch Gen Psychiatry. 2005;62:1228–1236. doi: 10.1001/archpsyc.62.11.1228. [DOI] [PubMed] [Google Scholar]
- Tsai YT, Lin CI, Chen HK, Lee KM, Hsu CY, Yang SJ, Yeh NH. Chromatin tethering effects of hNopp140 are involved in the spatial organization of nucleolus and the rRNA gene transcription. J Biomed Sci. 2008;15:471–486. doi: 10.1007/s11373-007-9226-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tsapakis EM, Soldani F, Tondo L, Baldessarini RJ. Efficacy of antidepressants in juvenile depression: meta-analysis. Br J Psychiatry. 2008;193:10–17. doi: 10.1192/bjp.bp.106.031088. [DOI] [PubMed] [Google Scholar]
- Voo KS, Carlone DL, Jacobsen BM, Flodin A, Skalnik DG. Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shares a CXXC domain with DNA methyltransferase, human trithorax, and methyl-CpG binding domain protein 1. Mol Cell Biol. 2000;20:2108–2121. doi: 10.1128/mcb.20.6.2108-2121.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DL, Han MH, Graham DL, Green TA, Vialou V, Iñiguez SD, Cao JL, Kirk A, Chakravarty S, Kumar A, Krishnan V, Neve RL, Cooper DC, Bolaños CA, Barrot M, McClung CA, Nestler EJ. CREB regulation of nucleus accumbens excitability mediates social isolation-induced behavioral deficits. Nat Neurosci. 2009;12:200–209. doi: 10.1038/nn.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AH, Grégoire S, Zika E, Xiao L, Li CS, Li H, Wright KL, Ting JP, Yang XJ. Identification of the ankyrin repeat proteins ANKRA and RFXANK as novel partners of class IIa histone deacetylases. J Biol Chem. 2005;280:29117–29127. doi: 10.1074/jbc.M500295200. [DOI] [PubMed] [Google Scholar]
- Willner P. Dopamine and depression: a review of recent evidence. I. Empirical studies. Brain Res. 1983;287:211–224. doi: 10.1016/0165-0173(83)90005-x. [DOI] [PubMed] [Google Scholar]
- Willner P, Muscat R, Papp M. An animal model of anhedonia. Clin Neuropharmacol. 1992;15(Suppl 1 Pt A):550A–551A. doi: 10.1097/00002826-199201001-00286. [DOI] [PubMed] [Google Scholar]
- Yan Q, Malashkevich VN, Fedorov A, Fedorov E, Cao E, Lary JW, Cole JL, Nathenson SG, Almo SC. Structure of CD84 provides insight into SLAM family function. Proc Natl Acad Sci U S A. 2007;104:10583–10588. doi: 10.1073/pnas.0703893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R. Risk and resilience in posttraumatic stress disorder. J Clin Psychiatry. 2004;65(Suppl 1):29–36. [PubMed] [Google Scholar]
- Zacharko RM, Anisman H. Stressor-induced anhedonia in the mesocorticolimbic system. Neurosci Biobehav Rev. 1991;15:391–405. doi: 10.1016/s0149-7634(05)80032-6. [DOI] [PubMed] [Google Scholar]
- Zheng M, Barrera LO, Ren B, Wu YN. ChIP-chip: data, model, and analysis. Biometrics. 2007;63:787–796. doi: 10.1111/j.1541-0420.2007.00768.x. [DOI] [PubMed] [Google Scholar]