

Abstract
Recently, a mutation in the mitochondrial protease Omi/HtrA2, G399S, was found in sporadic Parkinson's disease (PD) patients, leading to the designation of Omi/HtrA2 as PD locus 13 (PARK13). G399S reportedly results in reduced Omi protease activity. In vitro studies have suggested that Omi/HtrA2 acts downstream of PINK1, mutations in which mediate recessive forms of PD. We, as well as other, have previously shown that the Drosophila homologs of the familial PD genes, PINK1 (PARK6) and PARKIN (PARK2), function in a common genetic pathway to regulate mitochondrial integrity and dynamics. Whether Omi/HtrA2 regulates mitochondrial integrity and whether it acts downstream of PINK1 in vivo remain to be explored. Here, we show that Omi/HtrA2 null mutants in Drosophila, in contrast to pink1 or parkin null mutants, do not show mitochondrial morphological defects. Extensive genetic interaction studies do not provide support for models in which Omi/HtrA2 functions in the same genetic pathway as pink1, or carries out partially redundant functions with pink1, at least with respect to regulation of mitochondrial integrity and dynamics. Furthermore, Omi/HtrA2 G399S retains significant, if not full, function of Omi/HtrA2, compared with expression of protease-compromised versions of the protein. In light of recent findings showing that G399S can be found at comparable frequencies in PD patients and healthy controls, we do not favor a hypothesis in which Omi/HtrA2 plays an essential role in PD pathogenesis, at least with respect to regulation of mitochondrial integrity in the pink1/parkin pathway.
Keywords: Parkinson's disease, mitochondrial protease, Omi/HtrA2, Pink1, genetic interactions, Drosophila
Introduction
Parkinson's disease (PD) is characterized by degeneration of nigrostriatal dopaminergic neurons in the midbrain (Dauer and Przedborski, 2003), and genetic forms of the disease have provided insight into PD pathogenesis (Hardy et al., 2006). Mutations in PINK1 (PARK6), a nuclear gene encoding a mitochondrial serine-threonine kinase, and PARKIN (PARK2) cause recessively inherited forms of PD/parkinsonism (Kitada et al., 1998; Valente et al., 2004). Drosophila homologs of PINK1 and PARKIN act in a common genetic pathway (Clark et al., 2006; Park et al., 2006; Yang et al., 2006) to promote mitochondrial fission and/or inhibit mitochondrial fusion in multiple tissues, including dopaminergic neurons (Deng et al., 2008; Poole et al., 2008; Yang et al., 2008). Consistent with findings in Drosophila, patients with PINK1 or PARKIN mutations have indistinguishable clinical features, and also show mitochondrial defects (Ibáñez et al., 2006; Dodson and Guo, 2007). Recent studies also suggest that PINK1 and PARKIN regulate mitochondrial functions in mammals (Exner et al., 2007; Gautier et al., 2008; Piccoli et al., 2008; Wood-Kaczmar et al., 2008).
Omi/HtrA2 encodes a serine protease localized to mitochondrial intermembrane space. Although overexpression of Omi/HtrA2 leads to apoptosis after its release into the cytosol (for review, see Vande Walle et al., 2008), mice lacking Omi/HtrA2 or mice harboring a mutation in Omi/HtrA2 disrupting protease function (Jones et al., 1993, 2003) show loss of nondopaminergic neurons in the striatum, but not loss of apoptosis (Rathke-Hartlieb et al., 2002; Martins et al., 2004). These studies underscore the importance of studying the in vivo functions of Omi/HtrA2 using loss-of-function studies.
Recent reports have suggested links between Omi/HtrA2 and PD (Strauss et al., 2005; Bogaerts et al., 2008). One mutation in Omi/HtrA2, G399S, was identified in sporadic PD patients and reportedly impairs activation of protease activity. In addition, Omi/HtrA2 can physically bind to PINK1 in vitro, and Omi/HtrA2 can be phosphorylated by a serine-threonine kinase, p38, with this phosphorylation being dependent on PINK1. Furthermore, substitution of a putative PINK1-dependent phosphorylation site with a nonphosphorylatable moiety (S400A) markedly reduced protease activity (Plun-Favreau et al., 2007). Thus, it has been suggested that Omi/HtrA2 functions downstream of PINK1, with PINK1 positively regulating Omi/HtrA2 (Plun-Favreau et al., 2007). Based on these intriguing links between Omi/HtrA2 and PD, Omi/HtrA2 was recently designated as Parkinson disease-13 locus (PARK13).
However, two recent human genetic studies report no association of Omi/HtrA2 with PD (Ross et al., 2008; Simón-Sánchez and Singleton, 2008), with the G399S allele detected in both PD patients and healthy controls at a similar frequency. Because of these conflicting results, it is unclear whether Omi/HtrA2 acts as a true PD gene and whether it performs a major function downstream of PINK1. Resolution of these questions is crucial for understanding PD pathogenesis. Studies on the endogenous function of Omi/HtrA2 as it relates to PINK1 function are required to address these questions. Here, we report studies on loss-of-function and disease-associated mutants of Drosophila omi, and the results of extensive genetic interaction studies between pink1 and omi.
Materials and Methods
Molecular biology.
To generate UAS-omi, GMR-omi, and TMR-omi, the omi cDNA (EST clone from Drosophila Genome Research Center, AT14262) was subcloned into each vector (Brand and Perrimon, 1993; Hay et al., 1994; Huh et al., 2004). To generate Drosophila Omi mutants analogous to human Omi/HtrA2 mutations, S276C, S306A, G399S, and S400A, site-specific mutagenesis of S236C, S266A, G363S, and S364A of Omi was carried out, and the altered cDNAs were subcloned into pUASt and pTMR vectors, respectively. A fly mutation corresponding to the human PINK1G309D mutation, Pink1G426D, was generated by site-specific mutagenesis (made by I.E.C. and Atsushi Yamaguchi, Novato, CA). To silence omi, the omi transcript corresponding to the coding region was targeted using a microRNA-based technology (Chen et al., 2007) (Ganguly et al., 2008), and PCR products of these microRNA precursors were cloned into pUASt. To generate CaSpeR-pink1G426D, site-specific mutagenesis in the backbone of CaSpeR-pink1 was performed and the product subcloned into pCaSpeR4 vector. To generate CaSpeR-omi, a 2.5 kb PCR product, generated using the following primers, was subcloned into pCaSpeR4 vector (a gift from Nic Tapon, London, UK): 5′ primer: CAACTCGAGGAAGTACATTGGGCGGGTC; 3′ primer: GGGACTAGTGGGTTTGTCAGCGATTTC. All cloned PCR products were confirmed by DNA sequencing.
Drosophila genetics and strains.
EMS mutations were recovered using the Drosophila Tilling Service (Fred Hutchinson Cancer Research Center). These alleles were generated in a previous screen (Koundakjian et al., 2004). omiNSO is a nonsense allele resulting from a base substitution of C to T, leading to generation of a stop codon at Q196, and omiV110E is a missense allele resulting from substitution of T to A. We independently confirmed these alleles by sequencing. Prosα6T-GFP flies (Zhong and Belote, 2007) were obtained from J. M. Belote (Syracuse, NY), and UAS-mitoGFP and Df(3R)ED5644 flies were obtained from the Bloomington Drosophila Stock Center. pink15, TMR-pink1, UAS-pink1, and CaSpeR-pink1–9myc (Clark et al., 2006), UAS-parkin (Greene et al., 2003), and Mef2-Gal4 (Deng et al., 2008) flies have been previously described. For experiments involving transgenic flies, multiple independent fly lines were generated (Rainbow Transgenic Flies) and tested for each transgene. Drosophila strains were maintained in an 18°C, 25°C, or 29°C humidified incubator, or at room temperature.
Male and female fertility tests.
Recently eclosed individual male flies were placed with four virgin females in vials. Single 0- to 3-d-old females were placed in a vial supplemented with dry yeast along with five sibling males and maintained at 25°C. Males or females were scored as sterile if they failed to produce progeny by day 6.
Phase-contrast, immunofluorescence, and confocal microscopy.
For light microscopic analysis of the male germline, testes were dissected from recently eclosed males, squashed in PBS buffer, and imaged using an Olympus BX51 microscope equipped with phase-contrast optics. For analysis of muscle, nota of adult flies were dissected and fixed in 4% paraformaldehyde, and indirect muscle fibers were isolated and imaged by a Zeiss LSM5 confocal microscope. For analysis of dopaminergic neurons, anti-tyrosine hydroxylase (TH) (Immunostar) antibodies were used and imaged by a Zeiss LSM5 confocal microscope, and only clearly stained anti-TH-positive cells were counted. Wild-type, pink1 mutant, or omi mutant brains were counted blindly. The Immunofluorescent staining was performed as previously described (Guo et al., 1996). Phalloidin was used 1:1000 for testes staining (Invitrogen). Anti-Omi antibodies were a kind gift from M. Miura (Tokyo, Japan) and were used 1:300 for immunocytochemistry.
Transmission electron microscopy.
Testes and muscle were dissected, fixed in paraformaldehyde/glutaraldehyde, postfixed in osmium tetroxide, dehydrated, and embedded in Epon. Toluidine blue was used to stain 1.5-μm-thick tissue sections. Thin sections (80 nm thick) were stained with uranyl acetate and lead citrate, and examined using a JEOL 100C transmission electron microscope (University of California, Los Angeles Brain Research Institute Electron Microscopy Facility). At least three testes or thoraces of each genotype were examined by transmission electron microscopy (EM).
Scanning electron microscopy.
Freshly killed flies were mounted on their sides, placed on a platform under vacuum, and imaged at 180× magnification and 100 psi using a Hitachi 2460N scanning electron microscope. Analysis of eye phenotypes was performed as previously described (Guo et al., 2003; Gross et al., 2008).
Stress and longevity assays.
Zero- to three-day-old males were anesthetized on ice, aged for 48 h, starved for 6 h, and subjected to 5% sucrose plus each agent. Four vials of 30 flies were assayed simultaneously for each genotype. For longevity measurements, 120 males of each genotype were divided into six vials. Flies were maintained at 25°C and transferred to fresh food every 2 d.
Lysate preparation and Western blotting.
Heads or testes from age- and sex-matched adults were disrupted in lysis buffer containing complete protease inhibitor mixture (Roche) using a sonicator-3000 from MISONIX. Samples were boiled and centrifuged, and total protein was analyzed by Western blotting. Antibodies used were anti-Myc (Millipore) and anti-Omi (Igaki et al., 2007).
Northern blotting.
Standard protocols were used using a full-length pink1 probe as previously described (Clark et al., 2006).
Results
Overexpression-based genetic interactions of pink1 and omi in the eye
Omi/HtrA2 encodes a protein with a mitochondrial targeting sequence and a transmembrane domain, followed by a serine protease domain and a C-terminal PSD-95/DlgA/Zo-1 (PDZ) domain (Vande Walle et al., 2008). Drosophila melanogaster contains a single homolog of Omi/HtrA2 (CG8464, hereafter called omi), with 50% amino acid sequence identity, 68% similarity, and a domain structure similar to that of human Omi/HtrA2. To test the hypothesis that omi and pink1 function in the same pathway, we asked whether genetic interactions between these two genes could be observed in the Drosophila eye. The fly eye is dispensable for viability and fertility, and has been widely used as a system to study human neurodegenerative diseases [for review, see Bonini and Fortini (2003) and Marsh and Thompson (2006)]. We generated transgenic flies to carry out tissue-specific overexpression using the UAS-Gal4 system (Brand and Perrimon, 1993). When omi was overexpressed at high levels in the eye (25 or 29°C), small and rough eyes were observed (Fig. 1K compared with A), similar to a previously report (Igaki et al., 2007). These small eyes likely result from the ability of Omi to activate cell death when overexpressed (Challa et al., 2007; Igaki et al., 2007; Khan et al., 2008). The eye phenotypes resulting from omi overexpression were very sensitive to the level of omi expression. Flies expressing lower levels of omi (18°C) exhibited wild-type-appearing eyes, providing a sensitized genetic background for interaction studies (Fig. 1B). Eye-specific pink1 overexpression resulted in mild rough eyes (Fig. 1C) (Poole et al., 2008). However, flies overexpressing both pink1 and omi at 18°C exhibited smaller and rougher eyes than those associated with pink1 overexpression alone (Fig. 1F). This suggests that there is an overexpression-based interaction between pink1 and omi, which is consistent with a recent report (Whitworth et al., 2008) and has been interpreted, in conjunction with other observations, as indicating that omi acts downstream of pink1 in a common genetic pathway (Whitworth et al., 2008).
Figure 1.
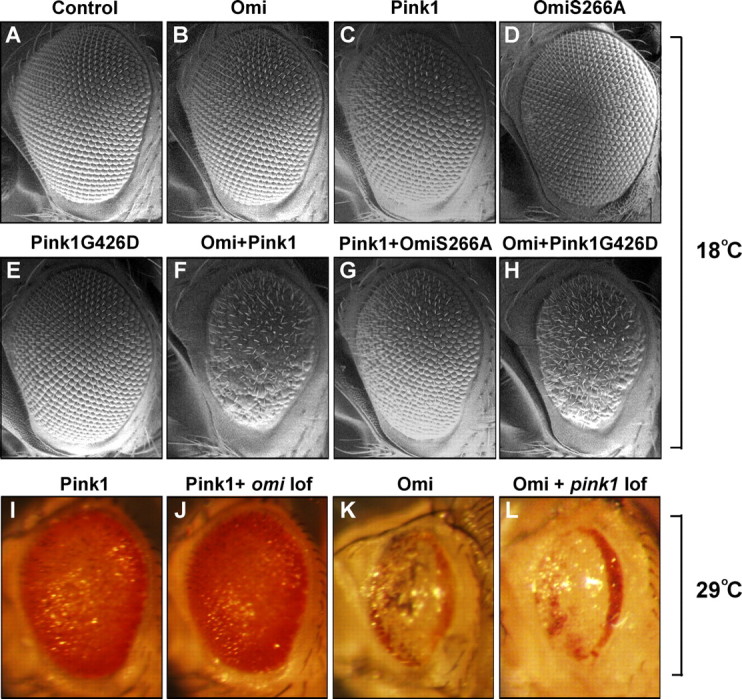
Overexpression-based genetic interactions between pink and omi. Scanning EM and light micrographs of Drosophila eyes are shown. At 18°C, omi overexpression (B) results in wild-type-appearing eyes, whereas overexpression of pink1 (C) leads to mild rough eyes. Overexpression of both omi and pink1 results in a small and rough eye (F). This pink1-omi overexpression interaction is abolished with expression of a protease-inactive version of Omi, S266A (G). Expression of OmiS266A by itself does not result in any eye phenotypes (D). Expression of a mutation analogous to the Pink1 disease mutant, G426D, has no phenotype (E); however, it still shows an interaction with omi overexpression (H). The phenotype of pink1 overexpression cannot be suppressed by loss of omi function induced by RNAi-omi, even when raised at 29°C (I, J). Silencing of omi function by RNAi-omi is strong because it completely suppresses the omi overexpression-induced eye phenotype (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Similarly, the eye phenotype resulting from omi overexpression cannot be modified by lack of pink1 (K, L). A–H are from flies raised at 18°C, whereas I–L are from flies raised at 29°C. Genotypes: Control: w; GMR-Gal4/+. Omi: w; GMR-Gal4, UAS-omi/+. Pink1: w; GMR-Gal4, UAS-pink1/+. Pink1G426D: w; GMR-Gal4/UAS-Pink1G426D. Omi+Pink1: w; UAS-omi/+; GMR-Gal4, UAS-pink1/+. Omi+Pink1G426D: w; GMR-Gal4, UAS-omi/UAS-Pink1G426D. Pink1+OmiS266A: w; UAS-OmiS266A/+; GMR-Gal4, UAS-pink1/+. Pink1+omi loss of function (lof): w; GMR-Gal4, UAS-pink1/UAS-RNAi-omi. Omi+pink1 lof: w pink15; GMR-Gal4, UAS-omi/+.
One possible explanation for the interaction observed when pink1 and omi are coexpressed is that overexpression of two mitochondrially targeted proteins causes competition for limited amounts of mitochondrial import machinery. In such a model, increased import of Pink1 could lead to excess Omi in the cytosol, resulting in a rough eye. Overexpression of a mitochondrial matrix-targeted green fluorescent protein (mitoGFP) with either pink1 or omi, however, did not lead to any enhancement of omi or pink1 overexpression phenotypes, suggesting that mitochondrial import is not limited (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The omi/pink1 co-overexpression interaction was dependent on the protease activity of Omi, because flies overexpressing a protease-dead version of Omi, Omi-S266A (Fig. 1D and see below), failed to show enhanced eye phenotypes when in conjunction with pink1 overexpression (Fig. 1G).
Further exploring the hypothesis that omi acts downstream of pink1, we found that pink1 overexpression-induced eye phenotypes could not be suppressed by loss of omi function (Fig. 1I,J). Similarly, the eye phenotype resulting from omi overexpression could not be modified by lack of pink1 (Fig. 1K,L). Thus, these results do not provide support for omi functioning downstream of pink1. We next explored the relationship between these interactions observed in the eye and the well characterized functions of pink in regulating mitochondrial morphology. We generated transgenic flies expressing Pink1G426D, a Drosophila mutation analogous to the PINK1 PD-associated mutation G309D. G309D alters a residue in the kinase domain (Valente et al., 2004) and has a significant reduction of PINK1 kinase activity, as assayed by in vitro autophosphorylation (Beilina et al., 2005). pink1 null mutant flies carrying G426D showed a largely abolished ability to rescue male sterility (<2% fertile, n = 60), muscle degeneration, and mitochondrial morphological defects of pink1 null mutants (Fig. 2), indicating that this mutant protein is strongly compromised with respect to normal pink1 function. Surprisingly, however, expression of Pink1G426D still led to a small and rough eye phenotype when combined with omi overexpression (Fig. 1H). These results suggest that pink1 functions required to mediate omi overexpression-based interactions in the eye are distinct from pink1 functions required to provide normal mitochondrial function. Together, although omi and pink1 displayed genetic interactions in overexpression-based assays, these results do not provide evidence to support models in which omi plays a major role in transducing pink1-dependent signals to regulate mitochondrial function.
Figure 2.
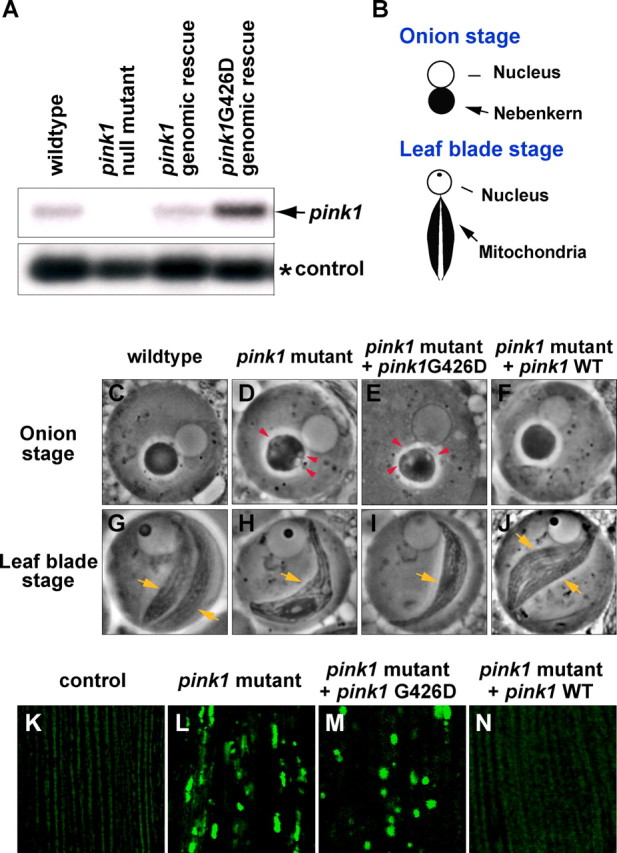
mRNA expression and phenotypic analysis of Pink1G426D, a mutation analogous to PD-associated mutation PINK1G309D. A, Northern blot of whole flies using full-length pink1 as a probe. Compared with wild type, pink1 null mutant (pink15) flies do not show full-length mRNA. Transgenic flies expressing pink1G426D mutants show comparable or higher expression of pink1 than is seen in flies expressing wild-type pink1. The arrow points to pink1 expression, and rp49 (*) serves as an RNA loading control. B–J, Schematic (B) and phase-contrast micrographs of spermatid mitochondria during the “onion stage” (C–F) and “the leaf blade stage” (G–J). Compared with wild type, pink1 mutants show vacuolation (red arrowheads) in the nebenkern during the onion stage (D), and one instead of the two mitochondrial derivatives (yellow arrows) seen in wild type at the leaf blade stage (H). A genomic rescue transgene carrying wild-type pink1 (CaSpeR-pink1) completely rescues the male sterility resulting from lack of pink1 (100% fertile, n > 120) (Clark et al., 2006), and spermatid phenotypes in both the onion stage and the leaf-blade stage (F, J). In contrast, pink1G426D (CaSpeR-pink1G426D) fails to rescue the sterility of pink1 males (<2% fertile, n = 60) or the spermatid phenotypes (E, I). Red arrowheads point to vacuolation of the nebenkern, and yellow arrows mark each mitochondrial derivative. K–N, Mitochondria of indirect flight muscle are labeled by mitoGFP. Compared with control (K), pink1 mutants display overall reduced levels of mitoGFP signal and large clumps of intense GFP signal (L), which can be completely rescued by overexpression of Pink1WT (N), but not Pink1G426D (M).
omi null mutants are male sterile, but show phenotypes distinct from those seen in pink1 or parkin null mutants
To further explore the roles of omi as it relates to pink1, we performed loss-of-function studies of omi mutants. The endogenous functions of omi in Drosophila have not been fully studied because of the absence of loss-of-function mutants. This analysis is more relevant to the role of omi as it relates to PD, because mutations in omi observed in PD patients are postulated to be loss-of-function or dominant-negative mutations, not resulting in increased activity. To obtain loss-of-function mutations in omi, we used TILLING (Till et al., 2003), a method for detecting ethyl methanesulfonate (EMS)-induced point mutations in a gene of interest after chemical mutagenesis. We obtained one nonsense mutation in omi, omiNSO, and one missense mutation, V110E (see below). The truncated protein encoded by omiNSO is predicted to lack the active site of the protease domain and the PDZ domain (Fig. 3A), and thus represents a null allele. Flies homozygous for omiNSO were semilethal. However, flies carrying omiNSO in trans to a deficiency in the region, Df(3R)ED5644, were fully viable, suggesting that the lethality associated with omiNSO is caused by a background mutation. Flies with ubiquitous expression of RNAi-omi using a tubulin-Gal4 driver were also viable. The silencing effect of RNAi-omi was confirmed by its ability to completely suppress omi overexpression-induced eye phenotypes (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). As expected, Western blotting using an anti-Omi antibody revealed no detectable Omi-positive bands in omiNSO/Df(3R)ED5644 flies (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 3.

omi null mutants show defects in spermatogenesis, but have normal mitochondrial morphology in testes. A, Schematic depicting domains of Omi. MTS, Mitochondrial targeting sequence. TM, Transmembrane domain. IBM, IAP-binding motif. The exact locations of omiNSO and omiV110E are depicted as blue and red asterisks, respectively. B, Sequence alignment of Omi/HtrA2 in various species in a highly conserved region, in which the conserved valine, mutated in omiV110E, is marked in red. C–E, C′–E′, Phase-contrast micrographs of testes. In contrast to a control fly (C, C′), in which the seminal vesicle (arrow) is full of sperm (phase dark), omi mutants (D, D′) show an empty seminal vesicle (bracket with an asterisk), which can be rescued by omi overexpression (E, E′) (arrow pointing to the seminal vesicle). C′–E′ are higher-magnification views of the seminal vesicle from C–E. F–H, Phalloidin staining of investment cones within one syncytial cyst. In contrast to controls, in which investment cones are well aligned, indicating synchronized movement (F), omi mutants show scattered investment cones in some of the cysts, indicative of a mild defect in individualization (G), which can be rescued by omi overexpression (H). I–O, Schematics and phase-contrast micrographs of spermatid mitochondria during the “onion stage” (I–L) and “the leaf blade stage” (I, M–O). Compared with wild type (J, M), omi mutants do not show any defects in either stage (K, N), whereas pink1 mutants show vacuolation (red arrowheads) in the nebenkern during the onion stage (L), and one instead of the two mitochondrial derivatives (yellow arrows) seen in wild type at the leaf blade stage (O). P–S, Schematic and transmission EM images of a portion of a postindividualization cyst. Each spermatid contains an axoneme (orange arrows) and mitochondrial derivative (red arrowheads) within an individual plasma membrane. The omi mutant cyst (R) shows disorganization of spermatids and occasional individualization defects (data not shown). However, compared with pink1 mutants, which show severe impairment in the size and morphology of mitochondria (S), omi mutant cysts show normal-appearing mitochondria (R). Genotypes: wild type: w/Y; control: w/Y; omiNSO/+; omi mutant: w/Y; omiNSO/Df(3R)ED5644; omi mutant + rescue: w/Y; TMR-omi/+; omiNSO/Df(3R)ED5644. Scale bars: C–E, 500 μm; Q–S, 500 nm.
omiNSO/Df(3R)ED5644 flies, hereafter called omi mutants, did not show any gross external defects. omi mutant females were fertile (96%, n = 50), but omi mutant males were sterile (100%, n = 110). In these males, seminal vesicles, which store mature sperm, were empty (Fig. 3D,D′), and no motile sperm were observed, suggesting defects in either production or transport to the seminal vesicles. To ensure that these phenotypes were attributable to lack of omi, we generated multiple transgenic fly lines expressing omi specifically in the male germline (TMR-omi). Overexpression of omi was confirmed using anti-Omi antibodies (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Many of these lines were male sterile. However, three of 10 lines were fertile. Those fertile lines show weaker overexpression of omi than the sterile lines (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). This suggests that the sterility is caused by a high level of overexpression, likely resulting in promiscuous activity of the Omi protease. Introduction of any of the fertile omi overexpression lines into the omi mutant background resulted in the presence of motile sperm in the seminal vesicles (Fig. 3E,E′) and restoration of fertility (95%, n = 100). A single copy of a genomic rescue transgene containing omi, but not surrounding genes, also fully rescued the male sterility of omi mutants (100%, n = 50). Together, these results demonstrate that omi is essential for spermatogenesis.
We also analyzed the second omi EMS allele. V110 corresponds to V154 in human Omi/HtrA2, which is located in a highly conserved region (Fig. 3A,B) predicted by structural studies to mediate homo-trimerization of Omi/HtrA2, which is required to activate its protease activity (Li et al., 2002). omiV110E/Df(3R)ED5644 and omiV110E/omiNSO mutant flies were also male sterile (0%, n = 45; 0%, n = 65), displayed empty seminal vesicles, and had no motile sperm, indicating that omiV110 is likely a null or strong hypomorphic allele. These results provide in vivo support for an important role for the trimerization motif for Omi function, and suggest that the protease activity of Omi is crucially important for its role in regulating spermatogenesis.
Because pink1 mutants also show male sterility, we asked whether omi mutant testes show defects in mitochondrial morphology, a prominent feature of pink1 mutants (Clark et al., 2006; Deng et al., 2008). During Drosophila spermatogenesis, mitochondria undergo significant morphological changes (Fuller, 1993). Stem cell differentiation is followed by mitosis and meiosis with incomplete cytokinesis, creating syncytial cysts of 64 spermatids. Early spermatids undergo mitochondrial aggregation and fusion, creating two giant mitochondria that form a spherical structure known as the nebenkern (Fuller, 1993). Under phase-contrast microscopy, such “onion stage” spermatids can be identified as having two adjacent spherical structures: the nucleus and the nebenkern (Fig. 3I,J). During subsequent spermatid elongation, the nebenkern begins to unfurl, creating two mitochondria at this “leaf-blade” stage (Fig. 3I,M). After elongation, spermatids undergo a process known as individualization, in which the cytoplasmic bridges that link the 64 spermatids within a cyst are broken and excess cytoplasm is extruded (Fuller, 1993). This individualization process requires synchronized movement of an actin-based structure known as the investment cone. After individualization, each spermatid tail consists largely of the axoneme, a microtubule-based structure required for motility, and mitochondrial derivatives (Fig. 3P,Q).
As expected for a protein with a mitochondrial targeting sequence, Omi localizes to nebenkerns (see Fig. 6A–C). In onion-stage spermatids, the nebenkerns of pink1 mutants show significant vacuolation (Fig. 3L), and during the subsequent leaf blade stage pink1 and parkin mutants contain only one mitochondrial derivative (Fig. 3O) rather than the two seen in wild type (Fig. 3M) (Clark et al., 2006; Riparbelli and Callaini, 2007; Deng et al., 2008). Surprisingly, mitochondria in omi mutants were indistinguishable from those in wild type. During the onion stage, the border of the nebenkern was smooth and no vacuolation was observed (Fig. 3K). At the leaf blade stage, omi mutant spermatids contained two mitochondrial derivatives instead of one observed in pink1 or parkin mutants (Fig. 3N). pink1 and parkin mutants also show dramatic defects in mitochondrial morphology during postindividualization stages, as visualized with transmission EM (Fig. 3S) (Greene et al., 2003; Clark et al., 2006; Riparbelli and Callaini, 2007; Deng et al., 2008). In contrast, mitochondria appeared normal in stage-matched omi mutants, although individual spermatids were somewhat disorganized within the cyst (Fig. 3R). In addition, investment cones in omi mutants were scattered (Fig. 3G compared with F), suggesting that movement of these structures is asynchronous. Such a phenotype is associated with individualization defects (Huh et al., 2004). Although the individualization defects were suppressed by testes-specific omi overexpression (Fig. 3H), we cannot rule out the possibility that defects in other postindividualization steps of spermatogenesis also contribute to sterility associated with omi mutants. This possibility seems particularly likely because the individualization defects observed in omi mutants appear relatively mild. In summary, omi mutant phenotypes in testes are distinct from those of pink1 or parkin mutants, in which defects in mitochondrial morphology are observed.
Figure 6.

Omi is localized to mitochondria, and its expression is not altered in pink1 null mutants; Pink1 expression is not altered in omi mutants either. A–F, Double labeling of onion-staged spermatids in wild type (A–C) and pink1 mutants (D–F) using Prosα6T-GFP (green), which labels the nucleus, and an anti-Omi antibody (red), which labels the nebenkern. In pink1 null mutants, Omi is still localized to the nebenkerns of spermatids. G, Western blotting (WB) of endogenous Pink1–9Myc expression using a genomic rescue transgene. Loss of omi function does not alter the cleavage pattern of Pink1.
In contrast to pink1 mutants, omi mutants do not show dopaminergic neuronal loss, muscle degeneration, or defects in mitochondrial integrity
Next, we asked whether omi mutants show phenotypes similar to those of pink1 mutants in other tissues and contexts. omi mutants were sensitive to treatments with multiple stress-inducing agents, including paraquat, a free radical inducer; rotenone, which impairs complex I activity in the mitochondrial respiratory chain (Przedborski and Ischiropoulos, 2005); protein folding inhibitors; and high concentrations of salt (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Thus, rather than being specifically sensitive to oxidative stress, omi mutant flies are generally stress sensitive. These results may suggest a general sickness of omi mutants, particularly because omi mutants had a shortened life span (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
An age-dependent decrease in the number of dopaminergic neurons has been reported in pink1 or parkin mutants (Meulener et al., 2005; Park et al., 2006; Yang et al., 2006) (Fig. 4A,B,D,E). In contrast, omi mutants did not show any dopaminergic neuronal loss in the brains of flies aged for 40 d (Fig. 4C,F). pink1 or parkin mutants also show striking indirect flight muscle degeneration and severely disrupted mitochondrial morphology with broken cristae, which are prominent in 1- to 2-d-old flies (Fig. 4I,J compared with G,H) (Greene et al., 2003; Pesah et al., 2004; Clark et al., 2006; Park et al., 2006; Yang et al., 2006). In contrast, although Omi is expressed in muscle and localized to mitochondria (data not shown), omi mutants did not exhibit any muscle degeneration, even when they were aged for 30 d (Fig. 4K). EM analysis of omi mutant muscle also failed to show any defects in mitochondrial integrity (Fig. 4L). Together, our data demonstrate that omi mutants, in contrast to pink1 mutants, do not display mitochondrial morphological defects in multiple tissues, including spermatids and muscle. omi mutants also fail to show dopaminergic neuronal loss seen in pink1 mutants.
Figure 4.
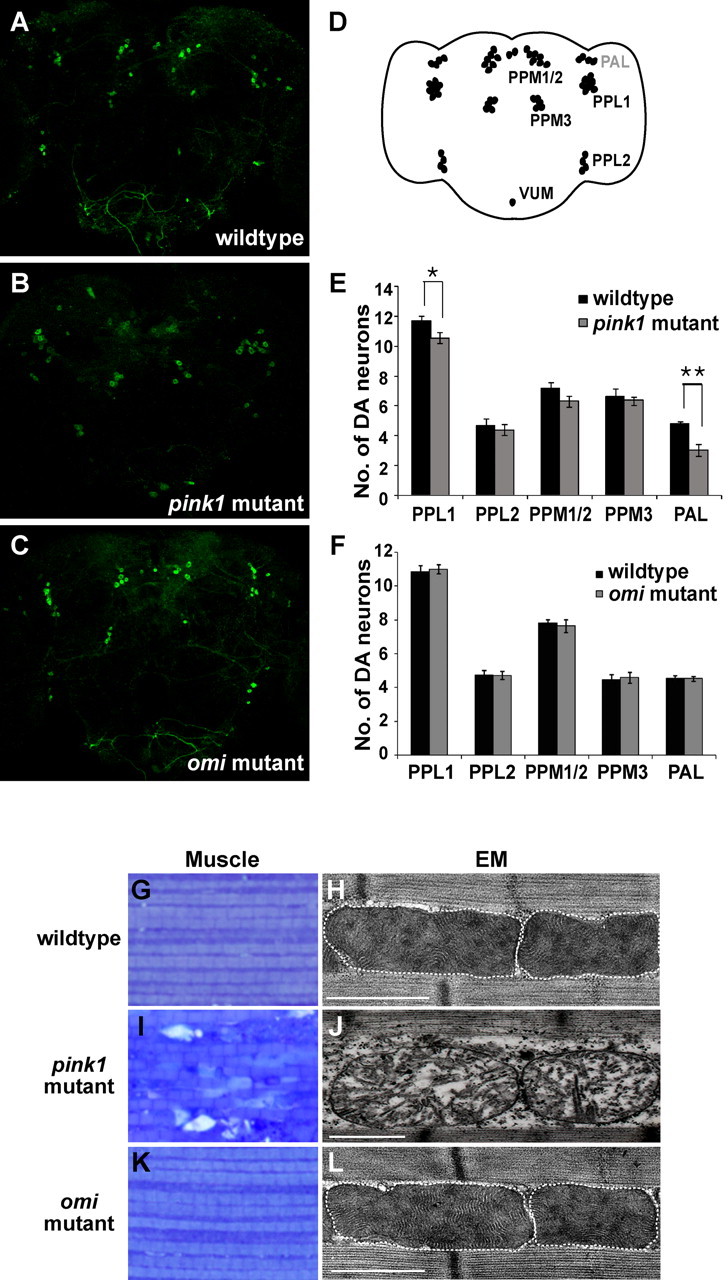
omi null mutants do not show dopaminergic neuronal loss, muscle degeneration, or mitochondrial morphological defects. A–C, Anti-tyrosine hydroxylase immunostaining of whole-mount brains from 40-d-old wild-type (A), pink1 mutant (B), and omi mutant (C) flies. Seventeen to twenty-seven individual flies (both hemispheres) were counted for each genotype. D, Schematic depicting locations of the major dopaminergic neuron clusters in the adult brain as designated by abbreviations (Nässel and Elekes, 1992). The major dopaminergic neuron clusters are located near the posterior surface of the brain, with the exception of the protocerebral anterior lateral (PAL), which is located near the anterior surface (labeled in gray). PPL, Protocerebral posterior lateral; PPM, protocerebral posterior medial; VUM, ventral unpaired medial. E, F, Quantification of dopaminergic neurons in each cluster in pink1 mutant flies and wild-type flies (E), and omi mutant flies and wild-type flies (F) aged for 40 d at 25°C. Error bars represent SDs, and Student's t test is used for statistical analysis. *p < 0.05; **p < 0.01. G–L, Toluidine blue staining of indirect flight muscle fibers (G, I, K) and EM studies of these muscles (H, J, L). The borders of mitochondria are marked with white dashed lines. In contrast to pink1 mutants (I, J), omi mutants do not show muscle degeneration or mitochondrial morphological defects (K, L), even when aged for 30 d. Genotypes: wild type: w/Y. omi mutant: w/Y; omiNSO/Df(3R)ED5644. pink1 mutant: w pink15/Y. Scale bars: H, J, L, 1 μm.
Loss-of-function studies fail to detect any genetic interactions between pink1 and omi
To further explore the hypothesis that omi and pink1 work together to regulate mitochondrial integrity, we searched for genetic interactions based on loss of function of these genes. Genetic interactions between pink1 and parkin provide an important reference for testing whether omi and pink1 act in a common pathway (Clark et al., 2006; Park et al., 2006). We, as well as others, have previously shown that Drosophila pink1 and parkin act in a common genetic pathway, with pink1 functioning upstream of parkin (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). This conclusion is based on several observations. Loss-of-function mutations in pink1 and parkin result in highly similar, if not identical, defects in mitochondrial integrity (Clark et al., 2006; Park et al., 2006; Yang et al., 2006; Deng et al., 2008; Poole et al., 2008). Although overexpression of parkin rescues pink1 null mutant phenotypes, overexpression of pink1 fails to rescue parkin null mutant phenotypes (Clark et al., 2006; Park et al., 2006). In addition, double mutants removing both pink1 and parkin show phenotypes identical to those of single mutants alone (Clark et al., 2006; Park et al., 2006).
In contrast to pink1 (Fig. 5C,H) or parkin overexpression (Clark et al., 2006), testes-specific omi overexpression did not rescue the male sterility or the mitochondrial phenotype of pink1 mutants (0% fertile, n = 65) (Fig. 5D,I). Expression of mutant versions of Omi analogous to S306A, S276C, G399S, or S400A (see below) also did not rescue male sterility of pink1 mutants (0% fertile, n > 30). Similarly, omi overexpression, which leads to massive loss of muscle integrity, also failed to rescue the muscle degeneration phenotypes seen in pink1 mutants. In addition, expression of a protease dead version of Omi, S266A, which does not result in loss of muscle integrity, also failed to rescue muscle phenotypes seen in pink1 mutants (data not shown). Consistent with the hypothesis that pink1 and omi function independently, neither the expression levels nor the mitochondrial localization of Omi was altered in pink1 mutants (Fig. 6D–F).
Figure 5.

Lack of genetic interactions between pink1 and omi in loss-of-function studies of mitochondrial morphology. A–J, Phase-contrast micrographs of spermatid mitochondria during the “onion stage” (A–E) and “the leaf blade stage” (F–J). pink1 null mutants show vacuolation of nebenkerns (B) and one single mitochondrial derivative (G). This phenotype can be completely suppressed by pink1 overexpression (overexpr.) (C, H), but not by omi overexpression (D, I). Double mutants removing both pink1 and omi function result in pink1 mutant-like phenotypes without any enhancement (E, J). Red arrowheads point to vacuolation of the nebenkern, and yellow arrows mark each mitochondrial derivative. K–N, Mitochondria of indirect flight muscle are labeled by mitoGFP. Compared with control (K), pink1 mutants display overall reduced levels of mitoGFP signal, and large clumps of intense GFP signal (L), which can be completely rescued by pink1 overexpression (M). Double mutants of pink1 and omi show pink1 mutant-like phenotypes (N). Genotypes: A, F, w. B, G, w pink15/Y. C, H, w pink15/Y; TMR-pink1/+. D, I, w pink15/Y; TMR-omi/+. E, J, w pink15/Y; omiNSO/Df(3R)ED5644. K, FM6/Y; Mef2-Gal4, UAS-mitoGFP/+. L, w pink15/Y; Mef2-Gal4, UAS-mitoGFP/+. M, w pink15/Y; Mef2-Gal4, UAS-mitoGFP/UAS-pink1. N, w pink15/Y; Mef2-Gal4, UAS-mitoGFP/UAS-RNAi-omi.
To investigate whether pink1 functions downstream of omi, we performed reverse rescue experiments. However, pink1 overexpression failed to rescue male sterility seen in omi mutants (0% fertile, n = 70). In addition, neither the protein levels nor cleavage patterns of Pink1 were altered in omi mutants (Fig. 6). Thus, we failed to find any positive evidence that omi functions either upstream or downstream of pink1 in a common pathway.
To test the hypothesis that omi might function in a parallel pathway with pink1 in a partially redundant manner, we generated double mutants that remove both pink1 and omi. These double mutant flies were viable and showed survival rates comparable to those of pink1 mutants alone. These animals were male sterile and exhibited mitochondrial morphological defects in spermatids and muscle that were indistinguishable from those of pink1 mutants alone, indicating that loss of omi function does not enhance pink1 mutant phenotypes (Fig. 5E,J,N; compare with B,G,L). Together, our loss-of-function in vivo studies do not provide support for the hypothesis that omi functions either upstream or downstream of pink1, or in parallel with pink1, at least with respect to the regulation of mitochondrial integrity.
PD-associated mutations in Omi, and a mutation abolishing a putative Pink1-dependent phosphorylation site, show distinct phenotypes from mutations impairing Omi protease function
Because we failed to detect any loss-of-function-based genetic interactions between pink1 and omi, we decided to examine the function of the PD disease-related omi mutations. The PD-associated polymorphism in Omi/HtrA2, A141S (detected in >1% of the normal population), and the mutation, G399S, have been reported to function as dominant-negative mutations, leading to a reduction of protease function of Omi/HtrA2 (Strauss et al., 2005). G399, which is located in the PDZ domain, is conserved in Drosophila, whereas A141, which is located in the IAP-binding domain of Omi/HtrA2, is not. Interestingly, S400, a residue next to G399, has been identified as a PINK1-dependent putative phosphorylation site for p38 (Plun-Favreau et al., 2007). This phosphorylation is reported to be important for Omi/HtrA2 activity, because S400A, a phosphorylation-incompetent mutation, markedly reduces protease activity (Plun-Favreau et al., 2007). To investigate whether these mutations affect Omi/HtrA2 function in vivo, we generated transgenic flies expressing Omi G363S or S364A, which are analogous to G399S or S400A in human Omi/HtrA2.
Both G399S and S400A reportedly compromise Omi protease activity in vitro (Strauss et al., 2005; Plun-Favreau et al., 2007). If this were true in vivo, G399S or S400A mutant forms of Omi would be expected to show similar phenotypes to protease-compromised Omi mutants. To test this hypothesis, we also generated two protease-impaired versions of Drosophila Omi, S266A and S236C. S266A is analogous to S306A in human Omi/HtrA2, which alters the active site serine in the protease domain and abolishes protease activity (Li et al., 2002), and S236C is analogous to the S276C mutation present in the mnd2 mice, which significantly reduces the protease function of Omi/HtrA2 (Jones et al., 2003) (Table 1). These mutants were expressed and assayed in multiple somatic tissues using the UAS-GAL4 system (Brand and Perrimon, 1993), and in the male germline using the TMR promoter (Huh et al., 2004; Clark et al., 2006). The actions of G363S and S364A were compared with that of wild-type Omi (Omi WT), as well as the protease-deficient S266A and S236C mutant forms.
Table 1.
Summary of phenotypic effects of various Omi mutants reported in this study and by others
| Genotype [fly (human)] | Molecular function from literature | Tissue-specific overexpression |
Muscle | Ability to rescue omi mutant sterility | ||
|---|---|---|---|---|---|---|
| Protease activity from literature | Testes | Eye | ||||
| Omi WT | Sterile | Small and rough | Disrupted | Rescue | ||
| S236C (S276C) | mnd2 mice | Reduced | Fertile | Wild type appearing | Wild type appearing | |
| S266A (S306A) | Protease active site | Protease dead | Fertile | Wild type appearing | Wild type appearing | Fail to rescue |
| G363S (G399S) | PD-associated mutation | Reduced | Sterile | Small and rough | Disrupted | Rescue |
| S364A (S400A) | pinkl-dependent phosphorylation site | Reduced | Sterile | Small and rough | Disrupted | |
In contrast to overexpression of Omi WT, which resulted in male sterility in most transgenic lines, all transgenic lines expressing S266A or S236C were male fertile (n > 13 transgenic lines tested for each mutant). Expression of S266A also failed to rescue the male sterility (0% fertile, n > 60) and empty seminal vesicle phenotypes resulting from lack of omi (Fig. 7B, Table 1). These data suggest that protease-compromised mutations result in loss of Omi function. Further supporting this hypothesis, eye-specific overexpression of S266A or S236C, in contrast to overexpression of Omi WT, resulted in wild-type-appearing eyes (Fig. 7E–G, Table 1). Similarly, muscle-specific overexpression of S266A or S236C, in contrast to Omi WT, did not affect muscle integrity (Table 1, data not shown). Expression of S266A and S236C were confirmed using an anti-Omi antibody (Fig. 7D). These results, together with those described earlier with the missense mutation, omiV110E, in the region responsible for activation of protease activity, suggest that Omi protease activity is important for its function in vivo.
Figure 7.
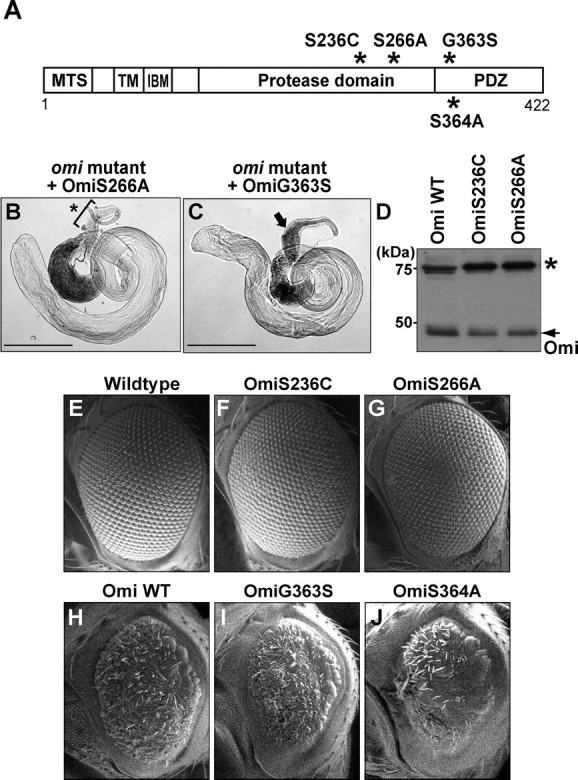
Functional analysis of Omi mutants. A, Schematic depicting positions of 4 mutations (*) with respect to domains in Omi. B, C, Phase-contrast micrographs. As with overexpression of Omi WT (Fig. 3E,E′), expression of G363S (C), but not S266A (B), restores the production of motile sperm in the seminal vesicle of omi mutants. An arrow in C indicates the presence of sperm (phase dark), whereas a bracket and an asterisk in B point to the absence of sperm. D, Both OmiS236C and OmiS266A are expressed at comparable levels compared with Omi WT, as detected by an anti-Omi antibody. Western blots of head lysates from flies overexpressing OmiWT, OmiS236C, or OmiS266A using anti-Omi antibodies are shown. Overexpression is accomplished using the eye-specific driver (GMR-Gal4), and flies are raised at 18°C to avoid cell death compromising recovery of proteins. A nonspecific band (*) serves as protein loading control. E–J, Scanning EM micrographs of Drosophila eyes. Compared with overexpression of Omi WT, which results in small and rough eyes at 25°C (H), overexpression of Omi G363S or S364A leads to similar rough eye phenotypes (I, J), whereas overexpression of Omi S236C or S266A results in wild-type-appearing eyes (F, G). GMR-Gal4 is used as an eye-specific driver. Scale bars: B, C, 500 μm.
In contrast, testes-specific expression of either OmiG363S or S364A resulted in significant male sterility, with only 3–4 lines of the 10–13 lines tested per construct giving fertile males, similar to what is seen with overexpression of Omi WT. These fertile lines likely represent those with lower expression levels. Using these fertile lines, we found that expression of G363S rescued the sterility and individualization phenotypes resulting from omi loss of function, as did those expressing Omi WT (Fig. 7C, Table 1). These results suggest that mutations analogous to G399S and S400A retain a significant amount of Omi activity. Further supporting this hypothesis, eye-specific overexpression of G363S or S364A resulted in small and rough eyes similar to those seen after overexpression of Omi WT (Fig. 7H–J, Table 1). Similarly, muscle-specific overexpression of G363S or S364A, or Omi WT, resulted in a massive loss of muscle integrity (Table 1, data not shown). Together, these observations (summarized in Table 1) suggest that Omi mutant proteins analogous to G399S and S400A behave similarly to Omi WT, but differently from those with compromised protease activity in vivo.
Discussion
The in vivo function of omi
Omi/HtrA2 has been studied extensively for its role in apoptosis (for review, see Vande Walle et al., 2008). However, whereas overexpression of Omi/HtrA2 induces apoptosis robustly in mammalian cells, mice lacking Omi/HtrA2 fail to show decreased apoptosis, but instead show nondopaminergic neuronal loss in the striatum (Martins et al., 2004). Omi/HtrA2 function is also implicated in regulating stress resistance (Vande Walle et al., 2008). Thus, determining the endogenous function of Omi/HtrA2 is crucially important to understanding its roles in both health and disease. Using Drosophila as a model, we have dissected the in vivo function of Omi. We find that omi is essential for spermatogenesis, stress resistance, and maintaining a normal life span. Furthermore, the protease activity of Omi is crucial for its function.
We have identified an essential role of omi during spermatogenesis. However, although Omi is localized to mitochondria in both testes and muscle, no mitochondrial morphology defects are observed in omi null mutants in either of these tissues. It is possible that Omi is responsible for some aspects of mitochondrial function, such as chaperone activity or modulation of respiratory chain function, which do not affect mitochondrial morphology, and thus are not detected in our assays. It is also possible that omi is required only in certain contexts, such as during exposure to oxidative stress, and that mitochondrial defects may be revealed in omi mutants under these conditions. Alternatively, Omi may function in the cytosol rather than in the mitochondria, with mitochondria serving to regulate the release of Omi into the cytosol. Future studies are required to distinguish these possibilities.
Interaction of Omi and Pink1
The genetic interactions observed between pink1 and parkin serve as an important reference for tests of the hypothesis that omi and pink1 act in a common pathway. In contrast to pink1 mutants, which show striking defects in mitochondrial integrity in muscle and testes, and a decrease in the number of dopaminergic neurons, omi mutants show normal mitochondrial morphology in both muscle and testes, and a normal number of dopaminergic neurons. Furthermore, in contrast to parkin overexpression, omi overexpression does not rescue pink1 mutant phenotypes. Overexpression of pink1 also fails to rescue male sterility resulting from omi loss of function. Lack of pink1 does not affect the levels or the subcellular localization of Omi, and Pink1 levels and processing are not altered in omi mutants. In addition, double mutants removing both pink1 and omi show identical phenotypes to pink1 mutants alone, suggesting that pink1 does not negatively regulate omi and that omi does not carry out partially redundant functions with pink1. Together, these data do not provide any in vivo evidence supporting the hypothesis that omi functions in the same pathway either upstream or downstream of pink1, or that it acts in a parallel manner to regulate mitochondrial morphology. These loss-of-function-based analyses are more relevant to PD than are omi overexpression-based analyses, because reported Omi/HtrA2 mutations associated with PD are proposed to represent loss-of-function or dominant-negative mutations (Strauss et al., 2005).
Genetic interactions between pink1 and omi have been observed by ourselves (see Results) and others (Whitworth et al., 2008) in eye-based overexpression studies: co-overexpression of pink1 with omi results in small eye phenotypes not associated with expression of either protein alone. Although in isolation, these results could be explained by a model in which omi and pink1 function in a common pathway (Whitworth et al., 2008), this model is difficult to reconcile with our loss-of-function data. The cellular basis of the pink1 overexpression-induced eye phenotype, and its relationship to the normal endogenous roles of pink1 in regulating mitochondrial function, is unclear. Although null mutants of pink1 and parkin show highly similar, if not identical, phenotypes in almost all assays tested, overexpression of pink1 results in a rough eye phenotype, whereas overexpression of parkin does not (data not shown) (Poole et al., 2008; Whitworth et al., 2008). Furthermore, a PD-causing, kinase-deficient mutant form of pink1, which fails to rescue pink1 null mutant phenotypes in multiple tissues, still interacts with omi in the eye-based overexpression assay, suggesting that Pink1 kinase activity is required for its mitochondrial functions but not for the genetic interaction with omi in this assay. Based on our findings, one is led to conclude that the functions of pink1 that mediate its co-overexpression interaction with omi are distinct from the functions of pink1 and parkin in regulating mitochondrial morphology. Such a mitochondrial integrity-independent role of pink may be important, but has yet to be identified in vivo. Alternatively, it is possible that the pink1-omi interaction observed in the eye is not physiologically relevant. Overexpression studies, as well as in vitro studies, can identify interactions that are forced to happen, but that do not normally occur. For example, either protein, when overexpressed, may act on inappropriate targets or act in inappropriate subcellular compartments, thus generating cellular toxicity. In combination, this toxicity may be augmented. This possibility is further suggested by the results of overexpression-based observations that place Rhomboid 7 as an upstream positive regulator of Pink1 (Whitworth et al., 2008). This conclusion is difficult to reconcile with more physiological loss-of-function-based observations showing that Drosophila rhomboid 7 functions to promote mitochondrial fusion (McQuibban et al., 2006), whereas both pink1 and parkin function to promote mitochondrial fission (Deng et al., 2008; Poole et al., 2008; Yang et al., 2008). In any case, our loss-of-function studies demonstrate that omi does not play an essential role in regulating mitochondrial integrity in the pink1/parkin pathway. They leave open the possibility that interactions between pink1 and omi are modulatory, or important in other contexts. However, these in vivo contexts remain to be identified.
Implications for Omi/HtrA2 as a PD gene
Omi/HtrA2 was recently designated as PARK13, based on a report identifying G399S mutations in sporadic PD patients (Strauss et al., 2005). Mammalian cell culture studies suggest that G399S results in a significant reduction in Omi/HtrA2 protease activity, providing a possible functional basis for disease association (Strauss et al., 2005). In contrast to previous in vitro observations (Strauss et al., 2005; Plun-Favreau et al., 2007), we find that both G399S and S400A retain significant, if not full, Omi function in vivo, leading to the conclusion that mutations previously thought to be associated with disease are functional in at least some contexts in vivo. Importantly, our conclusion is consistent with two recent reports showing that Omi G399S is found at similar frequencies in normal controls and PD patients (Ross et al., 2008; Simón-Sánchez and Singleton, 2008),
We cannot exclude the possibility that human Omi/HtrA2 has a dopaminergic neuron-specific function that is revealed under certain circumstances, nor can we exclude the possibility that Drosophila omi acts differently from human Omi/HtrA2. However, the extensive homology and conservation of key domain structures between fly and human Omi/HtrA2 suggest that it is likely that studies in Drosophila Omi are relevant to the function of Omi/HtrA2 in humans. Together with the observations that omi mutant phenotypes are distinct from those associated with loss of pink1 and parkin function, and that pink1 and omi fail to interact in loss-of-function-based assays, we favor a hypothesis in which Omi/HtrA2 does not play an essential role in PD pathogenesis.
Footnotes
J. H. Cao's present address: Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, CA 90095
R. B. Chowdhury's present address: Wales Cancer Trial Units, Neuadd Meirionnydd University Hospital of Wales, Cardiff CF14 4YS, UK.
I. E. Clark's present address: Department of Molecular, Cellular, and Developmental Biology, University of California, Los Angeles, Los Angeles, CA 90095.
This work was supported by a fellowship from the National Institutes of Health (NIH) National Research Service Award predoctoral fellowship to M.W.D., as well as grants and funds from NIH (R01, K02, K08), an American Parkinson Disease Association (APDA) grant, an APDA Center pilot grant, the Glenn Family Foundation, the Alfred P. Sloan Foundation, and the McKnight Foundation of Neuroscience to M.G. We are very grateful to M. Miura for antibodies, J. Belote for fly strains, N. Tapon and A. Whitworth for communicating unpublished results, A. Yamaguchi for generating one transgenic fly strain, B. A. Hay for insightful comments on this manuscript, F. Laski for use of his microtome, L. Cruz, H. Deng, and J. Olson for technical help, and the Guo laboratory members for discussions. J.Y. and J.H.C. performed experiments, M.W.D. performed most of the transmission electron microscopy studies, I.E.C., P.K., and R.B.C. contributed reagents, and M.G. performed experiments, supervised the project, and wrote the paper with helpful comments from M.W.D. and J.Y.
References
- BeilinaA, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR (2005) Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A 102:5703–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BogaertsV, Nuytemans K, Reumers J, Pals P, Engelborghs S, Pickut B, Corsmit E, Peeters K, Schymkowitz J, De Deyn PP, Cras P, Rousseau F, Theuns J, Van Broeckhoven C (2008) Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson disease. Hum Mutat 29:832–840. [DOI] [PubMed] [Google Scholar]
- BoniniNM, Fortini ME (2003) Human neurodegenerative disease modeling using Drosophila Annu Rev Neurosci 26:627–656. [DOI] [PubMed] [Google Scholar]
- BrandAH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401–415. [DOI] [PubMed] [Google Scholar]
- ChallaM, Malladi S, Pellock BJ, Dresnek D, Varadarajan S, Yin YW, White K, Bratton SB (2007) Drosophila Omi, a mitochondrial-localized IAP antagonist and proapoptotic serine protease. EMBO J 26:3144–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ChenCH, Huang H, Ward CM, Su JT, Schaeffer LV, Guo M, Hay BA (2007) A synthetic maternal-effect selfish genetic element drives population replacement in Drosophila Science 316:597–600. [DOI] [PubMed] [Google Scholar]
- ClarkIE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–1166. [DOI] [PubMed] [Google Scholar]
- DauerW, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39:889–909. [DOI] [PubMed] [Google Scholar]
- DengH, Dodson MW, Huang H, Guo M (2008) The Parkinson's disease genes pink1 and parkin promotes mitochondrial fission and/or inhibits mitochondrial fusion. Proc Natl Acad Sci U S A 105:14503–14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DodsonMW, Guo M (2007) Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol 17:331–337. [DOI] [PubMed] [Google Scholar]
- ExnerN, Treske B, Paquet D, Holmström K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Krüger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C (2007) Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 27:12413–12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FullerMT (1993) Spermatogenesis. In: The Development of Drosophila melanogaster, pp 71–147. Woodbury, NY: Cold Spring Harbor Laboratory.
- GangulyA, Feldman RM, Guo M (2008) ubiquilin antagonizes presenilin and promotes neurodegeneration in Drosophila Hum Mol Genet 17:293–302. [DOI] [PubMed] [Google Scholar]
- GautierCA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105:11364–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GreeneJC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 100:4078–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrossGG, Feldman RM, Ganguly A, Wang J, Yu H, Guo M (2008) Role of X11 and ubiquilin as in vivo regulators of the amyloid precursor protein in Drosophila PLoS ONE 3:e2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GuoM, Jan LY, Jan YN (1996) Control of daughter cell fates during asymmetric division: interaction of Numb and Notch. Neuron 17:27–41. [DOI] [PubMed] [Google Scholar]
- GuoM, Hong EJ, Fernandes J, Zipursky SL, Hay BA (2003) A reporter for amyloid precursor protein gamma-secretase activity in Drosophila Hum Mol Genet 12:2669–2678. [DOI] [PubMed] [Google Scholar]
- HardyJ, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A (2006) Genetics of Parkinson's Disease and parkinsonism. Ann Neurol 60:389–398. [DOI] [PubMed] [Google Scholar]
- HayBA, Wolff T, Rubin GM (1994) Expression of baculovirus P35 prevents cell death in Drosophila Development 120:2121–2129. [DOI] [PubMed] [Google Scholar]
- HuhJR, Vernooy SY, Yu H, Yan N, Shi Y, Guo M, Hay BA (2004) Multiple apoptotic caspase cascades are required in nonapoptotic roles for Drosophila spermatid individualization. PLoS Biol 2:E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IbáñezP, Lesage S, Lohmann E, Thobois S, De Michele G, Borg M, Agid Y, Dürr A, Brice A (2006) Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain 129:686–694. [DOI] [PubMed] [Google Scholar]
- IgakiT, Suzuki Y, Tokushige N, Aonuma H, Takahashi R, Miura M (2007) Evolution of mitochondrial cell death pathway: proapoptotic role of HtrA2/Omi in Drosophila Biochem Biophys Res Commun 356:993–997. [DOI] [PubMed] [Google Scholar]
- JonesJM, Albin RL, Feldman EL, Simin K, Schuster TG, Dunnick WA, Collins JT, Chrisp CE, Taylor BA, Meisler MH (1993) mnd2: a new mouse model of inherited motor neuron disease. Genomics 16:669–677. [DOI] [PubMed] [Google Scholar]
- JonesJM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnóczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES (2003) Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 425:721–727. [DOI] [PubMed] [Google Scholar]
- KhanFS, Fujioka M, Datta P, Fernandes-Alnemri T, Jaynes JB, Alnemri ES (2008) The interaction of DIAP1 with dOmi/HtrA2 regulates cell death in Drosophila Cell Death Differ 15:1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KitadaT, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. [DOI] [PubMed] [Google Scholar]
- KoundakjianEJ, Cowan DM, Hardy RW, Becker AH (2004) The Zuker collection: a resource for the analysis of autosomal gene function in Drosophila melanogaster Genetics 167:203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LiW, Srinivasula SM, Chai J, Li P, Wu JW, Zhang Z, Alnemri ES, Shi Y (2002) Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat Struct Biol 9:436–441. [DOI] [PubMed] [Google Scholar]
- MarshJL, Thompson LM (2006) Drosophila in the study of neurodegenerative disease. Neuron 52:169–178. [DOI] [PubMed] [Google Scholar]
- MartinsLM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL, Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz JB, Mak T, Downward J (2004) Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol 24:9848–9862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuibbanGA, Lee JR, Zheng L, Juusola M, Freeman M (2006) Normal mitochondrial dynamics requires rhomboid-7 and affects Drosophila lifespan and neuronal function. Curr Biol 16:982–989. [DOI] [PubMed] [Google Scholar]
- MeulenerM, Whitworth AJ, Armstrong-Gold CE, Rizzu P, Heutink P, Wes PD, Pallanck LJ, Bonini NM (2005) Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson's disease. Curr Biol 15:1572–1577. [DOI] [PubMed] [Google Scholar]
- NässelDR, Elekes K (1992) Aminergic neurons in the brain of blowflies and Drosophila: dopamine- and tyrosine hydroxylase-immunoreactive neurons and their relationship with putative histaminergic neurons. Cell Tissue Res 267:147–167. [DOI] [PubMed] [Google Scholar]
- ParkJ, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441:1157–1161. [DOI] [PubMed] [Google Scholar]
- PesahY, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G (2004) Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131:2183–2194. [DOI] [PubMed] [Google Scholar]
- PiccoliC, Sardanelli A, Scrima R, Ripoli M, Quarato G, D'Aprile A, Bellomo F, Scacco S, De Michele G, Filla A, Iuso A, Boffoli D, Capitanio N, Papa S (2008) Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem Res 33:2565–2574. [DOI] [PubMed] [Google Scholar]
- Plun-FavreauH, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J (2007) The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol 9:1243–1252. [DOI] [PubMed] [Google Scholar]
- PooleAC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A 105:1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PrzedborskiS, Ischiropoulos H (2005) Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson's disease. Antioxid Redox Signal 7:685–693. [DOI] [PubMed] [Google Scholar]
- Rathke-HartliebS, Schlomann U, Heimann P, Meisler MH, Jockusch H, Bartsch JW (2002) Progressive loss of striatal neurons causes motor dysfunction in MND2 mutant mice and is not prevented by Bcl-2. Exp Neurol 175:87–97. [DOI] [PubMed] [Google Scholar]
- RiparbelliMG, Callaini G (2007) The Drosophila parkin homologue is required for normal mitochondrial dynamics during spermiogenesis. Dev Biol 303:108–120. [DOI] [PubMed] [Google Scholar]
- RossOA, Soto AI, Vilariño-Güell C, Heckman MG, Diehl NN, Hulihan MM, Aasly JO, Sando S, Gibson JM, Lynch T, Krygowska-Wajs A, Opala G, Barcikowska M, Czyzewski K, Uitti RJ, Wszolek ZK, Farrer MJ (2008) Genetic variation of Omi/HtrA2 and Parkinson's disease. Parkinsonism Relat Disord 14:539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-SánchezJ, Singleton AB (2008) Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum Mol Genet 17:1988–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- StraussKM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Müller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Krüger R (2005) Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet 14:2099–2111. [DOI] [PubMed] [Google Scholar]
- TillBJ, Reynolds SH, Greene EA, Codomo CA, Enns LC, Johnson JE, Burtner C, Odden AR, Young K, Taylor NE, Henikoff JG, Comai L, Henikoff S (2003) Large-scale discovery of induced point mutations with high-throughput TILLING. Genome Res 13:524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ValenteEM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, et al (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304:1158–1160. [DOI] [PubMed] [Google Scholar]
- Vande WalleL, Lamkanfi M, Vandenabeele P (2008) The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ 15:453–460. [DOI] [PubMed] [Google Scholar]
- WhitworthAJ, Lee JR, Ho VM, Flick R, Chowdhury R, McQuibban GA (2008) Rhombiod-7 and HtrA2/Omi act in a common pathway with the Parkinson's disease factors Pink1 and Parkin. Dis Model Mech 1:168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood-KaczmarA, Gandhi S, Yao Z, Abramov AS, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, Downward J, Mansfield L, Jat P, Taylor J, Heales S, Duchen MR, Latchman D, Tabrizi SJ, Wood NW (2008) PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 3:e2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YangY, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A 103:10793–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YangY, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B (2008) Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A 105:7070–7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZhongL, Belote JM (2007) The testis-specific proteasome subunit Prosalpha6T of D. melanogaster is required for individualization and nuclear maturation during spermatogenesis. Development 134:3517–3525. [DOI] [PubMed] [Google Scholar]