

Abstract
The phosphatidylinositide 3-kinase pathway is frequently deregulated in human cancers and inhibitors offer considerable therapeutic potential. We previously described the promising tricyclic pyridofuropyrimidine lead and chemical tool compound PI-103. We now report the properties of the pharmaceutically optimized bicyclic thienopyrimidine derivatives PI-540 and PI-620 and the resulting clinical development candidate GDC-0941. All four compounds inhibited phosphatidylinositide 3-kinase p110α with IC50 ≤ 10 nmol/L. Despite some differences in isoform selectivity, these agents exhibited similar in vitro antiproliferative properties to PI-103 in a panel of human cancer cell lines, with submicromolar potency in PTEN-negative U87MG human glioblastoma cells and comparable phosphatidylinositide 3-kinase pathway modulation. PI-540 and PI-620 exhibited improvements in solubility and metabolism with high tissue distribution in mice. Both compounds gave improved antitumor efficacy over PI-103, following i.p. dosing in U87MG glioblastoma tumor xenografts in athymic mice, with treated/control values of 34% (66% inhibition) and 27% (73% inhibition) for PI-540 (50 mg/kg b.i.d.) and PI-620 (25 mg/kg b.i.d.), respectively. GDC-0941 showed comparable in vitro antitumor activity to PI-103, PI-540, and PI-620 and exhibited 78% oral bioavailability in mice, with tumor exposure above 50% anti-proliferative concentrations for >8 hours following 150 mg/kg p.o. and sustained phosphatidylinositide 3-kinase pathway inhibition. These properties led to excellent dose-dependent oral antitumor activity, with daily p.o. dosing at 150 mg/kg achieving 98% and 80% growth inhibition of U87MG glioblastoma and IGROV-1 ovarian cancer xenografts, respectively. Together, these data support the development of GDC-0941 as a potent, orally bioavailable inhibitor of phosphatidylinositide 3-kinase. GDC-0941 has recently entered phase I clinical trials.
Introduction
The phosphatidylinositide 3-kinase family consists of 15 members that are divided into four distinct classes based on their structure and biological properties (1-4). This highly conserved family of enzymes is involved in various aspects of cellular homeostasis and is deregulated in a number of pathophysiologic conditions. Consequently, phosphatidylinositide 3-kinases have become the focus of concerted drug discovery efforts in several disease areas, including immunity, inflammation, cardiology, and cancer (5). The class I, II, and III enzymes are lipid kinases, whereas the class IV enzymes are protein kinases (DNA-PK, ATM or ATR, and mTOR; refs. 6-8). The class I lipid kinases catalyze phosphorylation of the 3-hydroxyl position of phosphatidylinositols, mainly converting phosphatidylinositol (4, 5) diphosphate into phosphatidylinositol (3,4,5) triphosphate (6). The formation of phosphatidylinositol (3,4,5) triphosphate results in recruitment of a number of protein effectors to the plasma membrane, whereby they become activated, resulting in the assembly of signaling complexes and activation of downstream pathways leading to cell proliferation, motility, invasion, and angiogenesis, all of which are deregulated in cancer (7-12). Class IA enzymes are activated by receptor tyrosine kinases and cytokine receptors, which are frequently overexpressed or have activating mutations in many malignancies (12, 13). In addition, the PIK3CA gene that encodes the class IA p110α isoform is mutated or amplified in 15% of cancers overall, and the opposing negative regulator, the phosphatidylinositol (3,4,5) triphosphate phosphatase PTEN, is mutated, deleted, or silenced in a high proportion of malignancies (14-17). Furthermore, persistent signaling through the phosphatidylinositide 3-kinase/AKT pathway has been implicated as a major mechanism of resistance to chemotherapeutic agents, as well as those targeting the epidermal growth factor receptor family (18). Finally, recent data show that inhibition of MAP kinase extracellular signal-regulated kinases 1 and 2 (MEK 1/2), which has also been the focus of much drug discovery effort, causes activation of phosphatidylinositide 3-kinase signaling, suggesting that phosphatidylinositide 3-kinase inhibition may be valuable even in those tumors that do not have a primary activation of the phosphatidylinositide 3-kinase pathway (19). The evidence that so many diverse cancers may benefit from phosphatidylinositide 3-kinase inhibition has fuelled the development of inhibitors, with the ultimate aim of identifying clinical drug candidates.
The natural product wortmannin and the flavone LY294002 have been important laboratory tools that have contributed to our understanding of the importance of the phosphatidylinositide 3-kinase pathway and indicated the therapeutic potential of small molecule inhibitors (20-22). There has been considerable progress recently in the discovery and development of phosphatidylinositide 3-kinase inhibitors with improved pharmaceutical properties and various patterns of isoform selectivity (23, 24).
With our collaborators Hayakawa et al. (25-28), we have previously reported the discovery of three new series of phosphatidylinositide 3-kinase inhibitors and described the detailed pharmacologic properties of a novel synthetic lead compound of the tricyclic pyridofuropyrimidine class, PI-103 (29, 30). PI-103 is a potent and selective inhibitor of class I phosphatidylinositide 3-kinases, and also of mTOR and DNA-PK, which blocked the proliferation of human cancer cells in vitro and caused pharmacodynamic biomarker effects consistent with target inhibition (29-31). PI-103 showed therapeutic activity against a range of human tumor xenografts, exhibiting inhibition of angiogenesis, invasion, and metastasis, as well as direct antiproliferative effects (29, 31, 32). Although PI-103 provided in vivo proof of concept for the therapeutic potential of the pyridofuropyrimidine series, this compound suffered from limited solubility and extensive metabolism. A multiparameter lead optimization program focusing on improving pharmaceutical, pharmacokinetic, and pharmacodynamic properties has resulted in the identification of the clinical development candidate GDC-0941 (33). Here, we describe in detail the properties of two pharmacologically optimized additional lead candidates, the bicyclic thienopyrimidines PI-540 and PI-620, together with those of GDC-0941. PI-540 and PI-620 exhibited improved solubility and reduced metabolism with high tissue distribution and showed antitumor activity in the U87MG human glioblastoma xenograft model, which is PTEN negative and has an activated phosphatidylinositide 3-kinase pathway. The high bioavailability of GDC-0941 resulted in oral efficacy against the U87MG glioblastoma and IGROV-1 human ovarian cancer xenograft models in athymic mice. This very potent, orally bioavailable class I phosphatidylinositide 3-kinase inhibitor is currently undergoing phase I clinical trials under the auspices of Genentech.
Materials and Methods
Compound Supply
The synthesis of PI-103 was described by Hayakawa et al. (28), and the syntheses of PI-540, PI-620, and GDC-0941 were based on schemes described by Folkes et al. (33). Chemical structures are shown in Fig. 1A.
Figure 1.

Chemical structures of phosphatidylinositide 3-kinase inhibitors and their molecular and cellular effects on cancer cells in vitro. A, chemical structures of PI-103, PI-540, PI-620, and GDC-0941. B, in vitro IC50 values for PI-103, PI-540, PI-620, and GDC-0941 against recombinant phosphatidylinositide 3-kinase enzymes. C, effects of agents on tumor cell and human umbilical vein endothelial cell (HUVEC) proliferation. Data shown as the mean of three independent determinations of GI50 following 96-h continuous exposure to compound. The tumor cell lines used exhibit a variety of mechanisms of deregulation of the phosphatidylinositide 3-kinase pathway: PC3 and U87MG are PTEN null; IGROV-1 has a hetT319F deletion and frameshift in PTEN, a p85 binding domain hetR38C mutation of p110α, and an additional hetX1069W mutation that extends the C-terminus of p110α by four amino acids; and Detroit 562 and SKOV-3 have a hetH1047R mutation of the p110α kinase domain. D, schematic of some key proteins in the phosphatidylinositide 3-kinase pathway. E, effects of agents on molecular biomarkers. Electrochemiluminescent immunoassay (Meso Scale Discovery) analysis of the phosphorylation of AKT Thr308, AKT Ser473, GSK3β Ser9, p70S6K Thr421/Ser424, and S6 ribosomal protein Ser235/Ser236. U87MG cells were treated for 2 or 8 h with 5 to 500 nmol/L phosphatidylinositide 3-kinase inhibitor or 10 to 2,500 nmol/L rapamycin. Values shown are the mean IC50 ±SD.
Enzyme Assays
Phosphatidylinositide 3-kinase inhibitory activity was determined using a scintillation proximity assay in the presence of 1 μmol/L ATP (29, 33). Inhibition of mTOR protein kinase was determined using a TR-FRET-based LanthaScreen method from Invitrogen. Compounds were assayed at a maximum concentration of 10 μmol/L in the presence of 1 μmol/L ATP, and IC50 values were determined using GraphPad Prism software (33).
Cell Culture
The human tumor cell lines U87MG, PC3, SKOV-3, IGROV-1, Detroit 562, HCT116, SNUC2B, and LoVo were obtained from the American Type Culture Collection. All cancer cell lines were grown in DMEM (Invitrogen) containing 2 mmol/L glutamine (Gibco-BRL), with 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco-BRL), and supplemented with 10% fetal bovine serum (PAA Laboratories) in 5% CO2 in air at 37°C. Human umbilical vein endothelial cells (pooled) and their appropriate growth medium and supplements were obtained from TCS CellWorks. Cells were cultured according to the supplier’s instructions and used at passages 3 to 8. Cell viability was routinely >90%, as judged by trypan blue exclusion. All cell lines routinely tested negative for mycoplasma by PCR. GI50 values (concentrations) causing 50% inhibition of proliferation for tumor cells were determined using an Alamar Blue or a sulforhodamine B assay and, for human umbilical vascular endothelial cells, by an alkaline phosphatase assay following 96 h continuous exposure to compounds (29, 33). Antibiotics were removed before this assay.
PTEN status was verified in most cell lines, including U87MG and IGROV-1, by protein expression using Western blotting in-house, and in addition, PIK3CA status was determined or verified experimentally by sequencing. In the case of the colon cancer cell lines, PTEN status was again confirmed experimentally in-house by Western blotting. In addition, single-nucleotide polymorphism (SNP) profiling was used to confirm that the genotypes matched those provided in the Cancer Genome Project Cosmic database3 from which data on KRAS and PIK3CA mutation status was subsequently obtained.
Translocation, Immunoblotting, and Phosphoprotein Immunoassay on Cell Lines
Forkhead translocation assays were done as described previously (29). Immunoblotting was done as follows (29, 30). Cells were seeded in six-well plates at 3 to 5 × 105 cells/well in 2 mL medium, allowed to attach overnight, and treated with phosphatidylinositide 3-kinase inhibitors for the times indicated. After the incubation period, the medium was carefully removed from wells, and ~150 μL ice-cold Cell Extraction Buffer (Biosource International) supplemented with Protease Inhibitor (Sigma) and Phosphatase Inhibitor (Roche Applied Science) was added to each well. Cell supernatant was collected after centrifugation at 14,000 × g at 4°C for 10 min, and its protein concentration was quantified using the BCA Protein Assay Kit (Pierce).
For Western blotting (29, 30), 30 μg of each lysate was separated by SDS-PAGE, electrotransferred onto nitrocellulose membranes, and probed with specific primary antibody and horseradish peroxidase-conjugated secondary antibody (Cell Signalling Cambridge). Signal was detected with enhanced chemiluminescence reagent (Amersham Biosciences). Glyceraldehyde-3-phosphate dehydrogenase was used as the loading control.
For the electrochemiluminescent immunoassays (34), a Meso Scale Discovery system was used. Multispot phospho-AKT Ser473/total AKT assays were done with 10 μg protein in duplicate. The protocol used was as recommended by the manufacturer, except that samples were incubated on the plates overnight before addition of secondary antibody. IC50 values were determined (30) for inhibition of the phosphorylation of Thr308-AKT (singleplex plate); Ser473-AKT; Ser9-GSK3β;Thr421/Ser424-p70S6K (triplex plate) and total AKT, GSK3β, and p70S6K (triplex plate); and Ser235/Ser236 and total-S6 ribosomal protein (duplex plate). Briefly, cells were seeded at 8 × 104 cells/mL in 96-well plates, and 48 h later, they were treated with compounds for 2 or 8 h. Medium was then removed, and 50 μL of lysis buffer was added. Plates were freeze thawed once at -80°C and 40 μL of lysate was transferred directly onto the Meso Scale Discovery plate, and analysis was completed as described previously (34). For each treatment condition, a single well from each of three independent plates was analyzed.
Pharmacokinetics and Metabolism
All animal experiments were done in accordance with local and national United Kingdom Co-ordinating Committee on Cancer Research guidelines (35). Female BALB/c mice (6-8 wk old; Charles River) were dosed i.v. and p.o. with 10 mg/kg PI-540 or PI-620 (free base) in 10% DMSO-0.5% Tween 20 in saline (10 mL/kg), which did not cause hemolysis. Blood was collected after serial bleeding and centrifuged, and the plasma was frozen at -80°C. Tissues were snap frozen in dry ice and kept at -80°C until analysis. Quantitative analysis was done by liquid chromatography tandem mass spectrometry using multiple reaction monitoring, as described previously (29). Pharmacokinetic linearity was examined following i.p. administration of 25, 50, and 100 mg/kg PI-540 (dimesylate salt) and 12.5, 25, and 50 mg/kg PI-620 (HCl salt) in water. GDC-0941 (dimesylate salt) was administered p.o. at 50 mg/kg to female CrTac:Ncr-Fox1 (nu) athymic mice bearing established U87MG human glioblastoma xenografts (see below). Sampling and analysis were done as detailed above.
Xenograft Tumor Efficacy and Pharmacodynamic Studies
Two million U87MG human glioblastoma cells were injected s.c., bilaterally, into female 6- to 8-wk-old CrTac: Ncr-Fox1(nu) [Ncr] athymic mice bred in-house. PI-540 was prepared in sterile saline, PI-620 in sterile water, and GDC-0941 in 10% DMSO, 5% Tween 20, and 85% sterile saline. Compounds were dosed in 0.1 mL/10 g body weight of vehicle once or twice daily (PI-540 and PI-620 i.p.; GDC-0941 p.o.). Control animals received an equivalent volume of appropriate vehicle. Dosing for therapy studies commenced when solid tumors were well established (~5 mm mean diameter) and continued according to the schedule indicated in the figure legends. Tumors were measured across two perpendicular diameters, and volumes were calculated according to the formula: V = 4/3π [(d1 + d2) / 4]3 (29). Animals were weighed regularly and observed for adverse effects. When the experiment was terminated, mice were bled, plasma samples prepared, and tumors excised and weighed. Values of the percentage treated/control (T/C) were calculated from the treated versus control final tumor weights. Tumor samples were snap frozen for pharmacokinetic and/or pharmacodynamic analysis at intervals after the last dose. For dedicated pharmacodynamic studies, animals were dosed for 4 d and samples obtained as before. Plasma and tumor samples were analyzed for compound concentrations and tumor samples assessed for evidence of biomarker modulation by Meso Scale Discovery electrochemiluminescence immunoassay and/or immunoblot, as previously described (29, 34). In some experiments, IGROV-1 human ovarian cancer xenografts were studied using similar methods to those for U87MG.
Results
In vitro Potency against Phosphatidylinositide 3-Kinase and mTOR
Figure 1A shows the chemical structures and Fig. 1B illustrates the potency of PI-103, PI-540, PI-620, and GDC-0941 against each of the class I phosphatidylinositide 3-kinase enzymes and the class IV protein kinases mTOR and DNA-PK. All four compounds potently inhibited p110α with IC50 ≤ 10 nmol/L. PI-103 was at least an order of magnitude more potent against p110β. PI-540 and PI-620 had relatively low potency against p110γ with IC50 > 300 nmol/L, whereas PI-103 and GDC-0941 exhibited potencies of 15 and 75 nmol/L, respectively. PI-103 and PI-540 were more potent against mTOR than PI-620 and GDC-0941, and PI-103 was more potent than all the others against DNA-PK. Each of the compounds showed a similar high degree of selectivity toward class I phosphatidylinositide 3-kinases when profiled against a large panel of >70 protein kinases (refs. 29 and 33; data not shown).
Inhibition of Cell Proliferation In vitro
Figure 1C shows the cellular GI50 values of the four compounds evaluated in a panel of human cancer cell lines comprising prostate, ovary, glioblastoma, and oropharyngeal squamous carcinoma, together with human umbilical vein endothelial cells, following 96 hours continuous exposure. The tumor cell lines have different genetic abnormalities that can result in activation of the phosphatidylinositide 3-kinase pathway (see ref. 29 and legend to Fig. 1C). All compounds exhibited potent growth inhibition in each of the cell lines examined, with activity in the submicromolar range. PI-540 and PI-620 were less potent than PI-103 and GDC-0941 in some cell lines, for example, in IGROV-1 and human umbilical vein endothelial cells (Fig. 1C). However, in the Detroit 562 oropharyngeal cancer cells, the GI50 values were very similar (0.13-0.50 μmol/L) for all four compounds.
Target Modulation Following Treatment with Phosphatidylinositide 3-Kinase Inhibitors In vitro
We have previously reported inhibitory effects of PI-103 on the phosphatidylinositide 3-kinase pathway activity in various human cancer cells (29). We used immunoblotting to show pathway inhibition by PI-540 and PI-620 in U87MG glioblastoma and PC3 prostate cancer cells and, in addition, in A549 lung adenocarcinoma cells (Supplementary Fig. 1).4 Furthermore, 50% inhibition of forkhead transcription factor translocation was observed at 62 and 81 nmol/L for PI-540 and PI-620, respectively, compared with the previously reported 30 nmol/L for PI-103 (29).
Next, we examined the potency of the inhibitors in U87MG cells against various phosphorylated protein biomarkers of the phosphatidylinositide 3-kinase pathway using a set of electrochemiluminescent immunoassays (Meso Scale Discovery). Assays included phosphorylation at Thr308-AKT, Ser473-AKT, Ser9-GSK3β, Thr421/Ser424-p70S6K, and Ser235/Ser236-S6 ribosomal protein (Fig. 1D). In view of the activity of the compounds against mTOR kinase (Fig. 1B), the mTORC1 inhibitor rapamycin was also included for comparison. The results in Fig. 1E show very similar 2- and 8-hour IC50 values for PI-103, PI-540, PI-620, and GDC-0941 against each of the biomarkers of phosphatidylinositide 3-kinase pathway activity studied. The four phosphatidylinositide 3-kinase inhibitors were most potent against phosphorylation of AKT on both sites, with IC50 values in the range 10 to 40 nmol/L. Potency decreased by 7-to 12-fold with respect to phosphorylation of proteins further downstream of phosphatidylinositide 3-kinase. For example, PI-540 was 10-fold less potent in inhibiting phosphorylation of GSK3β Ser9 when compared with phosphorylation of AKT. Rapamycin treatment did not affect phosphorylation of AKT or GSK3β but inhibited phosphorylation of p70S6K and S6 ribosomal protein at 2 hours and, more potently, at 8 hours, an activity consistent with inhibition of mTORC1. In line with their relatively weaker effect on mTOR kinase activity, the 8-hour IC50 values of the four synthetic inhibitors on phosphorylation of ribosomal S6 protein on Ser235 was significantly less than that of rapamycin.
Given that the phosphatidylinositide 3-kinase inhibitors, especially GDC-0941, exhibited more potent antiproliferative activity against IGROV-1 ovarian cancer cells compared with U87MG glioblastoma cells (Fig. 1C), we examined the effects of PI-103 and GDC-0941 on the phosphorylation of AKT Ser473 as a sensitive biomarker of phosphatidylinositide 3-kinase inhibition in IGROV-1 cells and compared the results with those described above for U87MG cells (Fig. 1E). The IC50 values for the inhibition of phosphorylation of Ser473 on AKT in IGROV-1 cells following 2- or 8-hour exposure were 18 ± 2 and 17 ± 4 nmol/L, respectively, for PI-103 and 18 ± 1 and 38 ± 13 nmol/L, respectively, for GDC-0941. These values for the ovarian cancer line were remarkably similar to the values in the U87MG glioblastoma cells (Fig. 1E) despite the lower antiproliferative potency of the inhibitors in the glioblastoma line.
Finally, we compared the IC50 values for inhibition of Ser473 phosphorylation on AKT in three human colon cancer cell lines (Supplementary Table 1,4 which includes data on PTEN, PIK3CA, and KRAS status). Despite the fact that the antiproliferative GI50 values for PI-103 ranged >37-fold from 22 nmol/L (for LoVo) to 827 nmol/L (for SNUC2B), the IC50 values for the inhibition of phosphorylation of Ser473 on AKT after 2-hour exposure ranged only 2-fold from 18 nmol/L (for LoVo and SNU2CB) to 38 nmol/L for (HCT116). In the case of GDC-0941, the antiproliferative GI50 values ranged 9-fold from 180 nmol/L (for LoVo) to 1,627 nmol/L (for SNUC2B), whereas the IC50 values for inhibition of AKT phosphorylation on Ser473 following 2-hour treatment again ranged only 2-fold from 14 nmol/L (for HCT116) to 33 nmol/L (for SNUC2B). When these results for the colon cancer lines are considered together with the ovarian cancer and glioblastoma cell data (see above), it is clear that the degree of phosphatidylinositide 3-kinase inhibition is remarkably similar across all cancer cell lines, whereas the consequences in terms of antiproliferative potency are very different, indicating a differential antiproliferative response to a given degree of phosphatidylinositide 3-kinase blockade.
Pharmacokinetics of PI-540 and PI-620
The pharmacokinetics of PI-540 and PI-620 administered i.v. and p.o. to mice at 10 mg/kg are shown in Fig. 2A and B, respectively. Both compounds exhibited high plasma clearance with very large volumes of distribution (300 and 206 mL, respectively). The extensive distribution was confirmed by the high tissue concentrations, as shown by spleen to plasma ratios of 31 and 13.9, respectively, following i.v. dosing. Terminal half-lives after i.v. administration were short in plasma (0.24 and 0.9 hours) but longer in tissues (7.6 and 4.3 hours for PI-540 and PI-620, respectively). Both compounds were poorly orally bioavailable (33), with values <10% in each case, but they were well absorbed from the peritoneal cavity and showed linear pharmacokinetics at well-tolerated doses (Fig. 2C and D). This resulted in tumor concentrations above GI50 in athymic mice bearing U87MG glioblastoma xenografts for ~4 hours following 100 mg/kg PI-540 and 50 mg/kg PI-620 (Fig. 3A and B). Based on the tumor levels achieved, the concentrations would be expected to be above GI50 concentrations for ~ 4 hours following twice daily i.p. administration of 50 mg/kg PI-540 or 25 mg/kg PI-620. Also, concentrations were above GI50 for about 3.5h following 50 mg/kg PI-620.
Figure 2.

Pharmacokinetics of PI-540 and PI-620 in mice. A, plasma and spleen concentration-time profile of PI-540 following 10 mg/kg administration i.v. and p.o. to female BALB/c mice. Pharmacokinetic parameters are derived from WinNonlin noncompartmental analysis model 201 and 200 for i.v. and p.o., respectively. Concentrations were measured by liquid chromatography tandem mass spectrometry in A to D. B, plasma and spleen concentration versus time profile of PI-620 following 10 mg/kg administration i.v. and p.o. to female BALB/c mice. Pharmacokinetic parameters are derived from WinNonlin noncompartmental analysis model 201 and 200 for i.v. and p.o., respectively. C, plasma concentration versus time profiles following i. p. administration of PI-540 at 25, 50, and 100 mg/kg to female BALB/c mice. Pharmacokinetic parameters are derived from WinNonlin noncompartmental analysis model 200. D, plasma concentration versus time profiles following i.p. administration of PI-620 at 12.5, 25, and 50 mg/kg to female BALB/c mice. Pharmacokinetic parameters are derived from WinNonlin noncompartmental analysis model 200. Values are mean of n = 3 animals per time point.
Figure 3.
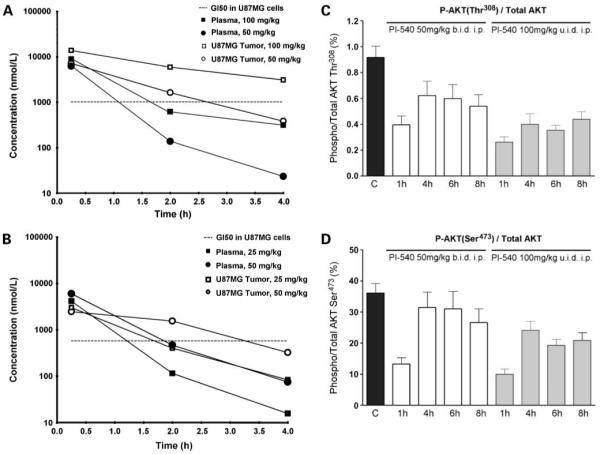
In vivo pharmacokinetic and pharmacodynamic properties of PI-540 and PI-620 in mice. A and B, concentrations of PI-540 (A) or PI-620 (B)in plasma and tumors of NCr athymic mice bearing well-established U87MG human glioblastoma xenografts. Animals were given a single i.p. administration of compound. Plasma and tumors were collected at 0.25, 2, and 4 h following compound administration. Concentrations were measured by liquid chromatography tandem mass spectrometry. C and D, U87MG human glioblastoma xenografts were grown s.c. bilaterally in female NCr athymic mice. Animals with well established tumors were given 4 d of treatment with PI-540 at 50 mg/kg b.i.d or 100 mg/kg u.i.d i.p. At 1, 4, 6, and 8 h after the last dose, tumors were excised, snap frozen, and assayed for levels of total and Thr308 phosphorylated AKT (C) and total and Ser473 phosphorylated AKT (D). Results were obtained by electrochemiluminescent immunoassay (Meso Scale Discovery). For pharmacokinetic data, results are mean of n = 3 animals per time point. For pharmacodynamic biomaker data, results are mean ± SE of n =6.
Target Modulation and Antitumor Activity of PI-540 and PI-620 in U87MG Glioblastoma Xenografts
Based on the above pharmacokinetic results, athymic mice bearing well-established U87MG glioblastoma xenografts received short courses of treatment with PI-540 (50 mg/kg b.i.d i.p. or 100 mg/kg u.i.d i.p) or PI-620 (25 mg/kg b.i.d i.p. or 50 mg/kg u.i.d. i.p.) for 4 days to examine their ability to inhibit the phosphatidylinositide 3-kinase pathway in tumor tissue in vivo. Electrochemiluminescence immunoassay analysis of the tumors showed that AKT phosphorylation was inhibited in a dose-dependent and time-dependent manner. Figure 3C and D show that phosphorylation on AKT Ser473 and AKT Thr308 was inhibited by >50% at 1 hour by PI-540 using both dose schedules. Levels remained below control values over the 8-hour time-course for the latter biomarker, although recovery was evident by 4 hours in the 50 mg/kg b.i.d schedule for phosphorylation of AKT Ser473. Downstream of AKT, both schedules gave more transient inhibition of the phosphorylation of P70S6K, but there was no detectable inhibition of phosphorylation of GSK3β (Fig. 4A and B). PI-620 also inhibited the phosphorylation of AKT at both sites at 1 hour, although recovery was complete by 4 hours at the lower doses used with this compound (Fig. 4C and D). Transient inhibition of phosphorylation of P70S6K and GSK3β was also seen (Fig. 5A and B).
Figure 4.
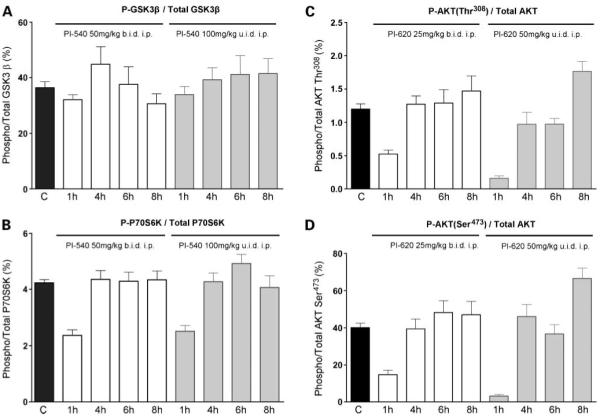
In vivo pharmacodynamic effects of PI-540 and PI-620 in mice. U87MG human glioblastoma xenografts were grown s.c. bilaterally in female NCr athymic mice. Animals with well-established tumors were given 4 d of treatment with PI-540 at 50 mg/kg b.i.d i.p. or 100 mg/kg u.i.d i.p. (A and B) and PI-620 at 25 mg/kg b.i.d i.p. or 50 mg/kg u.i.d i.p. (C and D). At 1, 4, 6, and 8 h after the last dose, tumors were excised, snap frozen, and assayed for levels of total and Ser9 phosphorylated GSK3β (A), total and Thr421/Ser424 phosphorylated P70S6K(B), total and Thr308 phosphorylated AKT (C), and total and Ser473 phosphorylated AKT (D). Results were obtained by electrochemiluminescent immunoassay (Meso Scale Discovery). Results are mean ± SE of n =6.
Figure 5.
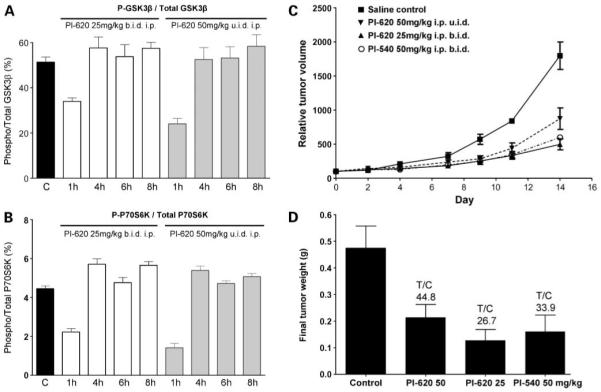
In vivo pharmacodynamic properties of PI-620 and antitumor activity of PI-540 and PI-620 in mice. A and B, U87MG human glioblastoma xenografts were grown s.c. bilaterally in female NCr athymic mice. Animals with well established tumors were given 4 d of treatment with PI-620 at 25 mg/kg b.i.d i.p. or 50 mg/kg u.i.d i.p. At 1, 4, 6, and 8 h after the last dose, tumors were excised, snap frozen, and assayed for levels of total and Ser9 phosphorylated GSK3β (A) or total and Thr421/Ser424 phosphorylated P70S6K(B). Results were obtained by electrochemiluminescent immunoassay (Meso Scale Discovery). C, relative mean tumor volume (percentage of initial volume before therapy) following daily administration of PI-540 (50 mg/kg b.i.d i.p.) and PI-620 (50 mg/kg u.i.d i.p and 25 mg/kg b.i.d i.p.) for 14 d. Established tumors were grown s.c. bilaterally in female NCr athymic mice, and controls received equivalent volumes of vehicle. D, mean final tumor weights following administration of PI-540 (50 mg/kg b.i.d i.p.) and PI-620 (50 mg/kg u.i.d i.p. and 25 mg/kg b.i.d i.p.) for 14 d. For pharmacodynamic biomarker data, results are mean ± SE of n = 6. For therapy data, the results are mean ± SE of n = 16.
In a subsequent efficacy study, PI-540 and PI-620 were dosed i.p. at 50 mg/kg once or twice a day and PI-620 was also dosed at 25 mg/kg twice a day for 14 days to athymic mice bearing established U87MG human glioblastoma xenografts. At these very well tolerated doses, the growth rate of the tumors was slowed significantly (Fig. 5C), and final T/C values were 33.9% (66.1% inhibition) for PI-540 and 44.8% and 26.7% (55.2% and 73.3% inhibition) for PI-620 dosed at 50 mg/kg u.i.d and 25 mg/kg b.i.d, respectively (Fig. 5D).
Pharmacokinetics of GDC-0941
The fast plasma and tissue clearance of PI-103 was the result of rapid glucuronidation of the phenol group (29). Despite decreases in mouse and human microsomal metabolism of PI-540 and PI-620 when compared with PI-103, significant in vivo glucuronidation was still observed (33). This accounts for the rapid clearance described in the previous section. To remove this metabolic liability, various phenol isosteres were synthesized and tested. The indazole derivative GDC-0941, which also contained the solubilizing sulfonyl piperazine, showed limited microsomal metabolism, resulting in 78% oral bioavailability (33), in addition to its potent inhibitory activity on the phosphatidylinositide 3-kinase pathway (Fig. 1B and E). Figure 6A shows the pharmacokinetics of GDC-0941 administered p.o. at 75 mg/kg to athymic mice bearing U87MG glioblastoma xenografts. GDC-0941 was very rapidly absorbed with Cmax achieved 30 minutes postadministration. Tumor distribution was equally rapid with Cmax reached at the same time. Although the tumor to plasma ratio was around 0.8, these properties resulted in tumor concentrations of compound well above the GI50 at 6 hours postadministration.
Figure 6.
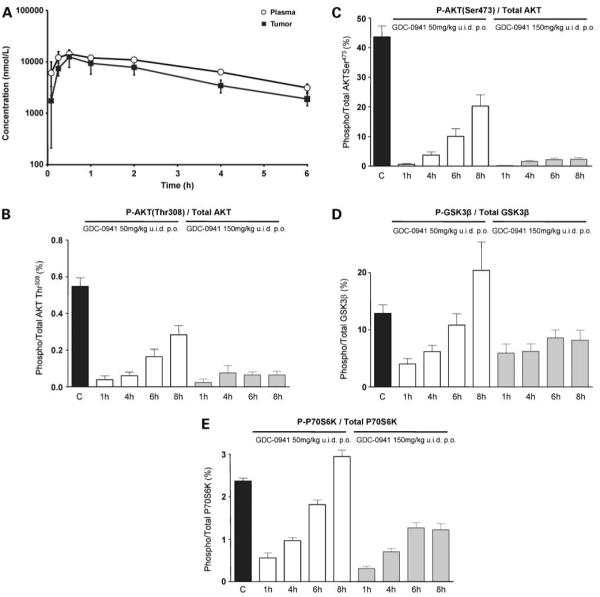
In vivo pharmacokinetic properties and pharmacodynamic effects of GDC-0941 in mice. A, mean plasma and tumor concentration versus time profile of GDC-0941 following a single oral administration of 50 mg/kg to female NCr athymic mice bearing s.c. U87MG human glioblastoma xenografts. B-E, U87MG human glioblastoma xenografts were grown s.c. bilaterally in female NCr athymic mice. Animals with well established tumors were given 4 d of oral treatment with GDC-0941 at 50 mg/kg or 150 mg/kg u.i.d. p.o. At 1, 4, 6, and 8 h after the last dose, tumors were excised, snap frozen, and assayed for total and Thr308 phosphorylated AKT (B), total and Ser473 phosphorylated AKT (C), total and Ser9 phosphorylated GSK3β (D), and total and Thr421/Ser424 phosphorylated P70S6K(E). Results were obtained by electrochemiluminescent immunoassay (Meso Scale Discovery). For pharmacokinetic data, results are mean ± SD of n = 3. For pharmacodynamic biomarker data, results are mean ± SE of n =6.
GDC-0941 Causes Sustained Inhibition of the Phosphatidylinositide 3-Kinase Pathway in U87MG Glioblastoma Xenografts
GDC-0941 was administered to athymic mice once daily p.o. at 50 mg/kg or 150 mg/kg for 4 days and phosphatidylinositide 3-kinase pathway activation in U87MG tumor xenografts measured as before by electrochemiluminescence immunoassay. Figure 6B and Cshow that both schedules resulted in dramatic reduction of levels of AKT phosphorylation (at both sites) and that inhibition was maintained for the 8-hour observation period, especially at the higher dose. Downstream in the phosphatidylinositide 3-kinase pathway, phosphorylation of GSK3β and P70S6K was also significantly inhibited. There was a gradual recovery to normal levels by 8 hours following 50 mg/kg doses; however, suppression was maintained at the 150 mg/kg dose (Fig. 6D and E).
Tumor Growth Inhibition and Pathway Modulation by GDC-0941 in U87MG Glioblastoma Xenografts
Based on its promising combination of potent phosphatidylinositide 3-kinase inhibitory activity and good oral bioavailability, we next investigated the antitumor activity of GDC-0941 following oral dosing. A dose-dependent inhibition of the growth of well established U87MG glioblastoma xenografts was observed when daily doses were administered p.o. to athymic mice for 19 days. Of note, at all doses above 25 mg/kg, the mean tumor volumes at day 19 were below the original volumes, indicating a degree of tumor regression (Fig. 7A). T/Cbased on final tumor weights ranged from 23.4% (76.6% inhibition) at 25 mg/kg to 2.3% (97.7% inhibition) at 150 mg/kg (Fig. 7B). The therapy was well tolerated, and all groups of mice gained weight at similar rates to controls (data not shown). Electrochemiluminescence immunoassay confirmed that the levels of activated AKT Ser473 at 4 hours after the last dose were reduced in a dose-dependent manner, being undetectable at the 150 mg/kg dose level. Phosphorylation of AKT had recovered by 8 hours following dosing at 25 mg/kg but remained partially (50 mg/kg) or completely (100 and 150 mg/kg) suppressed at the higher doses (Fig. 7C). We measured GDC-0941 concentrations in these tumor samples at 4 and 8 hours following the final dose and related them to drug levels measured in U87MG glioblastoma cells treated with GI50 concentrations of GDC-0941. The GDC-0941 was rapidly taken up into U87MG cells in vitro at 1 hour posttreatment and levels were relatively constant over 96 hours (24.8 ± 2.7 nmole/g protein). The results of the tumor uptake study are shown in Fig. 7D. Our findings suggested that, at doses of 100 and 150 mg/kg GDC-0941, tumor levels were above intracellular concentrations at GI50 levels for over 8 hours. In contrast, following 25 and 50 mg/kg, the tumor GDC-0941concentrations were higher than GI50 levels for <4 hours. These results were consistent with the pharmacodynamic biomarker modulation and antitumor activity described above.
Figure 7.
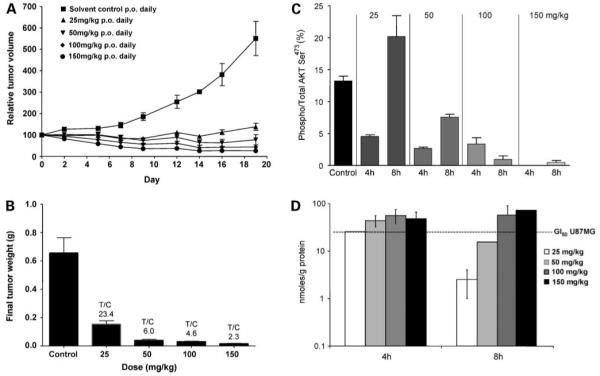
In vivo oral antitumor activity and associated pharmacokinetic and pharmacodynamic properties of GDC-0941 in U87MG human glioblastoma xenografts. A, relative mean tumor volume (percentage of initial volume before therapy) following 25, 50, 100, and 150 mg/kg daily doses of GDC-0941 p.o. for 19 d in the established human U87MG human glioblastoma xenograft model. Tumors were grown s.c. bilaterally in female NCr athymic mice, and controls received equivalent volumes of vehicle. B, mean final tumor weights for the U87MG human glioblastoma xenograft model following 25, 50, 100, and 150 mg/kg daily GDC-0941 p.o. for 19 d. C, pharmacodynamic effects in the U87MG human glioblastoma xenograft model. Tumors were from the same efficacy study as shown in A and B and were taken 4 and 8 h following the final dose. AKT phosphorylation Ser473 and total AKT were measured by electrochemiluminescent immunoassay (Meso Scale Discovery). D, tumor concentrations of GDC-0941 in the U87MG human glioblastoma xenograft model measured by liquid chromatography tandem mass spectrometry following 19 d daily oral dosing with 25, 50, 100, and 150 mg/kg in the efficacy study. Tumors were collected 4 and 8 h after the final dose. For therapy data, results are mean ± SE of n = 16. For pharmacodynamic biomarker data, results are mean ± SE of n = 6. For pharmacokinetic data, results are mean ± SD of n = 3.
Because evidence of regression was seen in U87MG glioblastoma xenografts treated with GDC-941, we looked for evidence of apoptosis. There was a clear increase in poly(ADP-ribose) polymerase cleavage in tumor samples taken 4 hours after oral dosing with 25 to 150 mg/kg GDC-0941, indicative of induction of apoptosis (Supplementary Fig. 2).4
Tumor Growth Inhibition and Pathway Modulation by GDC-0941 in IGROV-1 Ovarian Cancer Xenografts
Because IGROV-1 ovarian cancer cells were very sensitive to GDC-0941 in vitro (Fig. 1C), we determined the response in the setting of an in vivo solid tumor xenograft. The results showed that GDC-0941 exhibited marked dose-dependent antitumor activity by the oral route against well established IGROV-1 ovarian carcinoma xenografts (Supplementary Fig. 3A).4 The T/C values decreased from 50.5% (49.5% inhibition) at 25 mg/kg to 19.7% (80.3% inhibition) at 150 mg/kg (Supplementary Fig. 3B).4 Similar to results described in the previous section for the U87MG glioblastoma model, the inhibition of phosphorylation of AKT Ser47 was consistent with the antitumor efficacy, with both time-dependent and dose-dependent reduction of this biomarker of phosphatidylinositide 3-kinase inhibition clearly apparent (Supplementary Fig. 3C).4
Discussion
A substantial body of evidence shows the high frequency of genetic abnormalities that occur in the phosphatidylinositide 3-kinase pathway in human cancers and that are involved in the initiation, progression, and spread of tumors (5-17). As a result, drug discovery programs have been carried out with the aim of developing small molecule inhibitors of phosphatidylinositide 3-kinase. A number of agents have been described with varying levels of selectivity against class I phosphatidylinositide 3-kinase isoforms, DNA-PK, ATM, or mTOR (20-33). We have previously described PI-103, a small molecule pan-class I inhibitor that also targets DNA-PK and mTOR (29, 30). PI-103 showed that a relatively selective phosphatidylinositide 3-kinase inhibitor could show therapeutic activity in a number of human tumor xenograft models with various abnormalities in the phosphatidylinositide 3-kinase pathway. For example, PI-103 exhibited >50% growth inhibition in xenografts of the PTEN-null U87MG glioblastoma (29). These promising antitumor effects were observed despite the fact that the pharmacokinetic properties of PI-103 are suboptimal. This compound shows poor solubility because of its tricyclic core structure. In addition, it has a number of metabolic hotspots, especially the phenol ring, which we have shown to be extensively glucuronidated, resulting in rapid plasma and tissue clearance (29).
We show here the impact of the improvement in the pharmaceutical features on the overall pharmacologic behavior, pharmacokinetic and pharmacodynamic properties, and antitumor efficacy of the optimized compounds. The bicyclic thienopyrimidines PI-540 and PI-620 retain the phenol ring present in PI-103 and have solubilizing groups in position 6, namely, 4-methyl-piperazin-1-yl-methyl and 4-(2-hydroxyethyl)-piperazin-1-yl-methyl for PI-540 and PI-620, respectively. These compounds retained low nanomolar potency against p110α, being only 3-to 4-fold less potent than PI-103. In addition, they were 10- to 20-fold less potent than PI-103 against p110β. Inhibition of p110δ was very similar to that of PI-103, but these agents were generally less active against p110γ, mTOR, and DNA-PK. Selectivity for class I phosphatidylinositide 3-kinases versus a large number of protein kinases was very high. Despite the differences in selectivity patterns within the class I phosphatidylinositide 3-kinases, PI-540 and PI-620 retained submicromolar potency against human cancer cell lines with various activating abnormalities of the phosphatidylinositide 3-kinase pathway. The inhibitory activity on the phosphatidylinositide 3-kinase pathway in human cancer cells was shown by immunoblotting, quantitative electrochemiluminescence immunoassays, and forkhead translocation assays. Microsomal metabolism was significantly decreased for these compounds (33), although their plasma clearances remained high as a result of metabolism and tissue distribution. Despite the rapid clearance of PI-540 and PI-620, the high volume of distribution and high tumor-to-plasma ratios were sufficient to allow phosphatidylinositide 3-kinase pathway modulation and antitumor activity in the U87MG glioblastoma xenograft model. Thus, PI-540 and PI-620 gave 66% and 73% inhibition of U87MG tumor growth, which is greater than that seen with PI-103.
Replacement of the phenol by an indazole in GDC-0941 eliminated the glucuronidation seen with PI-540 and PI-620 (33), and as a result this agent showed a low plasma clearance (less than half that of liver blood flow in mice) and exhibited 78% oral bioavailability at 10 mg/kg. GDC-041 showed very similar potency to PI-103 against p110α and p110δ but was less active against p110β (10-fold) and p110γ (5-fold). In addition, GDC-0941 was much less potent on mTOR and DNA-PK. Importantly, the activity of GDC-0941 against the panel of human tumor cell lines was generally similar to that of PI-103, suggesting that high potency against mTOR and/or DNA-PK was not essential for the inhibition of cell proliferation. In addition, GDC-0941 potently inhibited growth of activated human endothelial cells, suggesting potential for antiangiogenic activity, as we previously reported for PI-103 (29).
The pattern of biomarker modulation in vitro following treatment of cells with all four compounds was similar, with potent IC50 values against phosphorylation of AKT on Ser473 and Thr308. However, differences in biomarker modulation and antitumor potency in vivo were seen as a result of improved pharmaceutical properties for PI-540, PI-620, and GDC-0941. For example, in U87MG glioblastoma xenografts, at best 50% inhibition of phosphorylation of AKT Ser473 was observed for a short time following PI-103 treatment (29), whereas GDC-0941 was able to maintain inhibition for in excess of 8 hours. This pharmacodynamic biomarker effect was consistent with compound exposure in tumor tissue. The antitumor activity improved in parallel with tumor exposure and the resulting biomarker modulation, with an enhancement from PI-103 to PI-540/620 and then from PI-540/620 to GDC-0941. GDC-0941 showed impressive dose-responsive therapeutic effects against established U87MG glioblastoma xenografts at doses of 25 to 150 mg/kg, with 98% growth inhibition seen at the highest dose. Tumor regression was also observed with evidence of apoptosis. Target modulation was time dependent and dose dependent as measured by inhibition of phosphorylation of AKT Ser473, and the pharmacokinetic-pharmacodynamic relationships were consistent with antitumor activity. Thus, the results provided a satisfactory pharmacologic audit trail (36). Prolonged tumor growth delay and phosphatidylinositide 3-kinase pathway biomarker modulation was also seen in established IGROV-1 ovarian cancer xenografts, a model that, like U87MG, also has a deregulated phosphatidylinositide 3-kinase pathway.
The main objective of the present paper was to describe the critical drug discovery activities in the optimization from PI-103 through PI-540 and PI-620 and leading to the clinical development candidate GDC-0941. It is beyond the scope of this article to address in detail the factors that may predispose cancer cells to sensitivity and resistance to the class or phosphatidylinositide 3-kinase inhibitors described herein. Previous studies with other phosphatidylinositide 3-kinase inhibitors have shown that these may be active in cancers with PIK3CA mutations or other phosphatidylinositide 3-kinase pathway abnormalities and that cancers driven by KRAS mutations may not be responsive, although in some cases, there is evidence that synergy may be achieved in KRAS mutant tumors by combining phosphatidylinositide 3-kinase and MEK 1/2 inhibitors (37-40). Because the present article describes key aspects of a drug discovery program, the cancer cell lines and xenograft models used were selected deliberately because they exhibited deregulated phosphatidylinositide 3-kinase signaling by mechanisms also found in human malignancies in the clinic. Nevertheless, initial tentative interpretations about effects of certain oncogenic abnormalities can be made from the pattern of responses to the thienopyrimidine class of agents studied here across the panel of cancer cell lines investigated thus far.
Firstly, it is clear that any differences in in vitro sensitivity to these agents between the various cancer cell lines studied here cannot be due to differences in the degree of phosphatidylinositide 3-kinase inhibition because this was shown to be remarkably similar, with IC50 values for inhibition of phosphorylation of Ser473 varying only around 2- to 3-fold across the cancer cell line panel compared with a much greater variation in GI50 values for the antiproliferative response. This clearly points to a differential antiproliferative response to a given degree of phosphatidylinositide 3-kinase blockade, indicating the involvement of additional factors. It is interesting to note that, as observed with PI-103 previously (30, 41), the quantitative IC50 values for phosphatidylinositide 3-kinase pathway inhibition are much lower than the GI50 values for the antiproliferative response. This suggests that >50% inhibition of the pathway is needed to arrest cancer cell growth by 50%.
Secondly, assessment of antiproliferative sensitivity in relation to PIK3CA, PTEN,or KRAS status suggests that there is no obvious simple picture emerging to date for the class of thienopyrimidine phosphatidylinositide 3-kinase inhibitors studied here. For example, in the small panel of three human colon cancer cell lines studied in the present article, the LoVo line (wild-type PTEN, wild-type PIK3CA, mutant KRAS)has alower GI50 for GDC-0941 (180 nmol/L) than HCT116 (wild-type PTEN, mutant PIK3CA, mutant KRAS), which has a GI50 of 905 nmol/L, although SNUC2CB (wild-type PTEN, wild-type PIK3CA,mutant KRAS) does have the highest GI50 of 1,627 nmol/L. Also of note is that there is an overlap in sensitivity between the three colon tumor lines, which all have mutant KRAS, and that of the other cancer cell lines studied here (Fig. 1 and C; Supplementary Table 1).4 Interestingly, in an independent study on a panel of cancer lines, there was again no obvious pattern relating in vitro sensitivity to GDC-0941 to mutation status of genes such as PIK3CA, PTEN,or KRAS, and among additional human tumor xenografts that responded to GDC-0941 was a non-small cell lung cancer with mutant KRAS (42). Finally, it should be highlighted that nonmalignant human umbilical vein endothelial cells are shown here to be very sensitive to the phosphatidylinositide 3-kinase inhibitors, indicating a dependence on phosphatidylinositide 3-kinase activity. It is therefore quite likely that the in vivo response that is seen in an animal tumor model could be affected by an antiangiogenic component of phosphatidylinositide 3-kinase inhibition, as we noted previously for PI-103 (29). Finding predictive biomarkers that can identify patients who will be most responsive to phosphatidylinositide 3-kinase inhibitors of various types, in addition to the proof of mechanism, target inhibition biomarkers of the type described here, will clearly be an important future goal (24, 43), together with evaluation of GDC-0941 in a broader panel of tumors with different molecular pathologies.
In summary, the present article has shown a progression in the multiparametric optimization of the molecular and pharmaceutical properties of a series of phosphatidylinositide 3-kinase inhibitors from PI-103 to PI-540 and PI-620 and then to GDC-0941. Class I phosphatidylinositide 3-kinase activity was retained, including especially high potency for GDC-0941 against p110α and p110δ, and much greater selectivity for these Class I phosphatidylinositide 3-kinase targets versus mTOR and DNA-PK was seen. A high degree of selectivity versus protein kinases was maintained. At the same time, pharmaceutical properties such as solubility and metabolism were enhanced. Despite relatively fast plasma clearance, PI-540 and PI-620 exhibited high tumor to plasma ratios and high absolute inhibitor concentrations in tumor compared with antiproliferative GI50 values in vitro, resulting in greater antitumor activity than PI-103 in the PTEN negative U87MG glioblastoma model. The enhanced metabolic stability of GDC-0941 reduced the systemic clearance and increased oral bioavailability leading to sustained tumor compound levels despite the lower tumor to plasma ratios, resulting in excellent pharmacologic phosphatidylinositide 3-kinase pathway biomarker modulation and even greater antitumor activity than was seen than with PI-540 and PI-620. Antitumor activity for GDC-0941 was confirmed in the PTEN mutant and PIK3CA mutant IGROV-1 ovarian cancer xenograft. Based on its molecular pharmacologic properties, oral bioavailability and promising oral antitumor activity, GDC-0941 has entered phase I clinical trials in cancer patients.
Supplementary Material
Effect of PI-540 or PI-620 on molecular biomarkers in human cancer cells; A549 lung adenocarcinoma, U87MG glioblastoma and PC3 prostate. Cells were treated with PI-540 or PI-620 for 2 h, lysates prepared and immunoblotting carried out for the proteins indicated. GAPDH was used as a loading control.
Effect of GDC-0941 on PARP cleavage in established human U87MG glioblastoma xenografts in athymic. Mice were treated with doses of 25, 50, 100 and 150 mg/kg p.o GDC-941 and tumors removed at 4h post-dosing. Control animals (marked 0) received drug vehicle alone. The positive control (marked +ve) was HT29 colon cancer cells treated with 50 nM SN38. GAPDH was used as the loading control.
In vivo oral antitumor activity and associated pharmacodynamic effects of GDC-941 in IGROV-1 ovarian cancer xenografts.
Relative mean tumor volume (% of initial volume prior to therapy) in the established IGROV-1 human ovarian cancer xenograft model following 25, 50, 100 and 150mg/kg oral daily doses of GDC-941 for 13 days. Established tumors were grown s.c. bilaterally in femal NCr athymic mice and controls received equivalent volumes of vehicle.
Mean final tumor weights in the IGROV-1 human ovarian cancer xenograft model following oral daily dosing with 25, 50, and 100 and 150 mg/kg oral daily doses GDC-941 for 13 days.
Pharmacodynamic effects measured in IGROV-1 human ovarian cancer xenografts from the above efficacy study 4h and 8h following the final dose. Phosphorylation of AKT Ser473 and total AKT were measured by electrochemiluminescent immunoassay (MSD).
Effect of GDC-0941 and PI-103 on proliferation and phosphorylation of AKT at Ser473 in human colon cancer cell lines. HCT116 has wild type PTEN, mutant PIK3CA and mutant KRAS. SNUC2B has wild type PTEN, wild type PIK3CA and mutant KRAS. LoVo has wild type PTEN, wild type PIK3CA and mutant KRAS.
Acknowledgments
We thank Dr. Wendy Alderton for her contribution to the project. We also thank David Knowles, Michael Moore, Peter Parker, and Mike Waterfield for their support and encouragement; Lori Friedman for helpful comments on the manuscript; and Swee Sharp and Val Cornwell for assistance with manuscript preparation.
Grant support: Cancer Research UK program grant numbers C309/A8274 and C309/A2187 and National Health Service funding to the National Institute for Health Biomedical Research Centre.
Footnotes
Supplementary material for this article is available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
Florence I. Raynaud, Suzanne A. Eccles, Sonia Alix, Gary Box, Sharon Gowan, Alex De Haven Brandon, Francesca Di Stefano, Angela Hayes, Alan T. Henley, Edward McDonald, Peter Sheldrake, Melanie Valenti, Paul A. Clarke, and Paul Workman are or were employees of The Institute of Cancer Research, which has a commercial interest in the development of phosphatidylinositide 3-kinase inhibitors, including GDC-0941, and operates a rewards-to-inventors scheme. They have been involved in a commercial collaboration with Yamanouchi (now Astellas Pharma) and with Piramed Pharma, and intellectual property arising from the program has been licensed to Genentech. Sonal Patel, Irina Chuckowree, Adrian Folkes, Letitia Lensun, Giles Pergl-Wilson, Anthony Robson, Nahid Saghir, Alexander Zhyvoloup, Stephen Shuttleworth, and Nan Chi Wan were employees of Piramed Pharma, which has been acquired by Roche. Paul Workman was a founder of, consultant to, and Scientific Advisory Board member of Piramed Pharma. Florence I. Raynaud is a consultant for ELARA Pharmaceuticals.
References
- 1.Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22:267–72. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- 2.Rameh LE, Cantley LC. The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem. 1999;274:8347–50. doi: 10.1074/jbc.274.13.8347. [DOI] [PubMed] [Google Scholar]
- 3.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 4.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436:127–50. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 5.Crabbe T, Welham MJ, Ward SG. The PI3K inhibitor arsenal: choose your weapon! Trends Biochem Sci. 2007;32:450–6. doi: 10.1016/j.tibs.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253:239–54. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- 7.Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI 3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–9. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- 8.Liang J, Slingerland JM. Multiple roles of the PI 3-K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–45. [PubMed] [Google Scholar]
- 9.Franke TF, Kaplan DR, Cantley LC. PI 3-Kinase: downstream AKTion blocks apoptosis. Cell. 1997;88:435–7. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 10.Jimenez C, Portela RA, Mellado M, et al. Role of the PI 3-K regulatory subunit in the control of actin organization and cell migration. J Cell Biol. 2000;151:249–62. doi: 10.1083/jcb.151.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang BH, Liu LZ. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets. 2008;8:19–26. doi: 10.2174/156800908783497122. [DOI] [PubMed] [Google Scholar]
- 12.Brader S, Eccles SA. Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis. Tumori. 2004;90:2–8. doi: 10.1177/030089160409000102. [DOI] [PubMed] [Google Scholar]
- 13.Wymann MP, Zvelebil M, Laffargue M. Phosphoinositide 3-kinase signalling—which way to target? Trends Pharmacol Sci. 2003;24:366–76. doi: 10.1016/S0165-6147(03)00163-9. [DOI] [PubMed] [Google Scholar]
- 14.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27:5486–96. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shayesteh L, Lu Y, Kuo WL, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;21:99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- 17.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 18.She QB, Solit D, Basso A, Moasser MM. Resistance to gefitinib in PTEN-null HER-overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3′-kinase/Akt pathway signaling. Clin Cancer Res. 2003;9:4340–6. [PubMed] [Google Scholar]
- 19.Pratilas CA, Halivovic E, Persaud Y, et al. BRAF mutation predicts MEK-dependence in non-small cell lung cancer (NSCLC) Proc AACR-NCI-EORTC. 2007;C210:324. [Google Scholar]
- 20.Davol PA, Bizuneh A, Frackelton AR., Jr. Wortmannin, a phosphoinositide 3-kinase inhibitor, selectively enhances cytotoxicity of receptor-directed-toxin chimeras in vitro and in vivo. Anticancer Res. 1999;19:1705–13. [PubMed] [Google Scholar]
- 21.Arcaro A, Wymann MP. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem J. 1993;296:297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase,2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–8. [PubMed] [Google Scholar]
- 23.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–26. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 24.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Hayakawa M, Kaizawa H, Moritomo H, et al. Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110α inhibitors. Bioorg Med Chem. 2006;14:6847–58. doi: 10.1016/j.bmc.2006.06.046. [DOI] [PubMed] [Google Scholar]
- 26.Hayakawa M, Kaizawa H, Kawaguchi K, et al. Synthesis and biological evaluation of imidazo[1,2-a]pyridine derivatives as novel PI3 kinase p110α inhibitors. Bioorg Med Chem. 2007;15:403–12. doi: 10.1016/j.bmc.2006.09.047. [DOI] [PubMed] [Google Scholar]
- 27.Hayakawa M, Kawaguchi K, Kaizawa H, et al. Synthesis and biological evaluation of sulfonylhydrazone-substituted imidazo[1,2-a]pyridines as novel PI3 kinase p110α inhibitors. Bioorg Med Chem. 2007;15:5837–44. doi: 10.1016/j.bmc.2007.05.070. [DOI] [PubMed] [Google Scholar]
- 28.Hayakawa M, Kaizawa H, Kawaguchi K, et al. Synthesis and biological evaluation of pyrido[3′,2′:4,5]furo[3,2-d]pyrimidine derivatives as novel PI3 kinase p110α inhibitors. Bioorg Med Chem Lett. 2007;17:2438–42. doi: 10.1016/j.bmcl.2007.02.032. [DOI] [PubMed] [Google Scholar]
- 29.Raynaud FI, Eccles S, Clarke PA, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–50. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 30.Guillard S, Clarke PA, Te Poele R, et al. Molecular pharmacology of phosphatidylinositol 3-kinase inhibition in human glioma. Cell Cycle. 2009;8:443–53. doi: 10.4161/cc.8.3.7643. [DOI] [PubMed] [Google Scholar]
- 31.Knight ZA, Gonzalez B, Feldman M, et al. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell. 2006;125:733–47. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan QW, Knight ZA, Goldenberg DD, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioblastoma. Cancer Cell. 2006;9:341–9. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Folkes AJ, Ahmadi K, Alderton WK, et al. The identification of 2-(1H-Indazol-4)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 Kinase for the treatment of cancer. J Med Chem. 2008;51:5522–32. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 34.Gowan SM, Hardcastle A, Hallsworth AE, et al. Application of meso scale technology for the measurement phosphoproteins in human tumor xenografts. Assay Drug Dev Technol. 2007;5:391–401. doi: 10.1089/adt.2006.044. [DOI] [PubMed] [Google Scholar]
- 35.Workman P, Balmain A, Hickman JA, et al. United Kingdom Co-ordinating Committee on Cancer Research (UKCCR) guidelines for the welfare of animals in experimental neoplasia (second edition) Br J Cancer. 1997;77:1–10. doi: 10.1038/bjc.1998.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarker D, Workman P. Pharmacodynamic biomarkers for molecular cancer therapeutics. Adv Cancer Res. 2007;96:213–68. doi: 10.1016/S0065-230X(06)96008-4. [DOI] [PubMed] [Google Scholar]
- 37.Halilovic E, She Q-B, Ye Q, Solit D, Rosen N. Coexistent PI3 mutation in human tumours is associated with decreased dependency on mutant KRAS and MEK/ERK signalling for transformation. Proc AACR. 2008;49:4938. [Google Scholar]
- 38.Serra V, Markman B, Scaltriti M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–30. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 39.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ihle NT, Lemos R, Jr., Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143–50. doi: 10.1158/0008-5472.CAN-07-6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powis G, Ihle NT, Yung WK. Inhibiting PI-3-K for glioma therapy. Cell Cycle. 2009;8:335. doi: 10.4161/cc.8.3.7884. [DOI] [PubMed] [Google Scholar]
- 42.Friedman L. GDC-0941, a potent, selective, orally bioavailable inhibitor of class I PI3K. Proc American Association for Cancer Research. 2008:LB-110. [Google Scholar]
- 43.Ihle NT, Powis G. Take your PIK: phosphatidylinositol 3-kinase inhibitors race through the clinic and toward cancer therapy. Mol Cancer Ther. 2009;8:1–9. doi: 10.1158/1535-7163.MCT-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of PI-540 or PI-620 on molecular biomarkers in human cancer cells; A549 lung adenocarcinoma, U87MG glioblastoma and PC3 prostate. Cells were treated with PI-540 or PI-620 for 2 h, lysates prepared and immunoblotting carried out for the proteins indicated. GAPDH was used as a loading control.
Effect of GDC-0941 on PARP cleavage in established human U87MG glioblastoma xenografts in athymic. Mice were treated with doses of 25, 50, 100 and 150 mg/kg p.o GDC-941 and tumors removed at 4h post-dosing. Control animals (marked 0) received drug vehicle alone. The positive control (marked +ve) was HT29 colon cancer cells treated with 50 nM SN38. GAPDH was used as the loading control.
Relative mean tumor volume (% of initial volume prior to therapy) in the established IGROV-1 human ovarian cancer xenograft model following 25, 50, 100 and 150mg/kg oral daily doses of GDC-941 for 13 days. Established tumors were grown s.c. bilaterally in femal NCr athymic mice and controls received equivalent volumes of vehicle.
Mean final tumor weights in the IGROV-1 human ovarian cancer xenograft model following oral daily dosing with 25, 50, and 100 and 150 mg/kg oral daily doses GDC-941 for 13 days.
Pharmacodynamic effects measured in IGROV-1 human ovarian cancer xenografts from the above efficacy study 4h and 8h following the final dose. Phosphorylation of AKT Ser473 and total AKT were measured by electrochemiluminescent immunoassay (MSD).
Effect of GDC-0941 and PI-103 on proliferation and phosphorylation of AKT at Ser473 in human colon cancer cell lines. HCT116 has wild type PTEN, mutant PIK3CA and mutant KRAS. SNUC2B has wild type PTEN, wild type PIK3CA and mutant KRAS. LoVo has wild type PTEN, wild type PIK3CA and mutant KRAS.