

Abstract
Amyloid diseases are traditionally characterized by the appearance of inter- and intracellular fibrillar protein deposits, termed amyloid. Historically, these deposits have been thought to be the etiology of the disease. However, recent evidence suggests that small oligomers of the amyloidogenic protein/peptide are the origin of neurotoxicity. Although the importance of identifying the toxic oligomeric species is widely recognized, such identification is challenging because these oligomers are metastable, occur at low concentration, and are characterized by a high degree of heterogeneity. In this work, a fluorescently labeled β-amyloid(1–40) is used as a model amyloidogenic peptide to test the effectiveness of what we believe is a novel approach based on single-molecule spectroscopy. We find that by directly counting the photobleaching steps in the fluorescence, we can determine the number of subunits in individual β-amyloid(1–40) oligomers, which allows us to easily distinguish among different species in the mixtures. The results are further analyzed by comparison with Monte Carlo simulations to show that the variability seen in the size of photobleaching steps can be explained by assuming random dipole orientations for the chromophores in a given oligomer. In addition, by accounting for bias in the oligomer size distribution due to the need to subtract background noise, we can make the results more quantitative. Although the oligomer size determined in this work is limited to only small species, our single-molecule results are in good quantitative agreement with high-performance liquid chromatography gel filtration data and demonstrate that single-molecule spectroscopy can provide useful insights into the issues of heterogeneity and ultimately cellular toxicity in the study of amyloid diseases.
Introduction
Amyloid diseases, including neurodegenerative diseases (e.g., Alzheimer's and Parkinson's), nonneuropathic systemic diseases (e.g., AL amyloidosis), and nonneuropathic localized diseases (e.g., type II diabetes), are generally thought to be caused by amyloidogenic protein/peptide misfolding and aggregation (1–4). At least 34 different proteins/peptides have been identified to date (4) that can fold abnormally, form amyloid fibrils, and are associated with pathology. However, the mechanism by which these proteins/peptides gain their neurotoxic function upon misfolding and aggregation remains unclear. With evidence from recent experiments, it has been suggested that the toxic agents at the origin of amyloid diseases are oligomeric forms of the amyloidogenic proteins/peptides (5–7), contrary to the traditional view implicating fibrils. The toxic oligomeric form may consist of only a few monomeric units. Indeed, atomic force microscopy and scanning transmission electron microscopy studies have shown that small annular structures resembling the pores formed by bacterial toxins are generated by β-amyloid and α-synuclein (8–11).
Although identification of the toxic oligomer species has become an important topic in studies of amyloid disease, understanding the role of these species in toxicity has been challenging. These amyloidogenic proteins/peptides usually occur at extremely low physiological concentrations, often in the nano- to subnanomolar range. Moreover, the amyloidogenic oligomers are highly heterogeneous and metastable. In some cases there is evidence that the structures of the small oligomers are in dynamic equilibrium (12,13); hence, ensemble studies mask the details of these features.
Here we present what to our knowledge is a new approach for studying and identifying amyloidogenic oligomers based on single-molecule fluorescence spectroscopy (SMS). For our model system we use β-amyloid(1–40), a 40 amino acid peptide that has been identified as the major component of the extracellular plaques found in the brains of patients with Alzheimer's disease (14). Because of the high sensitivity of confocal fluorescence microscopy, single oligomers made of fluorescently labeled β-amyloid(1–40) peptide can be readily identified and differentiated from other oligomers containing a different number of subunits. We show that by counting the number of photobleaching steps (15–17), one can determine the number of monomers of each detected β-amyloid(1–40) oligomer and thus resolve different equilibrium mixtures of β-amyloid while working at a protein concentration below 1 nM. Monte Carlo simulations in which β-amyloid oligomers are modeled as structures with random dipole orientations of the fluorophore associated with each monomer account for observed variations in photobleaching trajectories, as well as for biasing due to background noise subtraction, allowing us to improve the quantitative interpretation of the data. In line with previous work by Dukes et al. (17), who successfully applied SMS to the study of early β-amyloid oligomers, in this study we further investigated the single-molecule photobleaching methodology by numerical simulations of orientation effects and comparison with high-performance liquid chromatography (HPLC) data.
Materials and Methods
β-Amyloid(1–40) preparation
HiLyte Fluor 488 β-amyloid(1–40) was obtained from AnaSpec (San Jose, CA) and kept frozen at −20°C. For the purpose of consistency, before any samples were used, the peptide was first redissolved in trifluoroacetic acid (Sigma-Aldrich, St. Louis, MO) at 1 mg/mL and then bath-sonicated for at least 30 min to break apart any preformed aggregates (18). The solution was then aliquoted into small samples, and finally lyophilized and stored at −20°C. To make a fresh β-amyloid(1–40) sample, the lyophilized powder was directly dissolved in 10 mM sodium phosphate, 100 mM sodium chloride, pH 7.4 buffer, followed by a 60-s bath sonication.
Transmission electron microscopy of fibril formation
Both unlabeled β-amyloid(1–40) and HiLyte Fluor 488 β-amyloid(1–40) were dissolved in 10 mM sodium phosphate, 100 mM sodium chloride, pH 7.4 buffer in siliconized tubes (Corning, Corning, NY) to a final protein concentration of 25 μM. After 6 days of incubation, small amounts of β-amyloid(1–40) fibrils were pipetted onto grids and stained with uranyl acetate. The samples were then imaged with a Philips CM12 transmission electron microscope (New York, NY).
Gel filtration HPLC
An aliquot of 20 μL of 5 μM fresh HiLyte Fluor 488 β-amyloid(1–40) sample was run through a gel filtration column (TOSOH Bioscience, South San Francisco, CA) at room temperature in 10 mM sodium phosphate, 100 mM sodium chloride, pH 7.4 buffer with a flow rate of 1 mL/min to obtain fractions of different oligomer species. The molecular mass for this column was calibrated by using the following proteins/molecules under the same separation conditions: thyroglobulin (669 kDa), γ-globulin (165 kDa), ovalbumin (43 kDa), RNase A (14 kDa), ubiquitin (8.6 kDa), aprotinin (6.5 kDa), and NATA (180 Da).
Confocal fluorescence microscopy
The fluorescence photobleaching trajectories of single HiLyte Fluor 488 β-amyloid(1–40) oligomers were collected with a custom-made inverted confocal scanning microscope (19,20) equipped with a 1.45 NA 60× oil-immersion objective (Olympus, Center Valley, PA). The excitation laser beam from an Ar+-ion laser (Melles-Griot, Carlsbad, CA) at 457.9 nm was passed through a dichroic mirror and focused onto a cover glass (Fisher Scientific, Pittsburgh, PA) coated with the sample at a laser power of 10 μW. The sample fluorescence was collected by the same objective and then passed through a 457.9 nm notch-plus filter (Kaiser, Ann Arbor, MI) and a 520/60nm band-pass filter (Chroma, Rockingham, VT), and finally detected by a single-photon-counting avalanche photodiode (Perkin-Elmer Optoelectronics, Waltham, MA).
Sample preparation for single-molecule photobleaching
For photobleaching experiments with the incubated HiLyte Fluor 488 β-amyloid(1–40) samples, the dissolved peptides were first diluted to 100 nM concentration. For the gel filtration chromatography-separated HiLyte Fluor 488 β-amyloid(1–40) monomer/dimer fraction sample, the peptide was used directly after fractionation. A further dilution of either sample to a final concentration of 0.1–1 nM was applied before the HiLyte Fluor 488 β-amyloid(1–40) oligomers were deposited onto a cover glass. Two droplets of this diluted peptide solution were spin-coated onto the cover glass at 2000 rpm until the cover glass became dry (∼30–40 s at room temperature). The cover glasses were precleaned by rinsing with Milli-Q water and then baked in a kiln at ∼500°C for 2 h.
After the sample-coated cover glass was mounted under the microscope, it was raster-scanned on a piezoelectric stage (Physik Instrumente, Auburn, MA) until a preset count rate threshold indicating the presence of a fluorescent oligomer was reached to initiate the recording of a photobleaching trajectory. The photobleaching trajectories were recorded in time steps of 50 ms.
Photobleaching trajectory analysis
Both noise-filtering and photon-counting histogram (PCH) techniques were used in the photobleaching trajectory analysis.
The forward-backward nonlinear digital filtering technique was initially developed to extract small signals from ion channel recordings, and was later introduced for application in the single-molecule field (21,22). The most significant feature of this noise-filtering technique is that it can effectively preserve fast transients in the signal. With minimum edge blurring, this filtering technique is very suitable for our photobleaching trajectory analysis. Note that the parameters of this digital filter need to be carefully chosen to avoid signal distortions.
The PCH approach has been widely used in fluorescence correlation spectroscopy to differentiate multiple fluorescence intensity levels (23,24). This technique has also been used in photobleaching experiments (15–17). The number of photons detected per unit time from fluorophores (at excitation levels below the saturation intensity) follows Poisson distributions. The construction of a PCH can help reveal multiple fluorescence intensities by showing the corresponding Poisson distributions.
As discussed further below, the combination of a noise-filtering technique and a PCH allows for improved resolution of different fluorescence intensities.
Simulation of photobleaching
A program to generate simulated stochastic photobleaching trajectories was written in LabVIEW (National Instruments, Austin, TX) and involves the following steps:
-
1.
A random number generator is used to generate an integer (with a preset upper limit) as the number of subunits in the oligomer.
-
2.
A photobleaching time is assigned to each of the fluorophores in the oligomer. These photobleaching times are randomly generated based on the exponential probability distribution , where τ is the preset photobleaching lifetime, reflecting the photostability of the fluorophores that are maximally excited.
-
3.
A fixed absorption dipole orientation angle θ (relative to the cover glass surface) is assigned to each of the fluorophores in the oligomer by a random number generator.
-
4.
A position γ relative to the center of the laser beam focus is assigned to the oligomer (we also assume that the intensity of the laser beam falls off as a Gaussian profile ). This oligomer position is generated randomly (with a preset upper limit) based on a uniform probability of the oligomer being deposited on the cover glass.
-
5.
According to the dipole orientation of each fluorophore and the position of the oligomer, each fluorophore's photobleaching time is adjusted by , and the fluorescence intensity is adjusted by , where ϑ is the angle between the absorption dipole and the emission dipole. For simplicity, and with no impact on the qualitative features of the simulation, we assume that ϑ = 0 since, although the relative angle between the absorption and emission dipoles of HiLyte Fluor 488 has not been determined, the strong mirror image relationship between absorption and emission spectra indicates that they reflect the same electronic transition.
-
6.
The fluorescence signals of all the fluorophores within the oligomer are added to yield a single photobleaching trajectory.
-
7.
Poisson noise is added to the trajectory.
Results
Comparison of fibril formation between HiLyte Fluor 488 labeled and unlabeled Aβ(1–40)
It is critical to confirm that the fluorescence labeling of β-amyloid(1–40) with HiLyte Fluor 488 does not alter its aggregation and fibril formation. According to the three-dimensional structure of β-amyloid(1–42) recently determined by NMR (25), the N-terminus where the fluorophore resides has a rather flexible structure, which suggests that the peptide's aggregation should not be significantly altered by the labeling.
Imaging of fibrils by transmission electron microscopy (TEM) is a common and direct method for studying the fibril formation of amyloidogenic proteins/peptides (26,27). We used TEM to image the fibrils formed by both unlabeled β-amyloid(1–40) and HiLyte Fluor 488-labeled β-amyloid(1–40), and compared them side by side (Fig. 1). These images confirm that HiLyte Fluor 488 β-amyloid(1–40) is able to form fibrils, and both the timescale of the fibril formation and the morphology of the fibrils are similar to those of the unlabeled β-amyloid(1–40).
Figure 1.
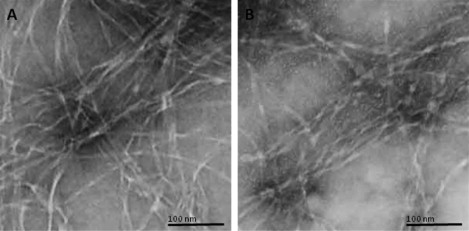
Comparison of unlabeled and HiLyte Fluor 488 labeled β-amyloid(1–40) fibril morphology. Both (A) unlabeled and (B) labeled β-amyloid(1–40) were incubated for 6 days and imaged with a transmission electron microscope.
We also obtained preliminary data on membrane permeabilization by β-amyloid(1–40) (data not shown). Both unlabeled β-amyloid(1–40) and HiLyte Fluor 488 β-amyloid(1–40) caused membrane permeabilization on plane membranes. This also confirms that the labeling of β-amyloid(1–40) does not qualitatively alter the pore formation ability of the peptide.
Gel filtration HPLC of HiLyte Fluor 488 Aβ(1–40) oligomers
An aliquot of 20 μL of 5 μM freshly dissolved HiLyte Fluor 488 β-amyloid(1–40) in 10 mM sodium phosphate, 100 mM sodium chloride, pH 7.4 buffer was run through a gel filtration column. The elution profile is shown in Fig. 2. Three elution peaks centered at 11.3, 11.8, and 12.4 min are clearly visible. According to the molecular mass calibration curve for this column, these peaks correspond to molecular masses of ∼14.5 kDa, 9.6 kDa, and 5.9 kDa, respectively. A fourth, minor peak elutes at 8.3 min, corresponding to a molecular mass of ∼150 kDa, representing a small amount of much larger oligomeric species. The gel filtration chromatography results clearly show that the β-amyloid(1–40) oligomers in solution are heterogeneous.
Figure 2.
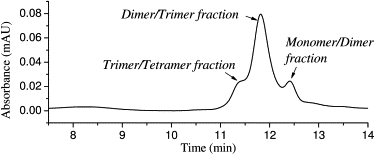
Elution profile of 5 μM HiLyte Fluor 488 β-amyloid(1–40) from the HPLC gel filtration column (see text for details).
Although the gel filtration chromatography clearly demonstrates the presence of at least three elution peaks, identification of the species based on the molecular mass standards is not straightforward, for two main reasons: 1), they do not correspond to the molecular masses of any specific oligomers, bur rather fall between two oligomers; and 2), the elution peaks are broader than those expected for pure single species. We conclude that these peaks are mixtures of two or more oligomeric species, as indicated by the peak assignments in the figure. This may be due to interaction between the oligomers and the column material, as suggested by the asymmetric long tail on the elution profile, or to some structural heterogeneity or some reequilibration among different β-amyloid(1–40) oligomer species during the column separation. Hence, while the HPLC data are instructive, it appears challenging to extract a molecular-level understanding of the sizes of the different oligomers of β-amyloid(1–40).
SMS determination of the size of fluorescently labeled Aβ(1–40) oligomers
The fluorescence signals from single HiLyte Fluor 488 β-amyloid(1–40) oligomers were monitored by a single-photon-counting avalanche photodiode with a nano- or subnanomolar concentration of labeled peptide deposited on a clean cover glass at a laser power of 10 μW. The number of transitions between the fluorophore ground state and excited singlet state is limited by photobleaching (typically less than 107 transitions for organic dye molecules) causing the fluorescence to disappear even though the excitation field remains on. The underlying mechanism of photobleaching may vary, but it is assumed to proceed through the excited triplet state, which is typically more reactive toward oxidation, and lasts much longer than the excited singlet state. Thus, the duration and intensity of fluorescence are controlled by the laser power, the absorption cross section of the fluorophore (a term that includes its orientation relative to the light's electric field), the fluorescence quantum yield, and the photooxidation rate. In the experiments described here, HiLyte Fluor 488 fluorophores typically photobleached within a few seconds, occasionally extending to >30 s.
Fig. 3 shows a typical fluorescence confocal scanning image of the cover glass surface coated with HiLyte Fluor 488-labeled β-amyloid oligomers, as well as a few typical photobleaching trajectories. Of all of the trajectories that were collected, ∼40% are “clean” trajectories (only shot noise present) with recognizable photobleaching events (for example, see Fig. 3, a and b, for trajectories of a dimer and a hexamer). The remaining 60% contain significant intensity fluctuations (much larger than shot noise) that lack any resolvable photobleaching features (Fig. 3 c is an example). This may be due to the fluorophores' dipole movement and/or interactions with the cover glass surface impurities, charges, etc. Although we determined that cleaning the cover glass reduces the number of trajectories containing these fluctuations, so far we have not been able to improve the performance beyond the current ∼40% “clean” trajectories. Since we could reliably use only the “clean” trajectories to determine the total number of photobleaching events and extract the oligomer size, only these were considered in our data analysis.
Figure 3.
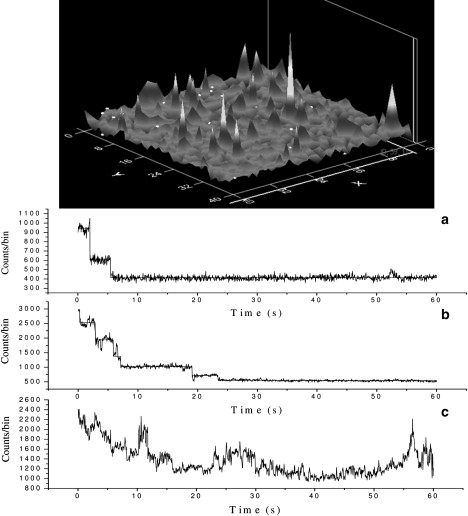
Typical fluorescence confocal scanning image (40 × 40 pixels for 20 × 20 μm2) and three photobleaching trajectories of HiLyte Fluor 488 Aβ(1–40) oligomers: (a) a dimer, (b) a hexamer, and (c) an example of a trajectory with fluctuations that make it difficult to determine the number of photobleaching steps. These trajectories are removed from the data set.
Determination of Aβ(1–40) oligomer size distributions
By using the photobleaching trajectories of a large number of individual HiLyte Fluor 488 β-amyloid(1–40) oligomers, it is possible to construct an oligomer size distribution by making a histogram of the number of photobleaching events in each trajectory, which represents the number of subunits in each photobleached oligomer. The determination of photobleaching events in a given trajectory involves both visual inspection and signal processing, including noise reduction and PCH (for more details see the Discussion section).
To demonstrate our ability to differentiate different oligomer distributions using the SMS approach, we used a sample of unresolved HiLyte Fluor 488 β-amyloid(1–40) and a sample of the monomer/dimer fraction collected from the HPLC gel filtration (indicated in Fig. 4, inset). The unresolved β-amyloid(1–40) sample is expected to be a mixture of oligomers of different sizes, whereas the gel filtration separated fraction is expected to contain only monomers and dimers.
Figure 4.
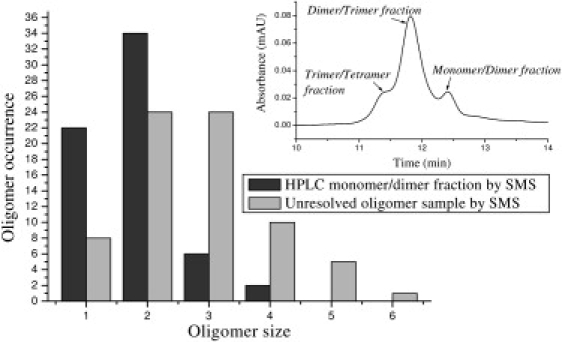
Oligomer size distribution comparison between an unresolved β-amyloid(1–40) sample and a gel filtration chromatography separated monomer/dimer sample (the inset presents the same data shown in Fig. 2, shown here for convenience). The monomer/dimer fraction was deposited onto a cover glass immediately after it was collected from the column. The SMS approach revealed significant contributions from larger oligomers compared to those separated by HPLC. Further analysis below shows improved quantitative analysis for the SMS data.
The histograms obtained for these two different samples are compared in Fig. 4. This comparison clearly confirms the higher degree of heterogeneity of the unresolved sample compared to the gel filtration separated sample. More importantly, the histogram shows that the SMS approach can distinguish between the two mixtures and that oligomers as large as hexamers are found in the unchromatographed mixture. The HPLC separation does not detect oligomers larger than tetramers, possibly because interactions with the column result in a breakup of the larger oligomers. As we show below in the Discussion section, this comparison can be made even more quantitative.
Discussion
Despite the intensive efforts and substantial progress made in amyloid disease studies in recent years, there remains a lack of understanding of how these amyloidogenic proteins/peptides cause cell death. Although recent studies point to early peptide oligomers as the key players in cytotoxicity, it has also been found that these oligomers are highly heterogeneous and exist in solution in a state of dynamic equilibrium (27–30). This makes it extremely challenging to apply traditional approaches to determine the toxic species and characterize the cytotoxicity mechanism. Traditional techniques, such as gel filtration chromatography, are powerful analytical tools for general protein studies, but in studies dealing with small quantities of heterogeneous and metastable protein/peptide assemblies, they are not adequate for quantitative interpretations.
The emerging field of SMS provides a new approach for characterizing early amyloidogenic oligomers. This study of HiLyte Fluor 488 β-amyloid(1–40) demonstrates that with the use of confocal fluorescence microscopy, one can identify single oligomers and determine their sizes. This allows individual oligomers and thus pure species, instead of an averaged ensemble, to be studied, thus enabling a more direct and potentially quantitative characterization.
To determine HiLyte Fluor 488 β-amyloid(1–40) oligomer photobleaching events in the recorded trajectories, we employ an analysis that includes visual inspection and signal processing. However, a common feature observed in all of the photobleaching trajectories is that bright fluorophores typically photobleach much more rapidly than dimmer ones. Based on this observation, we analyze our data in the following way: For the “large digital drop”-type photobleaching events, which usually correspond to bright fluorophores, visual inspection is adequate for counting the number of photobleaching steps. In contrast, for photobleaching trajectories in which only a small fluorescence change occurs (see Fig. 5, a and c), an analysis of noise reduction followed by fluorescence PCH provides an improved differentiation of two rather close fluorescence intensity levels (see Fig. 5, b and d). In Fig. 5 c, the PCH of the raw trajectory is well fitted with two Gaussian distributions, indicating that there are two close fluorescence intensity levels. After the forward-backward nonlinear filter is applied to the raw trajectory, these two fluorescence intensities are clearly separated on the PCH (Fig. 5 d). These close fluorescence intensity levels, resolved by noise reduction and PCH, usually correspond to dim fluorophores. Thanks to the typically long duration of these weak fluorescence signals, an adequate number of photons can still be collected to allow us to resolve these dim fluorophore photobleaching events.
Figure 5.
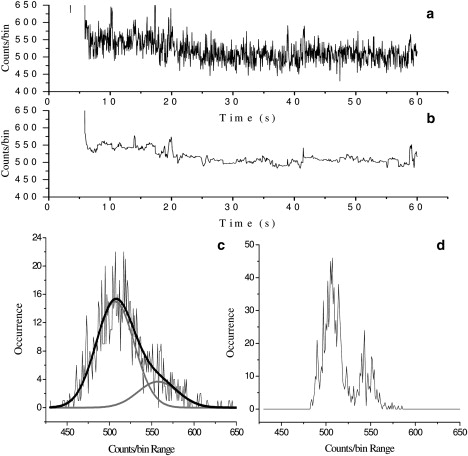
(a) Photobleaching trajectory showing two close fluorescence intensity levels. (b) The same trajectory after filtering by the forward-backward nonlinear filter. (c) The PCH of the raw trajectory. The solid curves show that this histogram is well fitted with two Gaussian distributions (with widths being equal to the square root of their means). (d) The PCH of the filtered trajectory. The two fluorescence levels are now clearly separated after the noise reduction.
It is important to note that despite the sample preparation and cover glass cleaning procedures employed, not all of the trajectories could be analyzed reliably. By following the above analysis, we were able to analyze ∼40% of the trajectories. The remaining 60% lack any distinct fluorescence intensity levels that can be resolved either visually or by the fluorescence PCH (see Fig. 3 c). These trajectories are noisy, with continuous fluorescence variations that on some occasions are even nonmonotonic. Thus, these were removed from the data set. Dark states are also occasionally observed in the photobleaching trajectories. These appear as photobleaching events, but recover (usually to the original fluorescence intensity level) after some time. The reason for this behavior is not clear, but it could be due to electron traps within the fluorophore local environment or to slow changes in the fluorophore dipole orientation, since the β-amyloid(1–40) oligomers were nonspecifically adsorbed onto the cover glass surface. Agarose gel was also tested as a matrix for immobilizing β-amyloid(1–40) oligomers for single-molecule experiments. However, the large “pore” size in agarose gel makes it very difficult to trap peptide monomers, and the high viscosity also makes an even distribution of oligomers in the agarose gel very unlikely. Alternative sample preparation methodologies, as well as modifications to the microscope, to minimize these complications remain under investigation.
In the analysis of the above trajectories, the relative fluorescence intensity was not used as a measure of the number of monomeric units, because it is not a faithful reporter of oligomer size. There are three main reasons for this: First, it is likely that different fluorophores, even in the same oligomer, will adopt different orientations for both the absorption and emission dipoles, thus yielding different fluorescence intensities. Second, even for fluorophores with the same dipole orientation, the fluorescence intensities may differ due to different interactions with surface impurities, charges, etc. Third, the excitation intensity of a fluorophore depends on where the oligomer is placed relative to the focus of the laser beam, which generally has a Gaussian intensity profile, producing different fluorescence intensities. Hence, there is no direct correlation between the observed fluorescence intensity and the oligomer size. Other sample immobilization techniques could make the use of intensity less sensitive to these issues.
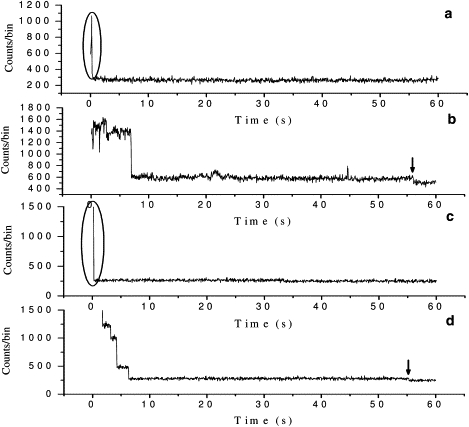
Further inspection of the analyzed trajectories indicates that they can be characterized by two features: 1), the photobleaching steps in fluorescence intensity in a given oligomer are not equal; and 2), the trajectories often have a large and fast initial fluorescence drop, which is often followed by a long and low fluorescence tail before photobleaching occurs (see Fig. 6, a and b, for examples). To understand these features better, we considered a simple dipole orientation model. The model was tested with the use of a stochastic simulation program.
Figure 6.
(a) Experimental photobleaching trajectory with a large and fast initial drop. (b) Experimental photobleaching trajectory with a long, low tail. (c) Simulated photobleaching trajectory with a large, fast initial drop. (d) Simulated photobleaching trajectory with a long, low tail (photobleaching events are highlighted by the circles and arrows).
Considering that our sample was spin-coated onto a cover glass, and the β-amyloid(1–40) oligomers were trapped on the glass surface by nonspecific interactions, it is reasonable to assume that the fluorophores, which are attached to the oligomers, do not adopt the same dipole orientation. The fact that the noise in these analyzable trajectories is nearly shot noise-limited indicates that these photobleached fluorophores have relatively fixed dipole orientations. The above two considerations suggest that fluorophores with a less favorably oriented transition dipole would be excited less frequently, emit fewer photons per unit time, and hence photobleach more slowly (since the excitation rate is lower).
To test this hypothesis, we simulated the photobleaching trajectory of an oligomer assuming that its individual fluorophores have random but fixed dipole orientations, and the excitation field is circularly polarized. Based on this model, it is anticipated that with a more parallel orientation of the fluorophore dipole relative to the cover glass surface, the fluorophore will be more frequently excited and the fluorescence intensity will be higher, and thus the time required for photobleaching will be shorter. Note: In the plane parallel to the cover glass, the laser was circularly polarized, so there was no dependence of the excitation on the absorption dipole angle projected onto the cover glass plane.
Using the stochastic simulation described above in Materials and Methods, and the random dipole orientation model, we produced photobleaching trajectories. Typical trajectories are shown in Fig. 6, c and d, and clearly resemble the experimental trajectories in Fig. 6, a and b, which represent a monomer and a trimer, respectively. Although such a simulation does not prove that the proposed model is the origin of the variability in the trajectories, it supports the notion that different dipole orientations are a major contributor to the variations in the photobleaching trajectories. In fact, we observed the same variability in fluorescence intensities and photobleaching behavior in the fluorophore-only control experiments (data not shown) as in the fluorophore-labeled β-amyloid experiments.
In addition to its likely important contribution to the variation in the photobleaching trajectories, the random dipole orientation model also suggests some possible systematic errors embedded in the data. For the photobleaching experiments, a threshold is preset such that when the detected fluorescence signal is higher than this threshold, the scanning is paused and the recording of a photobleaching trajectory is triggered. This helps to mitigate background fluorescence and prevent any unwanted triggering by the occasionally very high local background noise from the cover glass. However, thresholding may also bias the data set toward brighter fluorophores and thus against monomers and, to a lesser extent, other small oligomers.
To quantify the effect of thresholding, we performed another stochastic simulation. In this simulation, we used the same photobleaching trajectory generation program as described in Materials and Methods. Based on the discussion above, it is expected that, depending on the initial fluorescence intensities of these simulated trajectories, some will be recorded whereas others will be missed because they are below the preset threshold. Hence, for each oligomer species, we let the program continuously generate photobleaching trajectories until the number of the “observed” (above-threshold) trajectories matched that in our experimental single-molecule data. The total number of these generated trajectories includes both “recorded” and “missed” oligomers, and thus represents the corrected number for those specific oligomeric species. Because of the stochastic nature of this simulation, we repeated it 10,000 times for each oligomer species. Fig. 7 shows a comparison of the observed oligomer distribution from Fig. 4 and the corrected distribution of the same data.
Figure 7.
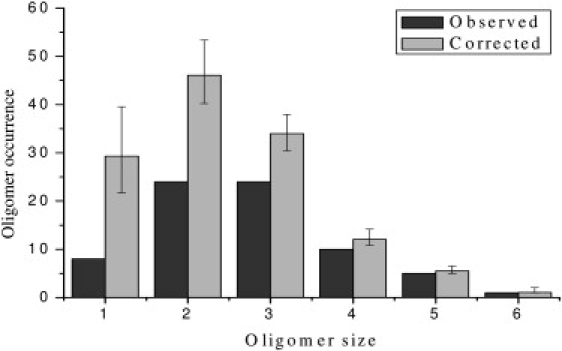
Comparison of the observed oligomer distribution and the corrected oligomer distribution.
Not surprisingly, the effect of the correction decreases with the increase in oligomer size, since the more fluorophores an oligomer contains, the less likely it is to be missed due to thresholding. The simulation results confirmed this. It should also be noted that in this simulation, an empirical value was used to estimate the maximum fluorescence emission rate from a single fluorophore under the microscope (This corresponds to the fluorophores that are at the center of the beam focus and whose dipole orientations are along the excitation field). Different values for this maximum emission rate yield slightly different corrected distributions, but the overall shapes of the corrected oligomer distributions remain similar.
Besides the threshold effect, there are other factors that might cause systematic errors in the estimation of oligomer size, such as self-quenching between fluorophores within an oligomer, which could result in an underestimate of this size. Also, the random dipole orientation assumption may not represent the true distribution for real samples on cover glass, and possible slow movements of the fluorophore dipoles on the cover glass may complicate the data analysis as well. In general, an immobilization of free rotating oligomers in an aqueous environment is preferable; this is a focus of ongoing work.
To test the validity of our procedure for correcting the photobleaching-derived distribution, we performed a control experiment on dye-labeled parvalbumin. The mass spectroscopy data of this parvalbumin sample indicate 48.6% single-labeling, 44.5% double-labeling, and 6.9% triple-labeling mixtures. Fig. 8 shows the comparison of the corrected SMS data with the mass spectroscopy data. (The smaller error bars here compared to those in Fig. 7 are due to an improved thresholding algorithm with a CCD camera used in the control experiment. The real errors, however, should be larger than those error bars because the uncertainties of the empirical estimates used in the simulation are not indicated in the figure.) It clearly demonstrates that our single-molecule approach can accurately reconstruct the content distribution of the studied sample.
Figure 8.
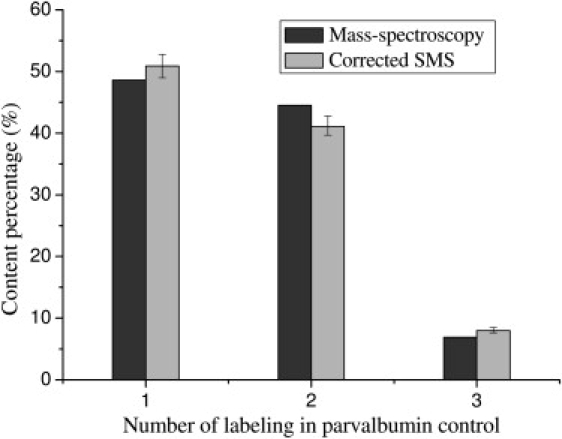
Comparison of the mass-spectroscopy distribution with the corrected single-molecule distribution in the parvalbumin control experiment.
We further analyzed our β-amyloid(1–40) sample by comparing the single-molecule data with the HPLC gel filtration-derived oligomer distribution of the same sample. This allowed us to gain more insight into this heterogeneous and dynamic mixture. To correlate these two sets of data, we applied a multiple Gaussian distribution fitting to the gel filtration elution profile, as shown in Fig. 9 a. Depending on the choice of the fitting windows and the initial parameters, this procedure yielded several different fits with only slightly different peak heights, widths, and positions, and with nearly the same quality of fit. Fig. 9 a shows a typical fit with four Gaussian peaks and a fifth, low and broad distribution describing the slowly varying background. According to their elution times, these four peaks most probably represent four different oligomer species: monomer, dimer, trimer, and tetramer, respectively.
Figure 9.
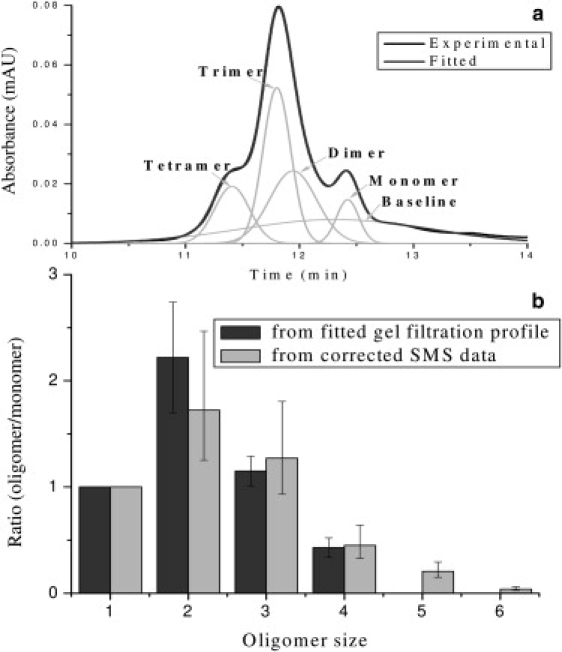
(a) Multiple Gaussian fitting was applied to the gel filtration elution profile. The “experimental” curve indicates the raw data of the elution profile, and the “fitted” curve indicates a typical good fitting result, which is the summation of monomer, dimer, trimer, tetramer, and baseline. These two curves closely match each other. (b) Comparison of the relative oligomer size distributions derived from different data sets: the gel filtration profile and the corrected single-molecule data. The data are normalized so that they give the same ratio for the monomer.
Integration of the area under each peak yields the total far-ultraviolet absorption for each β-amyloid(1–40) oligomer species. Assuming that the average absorptivity of the peptide backbone does not change upon oligomerization (i.e., that the average absorptivity of a dimer is twice that of a monomer), an oligomer size distribution of β-amyloid(1–40) was derived from the absorption of each species. This was done for each of the best four fits to the HPLC elution profile, and an averaged oligomer distribution was derived. A similar relative distribution was constructed from the corrected single-molecule data shown in Fig. 7. Fig. 9 b presents a comparison of these two relative distributions obtained with our two different methods.
The results show relatively good agreement in terms of oligomer size distribution between the single-molecule approach and the gel filtration chromatography approach. This confirms that our sample preparation for single-molecule detection did not distort the β-amyloid oligomer distribution. The agreement also indicates that, under our experimental conditions for gel filtration chromatography, the oligomer distribution does not reequilibrate very rapidly, and thus both approaches are able to quantitatively reconstruct the oligomer size distribution. However, the single-molecule approach has some other advantages. The single-molecule approach works at nanomolar or lower sample concentrations, which could be extremely beneficial for amyloidogenic studies. The determination of oligomer size is more direct in the single-molecule approach, whereas there is some ambiguity in determining the oligomer size in the gel filtration method since the positions of the elution peaks do not correspond to individual oligomers very well, and there is a broad baseline peak indicating the existence of an interaction between some labeled peptides and column material. In addition, the single-molecule approach successfully detected oligomers larger than tetramers, whereas the gel filtration method missed them. This suggests that β-amyloid(1–40) oligomers that are larger than a tetramer may not be stable during gel filtration. These large oligomers may have reequilibrated themselves into smaller species during the separation, contributing to the baseline peak.
Conclusions
In this work, an approach based on SMS was used to study the heterogeneity of HiLyte Fluor 488 β-amyloid(1–40) oligomers. A fluorescently labeled β-amyloid(1–40) oligomer mixture was “frozen” at a given time point by depositing it onto a cover glass. The surface-bound oligomers were then photobleached, and the number of subunits in individual oligomers was determined by counting the photobleaching events. By constructing a histogram of the number of subunits in all of the individual oligomers, we were able to extract an oligomer size distribution that was then corrected according to our computer simulations. As an indication of heterogeneity, the oligomer size distribution reveals the intrinsic properties of a given β-amyloid(1–40) oligomer mixture at a given time point. In addition to the work described above, the single-molecule approach can also be used to extract dynamic information. Real-time monitoring of oligomer formation is possible if the laser power is lowered and the observation time window extended. Hence, SMS potentially could be used to reveal transient events that might occur in such a dynamic and heterogeneous mixture and might be masked by any ensemble-averaging measurements.
For large oligomers (we have identified oligomers as large as octamer by photobleaching), the photobleaching events in the fluorescence trajectory may become more difficult to distinguish, and in some cases self-quenching among the fluorophores may increase as well. However, since most of the recent evidence points to small oligomers, such as the pore-like annular structures that are probably made of hexamer units, as the pathologically relevant species in Alzheimer's disease (8–11), the single-molecule approach appears very promising for studies of these small oligomers. We anticipate that the detailed information on the early oligomers of amyloidogenic proteins/peptides gained through SMS will provide a deeper understanding of amyloid diseases.
Acknowledgments
We are grateful to Professor Bardford Orr (Applied Physics Program, University of Michigan) for providing us access to the AFM instrument, and Professor Matthew Chapman (Department of Molecular, Cellular, and Developmental Biology, University of Michigan) for access to the TEM instrument. We also thank Pascale Leroueil and Xuan Wang for valuable technical support.
This work was supported by the National Institutes of Health (grant 1R21AG027370-01A1).
References
- 1.Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe D.J. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 3.Stefani M., Dobson C.M. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 4.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 5.Bitan G., Teplow D.B. Rapid photochemical cross-linking-a new tool for studies of metastable, amyloidogenic protein assemblies. Acc. Chem. Res. 2004;37:357–364. doi: 10.1021/ar000214l. [DOI] [PubMed] [Google Scholar]
- 6.Watson D., Castaño E., Kokjohn T.A., Kuo Y.-M., Lyubchenko Y. Physicochemical characteristics of soluble oligomeric Aβ and their pathologic role in Alzheimer's disease. Neurol. Res. 2005;27:869–881. doi: 10.1179/016164105X49436. [DOI] [PubMed] [Google Scholar]
- 7.Lansbury P.T. Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. USA. 1999;96:3342–3344. doi: 10.1073/pnas.96.7.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quist A., Doudevski I., Lin H., Azimova R., Ng D. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyubchenko Y.L., Sherman S., Shlyakhtenko L.S., Uversky V.N. Nanoimaging for protein misfolding and related diseases. J. Cell. Biochem. 2006;99:52–70. doi: 10.1002/jcb.20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Losic D., Martin L.L., Mechler A., Aguilar M.-I., Small D.H. High resolution scanning tunnelling microscopy of the β-amyloid protein (Aβ1–40) of Alzheimer's disease suggests a novel mechanism of oligomer assembly. J. Struct. Biol. 2006;155:104–110. doi: 10.1016/j.jsb.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Lashuel H.A., Petre B.M., Wall J., Simon M., Nowak R.J. α-Synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 12.Bitan G., Kirkitadze M.D., Lomakin A., Vollers S.S., Benedek G.B. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabella S., Quaglia M., Lanni C., Racchi M., Govoni S. Capillary electrophoresis studies on the aggregation process of β-amyloid 1–42 and 1–40 peptides. Electrophoresis. 2004;25:3186–3194. doi: 10.1002/elps.200406062. [DOI] [PubMed] [Google Scholar]
- 14.Wong C.W., Quaranta V., Glenner G.G. Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc. Natl. Acad. Sci. USA. 1985;82:8729–8732. doi: 10.1073/pnas.82.24.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shu D., Zhang H., Jin J., Guo P. Counting of six pRNAs of phi29 DNA-packaging motor with customized single-molecule dual-view system. EMBO J. 2007;26:527–537. doi: 10.1038/sj.emboj.7601506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ulbrich M.H., Isacoff E.Y. Subunit counting in membrane-bound proteins. Nat. Methods. 2007;4:319–321. doi: 10.1038/NMETH1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dukes K.D., Rodenberg C.F., Lammi R.K. Monitoring the earliest amyloid-β oligomers via quantized photobleaching of dye-labeled peptides. Anal. Biochem. 2008;382:29–34. doi: 10.1016/j.ab.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 18.Zagorski M.G., Yang J., Shao H., Ma K., Zeng H. Methodological and chemical factors affecting amyloid β peptide amyloidogenicity. Methods Enzymol. 1999;309:189–204. doi: 10.1016/s0076-6879(99)09015-1. [DOI] [PubMed] [Google Scholar]
- 19.Shi J., Palfey B.A., Dertouzos J., Jensen K.F., Gafni A. Multiple states of the Tyr318Leu mutant of dihydroorotate dehydrogenase revealed by single-molecule kinetics. J. Am. Chem. Soc. 2004;126:6914–6922. doi: 10.1021/ja038902y. [DOI] [PubMed] [Google Scholar]
- 20.Shi J., Dertouzos J., Gafni A., Steel D.G., Palfey B.A. Single-molecule kinetics reveals signatures of half-sites reactivity in dihydroorotate dehydrogenase A catalysis. Proc. Natl. Acad. Sci. USA. 2006;103:5775–5780. doi: 10.1073/pnas.0510482103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung S.H., Kennedy R.A. Forward-backward non-linear filtering technique for extracting samll biological signals from noise. J. Neurosci. Methods. 1991;40:71–86. doi: 10.1016/0165-0270(91)90118-j. [DOI] [PubMed] [Google Scholar]
- 22.Haran G. Noise reduction in single-molecule fluorescence trajectories of folding proteins. Chem. Phys. 2004;307:137–145. [Google Scholar]
- 23.Thompson N.L., Lieto A.M., Allen N.W. Recent advances in fluorescence correlation spectroscopy. Curr. Opin. Struct. Biol. 2002;12:634–641. doi: 10.1016/s0959-440x(02)00368-8. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y., Müller J.D., Ruan Q., Gratton E. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 2002;82:133–144. doi: 10.1016/S0006-3495(02)75380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antzutkin O.N. Amyloidosis of Alzheimer's Aβ peptides: solid-state nuclear magnetic resonance, electron paramagnetic resonance, transmission electron microscopy, scanning transmission electron microscopy and atomic force microscopy studies. Magn. Reson. Chem. 2004;42:231–246. doi: 10.1002/mrc.1341. [DOI] [PubMed] [Google Scholar]
- 27.Parbhu A., Lin H., Thimm J., Lal R. Imaging real-time aggregation of amyloid β protein (1–42) by atomic force microscopy. Peptides. 2002;23:1265–1270. doi: 10.1016/s0196-9781(02)00061-x. [DOI] [PubMed] [Google Scholar]
- 28.Bitan G., Lomakin A., Teplow D.B. Amyloid β-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J. Biol. Chem. 2001;276:35176–35184. doi: 10.1074/jbc.M102223200. [DOI] [PubMed] [Google Scholar]
- 29.Levine H. Soluble multimeric Alzheimer β(1–40) pre-amyloid complexes in dilute solution. Neurobiol. Aging. 1995;16:755–764. doi: 10.1016/0197-4580(95)00052-g. [DOI] [PubMed] [Google Scholar]
- 30.Bitan G., Teplow D.B. Preparation of aggregate-free, low molecular weight amyloid-β for assembly and toxicity assays. Methods Mol. Biol. 2005;299:3–9. doi: 10.1385/1-59259-874-9:003. [DOI] [PubMed] [Google Scholar]