

Abstract
The giant protein titin is responsible for the elasticity of nonactivated muscle sarcomeres. Titin-based passive stiffness in myocardium is modulated by titin-isoform switching and protein-kinase (PK)A- or PKG-dependent titin phosphorylation. Additional modulatory effects on titin stiffness may arise from disulfide bonding under oxidant stress, as many immunoglobulin-like (Ig-)domains in titin's spring region have a potential for S-S formation. Using single-molecule atomic force microscopy (AFM) force-extension measurements on recombinant Ig-domain polyprotein constructs, we show that titin Ig-modules contain no stabilizing disulfide bridge, contrary to previous belief. However, we demonstrate that the human N2-B-unique sequence (N2-Bus), a cardiac-specific, physiologically extensible titin segment comprising 572 amino-acid residues, contains up to three disulfide bridges under oxidizing conditions. AFM force spectroscopy on recombinant N2-Bus molecules demonstrated a much shorter contour length in the absence of a reducing agent than in its presence, consistent with intramolecular S-S bonding. In stretch experiments on isolated human heart myofibrils, the reducing agent thioredoxin lowered titin-based stiffness to a degree that could be explained (using entropic elasticity theory) by altered extensibility solely of the N2-Bus. We conclude that increased oxidant stress can elevate titin-based stiffness of cardiomyocytes, which may contribute to the global myocardial stiffening frequently seen in the aging or failing heart.
Introduction
Striated muscles generate passive tension in response to a stretch due largely to the presence of the giant elastic protein titin in their unitary building blocks, the sarcomeres. Titins are 3.0–3.7 MDa size molecules spanning half of a sarcomere, but only the molecular segment in the sarcomere's so-called I-band region is functionally extensible and assumes the function of a molecular spring (1,2). The cardiac titin springs show remarkable plasticity during heart development and disease in that their elasticity can be tuned via differential splicing of the titin gene, thus generating isoforms of different length and springiness (1,2). For instance, the failing human heart expresses a higher ratio of compliant titin isoforms (so-called N2BA) over stiffer titin isoform (so-called N2B) compared to the normal human heart (3–5). As both isoform types are coexpressed in the cardiac sarcomere, the higher N2BA:N2B expression ratio lowers myofibrillar passive tension and stiffness. Dramatic alterations in titin mechanics occur during pre- and perinatal heart development, during which the stiffness of the cardiac titin springs increases by up to an order of magnitude owing to titin-isoform switching in favor of the N2B isoform (6–10). Furthermore, recent evidence suggests that titin stiffness can also be modulated acutely through phosphorylation mediated by protein kinases, PKA (11–16) or PKG (16). Both PKA and PKG phosphorylate titin at a site located within a cardiac-specific segment, the N2-B domain (serine residue S469 of the human N2-B unique sequence (N2-Bus)), and this posttranslational modification reduces titin-based passive tension and stiffness (16). The distinct mechanisms of titin-stiffness adjustment, isoform-transition, and phosphorylation-dependent stiffness modulation may complement or oppose one another (17).
A previous study elucidating the atomic structure of the first immunoglobulin (Ig)-like domain in I-band titin, I1, proposed the presence of a disulfide (S-S) bond between β-strands C and E of this domain, formed under oxidizing conditions (18). However, only very rare S-S bonding was found in single-molecule mechanical experiments on I1 using atomic force microscopy (AFM) force spectroscopy (19). After analyzing the published primary structure of titin (20), Mayans et al. (18) suggested that many other Ig domains in I-band titin, particularly those located in the differentially spliced I-band Ig-domain segment (Fig. 1, inset), carry the potential for disulfide bridge formation. However, this proposal has not been verified experimentally. Of importance, a disulfide bridge would stabilize the Ig domain under conditions of oxidative stress and thus stiffen the titin springs in the sarcomeres.
Figure 1.
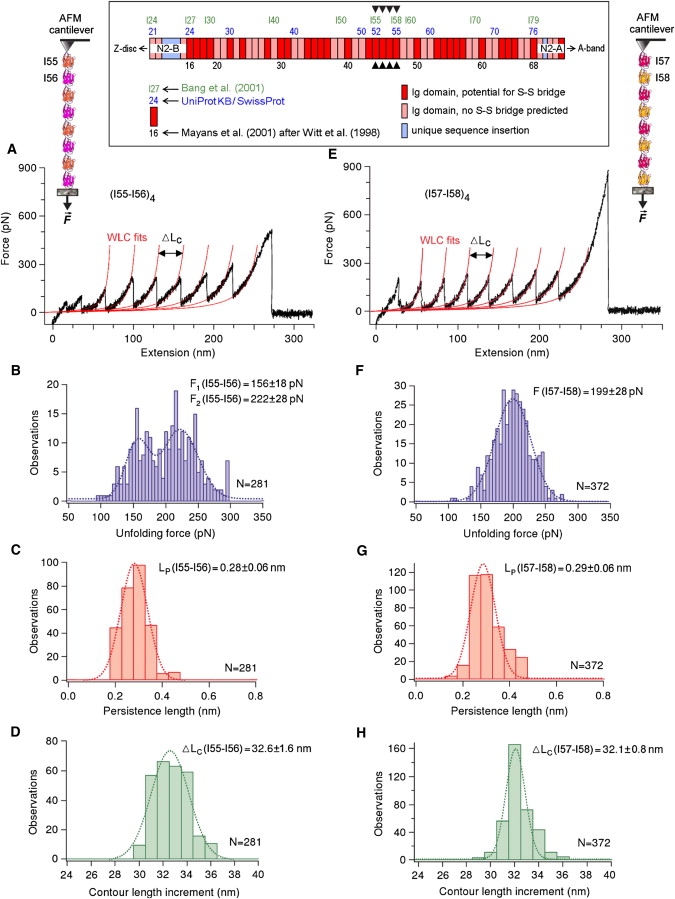
AFM force-extension experiments to test whether titin Ig domains can be stabilized by internal (S-S) bonding. (A and E) Exemplary recordings and WLC fits to Ig-unfolding peaks; (B and F) Ig-domain unfolding forces; (C and G) persistence length, Lp, of the (partially) unfolded polypeptide chain; and (D and H) contour-length increment upon Ig unfolding, ΔLc, for two different polyprotein constructs: (A–D) (I55-I56)4 and (E–H) (I57-I58)4. Values at the top of the histograms are mean ± SD; dotted lines are the best Gaussian fits. (Inset) Domain numbering for the differentially spliced I-band Ig-domain segment in human cardiac titin is according to Bang et al. (23), Mayans et al. (18) using the nomenclature of Witt et al. (20), or the UniProtKB/Swiss-Prot entry (Q8WZ42) for human titin. Predictions for S-S bridge potential in Igs were taken from Mayans et al. (18). Arrowheads point to the four Ig domains investigated by AFM.
In this study we tested the possibility that altered mechanical stability of titin domains is caused by redox state-dependent S-S bridge formation. Contrary to the theoretical predictions, single-molecule AFM force-extension measurements on recombinant Ig-domain polyprotein constructs failed to show evidence for domain-stabilizing disulfide bonding in the differentially spliced I-band Ig segment. However, we found that the cardiac-specific N2-Bus, a physiologically extensible titin region (21) that comprises 572 amino-acid residues in human titin and was previously thought to be an intrinsically disordered protein segment (22), is capable of S-S bonding under oxidizing conditions. In AFM force spectroscopy experiments, intramolecular bonding stabilized the N2-Bus mechanically. Entropic elasticity modeling and mechanical experiments on isolated human heart myofibrils showed that titin-based stiffness can be elevated in sarcomeres as a result of increased oxidant stress. Thus, disulfide bonding occurring in titin's N2-Bus, but not the Ig domains, may play a role in the increased global passive stiffness of the heart seen in various pathological conditions.
Materials and Methods
Recombinant titin constructs
To study whether a disulfide bridge can be detected in mechanical AFM experiments on titin Ig domains, we generated two different recombinant polyprotein constructs, each of which contained two neighboring Ig domains from the differentially spliced I-band titin segment: (I55-I56)4 and (I57-58)4 (after the nomenclature of Bang et al. (23)), which would be called (44-45)4 and (46-47)4 according to Witt et al. (20) and Mayans et al. (18), respectively, or (52-53)4 and (54-55)4 according to the UniProtKB/SwissProt server Ig-domain numbering (Fig. 1, inset). Both polyprotein constructs, whose constituting Ig domains have been suggested (18) to potentially contain a disulfide bond (Fig. 1, inset), were expressed in Escherichia coli (24) and purified under highly oxidative conditions in the presence of H2O2.
Two different recombinant constructs were engineered that contained the N2-Bus flanked by Ig domains. One construct type comprised three identical I91 domains (initially called I27 (25) in the single-molecule field) on either side of the N2-Bus, (I91)3-N2-Bus-(I91)3 (22), and the other contained the N2-Bus flanked by its naturally occurring Ig domains, (I24-I25)-N2-Bus-(I26-I27) (14). These constructs were used for AFM force-extension measurements in which the signals from the Ig domains served as a “fingerprint” to detect single-molecule tethers (26).
We also expressed in Escherichia coli the human N2-Bus without flanking Ig domains for use in Ellman's test (see below). This N2-Bus construct was purified in the absence of a reducing agent (dithiothreitol (DTT); Sigma Aldrich, Munich, Germany). In addition to the wild-type N2-Bus, two different mutants were generated: one with the cysteine at position 7 of the N2-Bus replaced by a serine (C7S), and one with the cysteine at position 100 replaced by a serine (C100S).
The expression of all recombinant constructs was monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis.
AFM force measurements
Both an MFP-3D atomic force microscope (Atomic Force F&E GmbH, Mannheim, Germany) and a custom-built atomic force microscope (14) were used. The spring constant of each individual cantilever (sharpened silicone nitride, MSCT-AUHW; Veeco, Santa Barbara, CA) was calibrated using the equipartition theorem, and was typically found to be ∼40 pNnm−1. In a given experiment, 2 μL of a 2 nM protein suspension were deposited onto a glass coverslip and after 5 min the glass slide was rinsed with 200 mM phosphate-buffered saline (PBS). AFM force spectroscopy with the Ig-domain constructs was done in 200 mM PBS buffer. AFM measurements with the N2-Bus-containing constructs were performed in 200 mM PBS in both the absence and presence of 10 mM DTT. After an equilibration time of 5 min, polyproteins were picked up randomly by adsorption to the AFM cantilever tip, facilitated by pressing down onto the sample for ∼1 s at high force (∼1.5 nN), before the stretch commenced. Surface protein density was optimized to ensure a low probability of tethering to the AFM tip (about one in 400 attempts) to minimize the chance of capturing two or more molecules and stretching them simultaneously. The pulling rate in all AFM force-extension measurements was 500 nm s−1. Experiments were performed at room temperature.
AFM data analysis
Protein elasticity was modeled using the wormlike chain (WLC) model of pure-entropic elasticity according to the following equation:
| (1) |
where F is the entropic-based force, Lp is the persistence length (a measure of the bending rigidity of the polymer chain), x is the end-to-end extension, Lc is the contour length, kB is the Boltzmann constant, and T is the absolute temperature (26). Adjustable parameters in the model calculations were Lp and Lc. Local force maxima (sawtooth-like force peaks) in force-extension curves were detected by means of a custom-made Igor procedure (WaveMetrics, Portland, OR), with the final force peak in each trace discarded as indicating detachment from the coverslip and/or AFM tip. Continuous interpeak segments in a sawtooth-like force-extension trace were fitted using the WLC model as follows: If the difference in Lc for consecutive segments fell within the range of 24–35 nm for a given fixed Lp, then the force peak of the preceding segment was assigned as an Ig-domain unfolding event (22,27). For the N2-Bus-containing constructs, we analyzed only those force-extension curves that showed at least three ((I24-I25)-N2-Bus-(I26-I27)) or four ((I91)3-N2-Bus-(I91)3) regularly spaced Ig-unfolding peaks, because only then could we be sure that the full N2-Bus was stretched. The Ig-unfolding peaks (regularly spaced owing to a similar or even identical increase in contour length upon each unfolding event) represent a “fingerprint” that is obtained simply by using the WLC model. No alternative “fingerprinting” approaches (28) were applied here. The entropic elasticity parameters for the N2-Bus were extracted from a WLC fit to the first Ig-unfolding peak, which likely represented the force-extension behavior of the N2-Bus.
Disulfide bridge predictions
S-S bonds in the N2-Bus were predicted by the DiANNA (DiAminoacid Neural Network Application) webserver (http://clavius.bc.edu/∼clotelab/DiANNA/), the CysPRED server (http://gpcr.biocomp.unibo.it/cgi/predictors/cyspred/pred_cyspredcgi.cgi), the PreCys server (http://bioinfo.csie.ntu.edu.tw:5433/Disulfide/), or the DISULFIND server (http://disulfind.dsi.unifi.it/). DiANNA finds the maximum likelihood bonding state assignment that satisfies the constraint for having an even number of disulfide bonded cysteines (ignoring interchain bonds (29)). CysPRED is a neural network-based predictor that uses evolutionary information (30). PreCys uses a two-level method of support vector machines and global information of protein structure (31). DISULFIND employs a bidirectional recursive neural network that is trained to predict the expected distance of candidate patterns from the real one (32).
Ellman's test
Ellman's test is a standard method for detecting free thiols in solution (33). Ellman's reagent, 5,5′-Dithio-bis(2-nitrobenzoic acid) (DTNB; Sigma Aldrich, Munich, Germany), binds to free sulfhydryl groups as NTB, which can be detected as a yellow color when measuring absorbance at 412 nm. Additionally, we used the property of a reducing agent, sodium borohydride (NaBH4; Sigma Aldrich, Munich, Germany), to denature a protein in solution, thereby cleaving any disulfide connections. Two different samples were prepared: 1), the control, which contained the N2-Bus (wild-type or mutants) and DTNB; and 2), the sample of interest that additionally contained the reducing agent, NaBH4. By measuring (at room temperature) the absorbance at 412 nm of the control solution (free thiol groups in nonreducing environment) and the protein-reagent complex (free thiol groups after breaking of disulfide connections), we were able to calculate the number of disulfide bridges in the N2-Bus using the following equation:
| (2) |
where N is the number of free thiols, MProtein is the molar mass of the N2-Bus (63.27 kg mol−1), Ereduced is the extinction coefficient of the NaBH4-containing solution (dimensionless), 12,000 [L mol−1cm−1] is the molar extinction coefficient of NTB in the solution, cProtein is the protein concentration, and d is the thickness of the cuvette (1.0 cm). To prevent oxidation of thiols after the disulfide connections were broken, the solution was covered with N2. For each protein type, Ellman's test was performed at least twice (the same construct was expressed at least twice). Since the results were essentially identical, only those obtained from the first measurement are shown.
Simulation of titin-based force
The titin force versus extension or sarcomere length (SL) relationship was simulated using a force-extension curve generated from the weighted sum of three WLC force-extension relations corresponding to the different extensible regions in cardiac titin (5,22): segments of tandem Ig, the PEVK region, and the N2-Bus. These segments are characterized by different contour and persistence lengths, i.e., the total titin extension, X, is given by
| (3) |
where the extension at a force F of the i th spring satisfies Eq. 1. Solutions for the equations were found with the use of a standard numerical interpolation technique.
Values for the number of Igs and PEVK residues for each WLC were as follows: I-band titin segment of N2B-isoform, 41 Ig domains, 186 residues in the PEVK region, and either 306 or 572 residues in the N2-Bus to account for its contour length in the presence and absence, respectively, of an S-S bond (which considers that bonding can occur between residues C100 (second cysteine) and C367 (fourth cysteine) of the N2-Bus). Assuming that each residue spans ∼0.36 nm, the resulting contour lengths were Lc = 110 nm (oxidizing conditions) and Lc = 205 nm (reducing conditions). For the I-band titin segment of the N2BA-isoform, a 986-residue PEVK region and additionally 40 Ig domains were assumed. Other parameters, including the persistence length values for each WLC, were as described previously (16). Titin-based myofibrillar passive tension was deduced from the force per titin, and SL was deduced from the I-band titin extension (and an A-band width of 1.6 μm) as described previously (22). Finally, since both N2B and N2BA are coexpressed in a human heart myofibril, the N2B and N2BA isoforms were weighted 65% and 35%, respectively (3).
Passive stiffness measurements on isolated cardiomyofibrils
Single human cardiomyofibrils were prepared and studied for passive force generation as previously described (34). Briefly, left ventricular tissue from human donor heart (16) was defrozen, homogenized in a “rigor” buffer without ATP (for composition, see Opitz et al. 34), and a drop of the myofibril suspension was deposited onto a coverglass under a Zeiss Axiovert 135 inverted microscope. Either end of a single myofibril was glued in ATP-containing “relaxing” buffer (for composition, see Opitz et al. 34) to the tip of a glass microneedle. One needle was connected to a custom-built force transducer with NanoNewton resolution (35), and the other was connected to a piezoelectric actuator (Physik Instrumente, Karlsruhe, Germany). Motor control and data acquisition were achieved with the use of custom-written LabView algorithms (National Instruments, Munich, Germany). Before conducting the actual experiment, we added 50 nM thioredoxin (Trx) reductase (from rat liver; Sigma Aldrich, Munich, Germany) and 2 mM β-nicotinamide-adenine-dinucleotide phosphate (NADPH; Sigma Aldrich) to the relaxing buffer. After equilibration, the SL of the myofibril (measured by video microscopy) was oscillated between slack (typically, 1.9 μm) and 140% slack at a physiological frequency of 1 Hz for 10 s. After each burst, the myofibril was allowed to recover at slack SL for 50 s. The same stretch protocol was repeated 10–12 times. Then, 8 μM of recombinant human Trx (kindly provided by Dr. A. Holmgren, Stockholm, Sweden) were added to this buffer and the stretch protocol was repeated again. Experiments were performed at room temperature. On the recorded traces we analyzed the peak-to-peak force amplitudes using LabView algorithms and calculated the mean amplitude value for each 10-s burst as a measure of passive stiffness. Data for Trx-treated myofibrils were expressed relative to controls (before addition of the Trx).
Statistics
Significant differences in myofibrillar passive stiffness were probed with the use of an unpaired Student's t-test; p-values < 0.05 were taken as indicating significant differences.
Results and Discussion
Differentially spliced Ig domains, I55–I58, are not stabilized by a disulfide bridge
We selected the differentially spliced I-band titin Ig domains I55, I56, I57, and I58 (Fig. 1, inset, arrowheads) as typical examples of Ig modules with a potential for S-S bonding, as predicted from sequence comparison with I1, a titin Ig domain with known atomic structure (18). The Ig domains I55–I58 (nomenclature of Bang et al. (23)) were called domains 44–47 by Mayans et al. (18), who used the nomenclature of Witt et al. (20). We designed polyproteins containing four identical copies of a pair of either I55-I56 (I55-I56)4 or I57-I58 (I57-I58)4 (Fig. 1). AFM force-extension experiments were performed on these polyproteins to determine their mechanical properties. Representative force traces, recorded in 200 mM PBS buffer, are shown in Fig. 1, A and E. The unfolding forces of (I57-I58)4 (Fig. 1 F) averaged around 200 pN (199 ± 28 pN; mean ± SD; n = 372), whereas those of (I55-I56)4 (Fig. 1 B) were more broadly distributed; a 2-Gaussian fit returned two different means of 156 ± 18 and 222 ± 28 pN (n = 281). These results show that I57 and I58 have similar mechanical stabilities, resembling that of the well-studied titin Ig domain, I91 (frequently also referred to as I27 according to Ig numbering before Bang et al.'s (23) publication). In contrast, I55 and I56 differed in their mechanical stabilities, but these fell within the range reported for the constitutively expressed Ig domains in I-band titin, I1-I15, and various differentially spliced Ig domains (22,26,36–38).
Fitting the WLC model (Eq. 1) to the individual force peaks returned the persistence length, Lp, of the unfolded Ig domains. The mean Lp values were 0.28 and 0.29 nm (Fig. 1, C and G), consistent with values expected for an unfolded polypeptide chain. The WLC fit also revealed the increase in contour length (ΔLc) for each Ig-unfolding event (Fig. 1, D and H). If these Ig domains contain disulfide bridge(s), the ΔLc should be significantly shorter than the ΔLc for a complete unfolding event (27–34 nm for titin Ig domains (22,27)) because AFM stretch forces do not break the covalent S-S bond (39–41). However, the ΔLc averaged 32.6 ± 1.6 nm (mean ± SD) for (I55-I56)4 and 32.1 ± 0.8 nm for (I57-I58)4, without any hint of a substantially shorter ΔLc. Previous AFM studies on the differentially spliced titin Ig domains (36–38) reported ΔLc values similar to ours. In contrast, short ΔLc values (18–25 nm; mean = ∼22 nm) have been observed, albeit very infrequently, in AFM force-extension experiments using I1 (19). Consistent with this, steered molecular-dynamics simulations showed that a disulfide bond in I1 bridging β-strands C and E would limit the maximum extension of this Ig domain to 22.0 nm (42). In this work, since we purified the Ig polyproteins in the presence of H2O2 (a strong oxidant) and conducted the AFM experiments in the absence of a reducing agent, S-S bond(s) should have been formed at least occasionally if the domains were capable of such bonding. The complete absence of short ΔLc values instead suggests that these Ig domains do not form a disulfide bridge under oxidizing conditions. In the natural environment of titin in the myocyte, the core of the (folded) Ig domains is presumably well shielded and S-S bond formation is even less likely to take place. Thus, in cardiac cells exposed to oxidant stress, titin-based stiffness is very unlikely to be altered because of titin Ig domains forming S-S bridges.
Oxidizing conditions promote S-S bonding in titin's cardiac-specific N2-B unique sequence
We previously reported (14) that web-based disulfide connectivity predictors indicated the potential for S-S bonding in a titin segment expressed only in the cardiac isoforms of this giant protein, specifically, the N2-B unique sequence (see Fig. 1, inset). Here we extended that analysis and applied several different webserver algorithms to search for the potential of S-S bridge formation in the 572-residue human N2-Bus, which contains six cysteine residues (Fig. 2 A). Indeed, three out of four algorithms returned a positive result (Fig. 2 A, bottom): DiANNA predicted three S-S bridges (connecting the C7–C445, C100–C367, and C264–C408 residues in the N2-Bus), CysPRED predicted one S-S bridge (C7–C264), and PreCys also predicted one S-S bridge (C7–C445). Disulfind predicted no S-S connectivity.
Figure 2.

S-S bonding predictions for the human N2-Bus and Ellman's test for the presence of free thiols in recombinant N2-Bus constructs. (A) Amino-acid sequence of the human N2-Bus (accession No. X90568 in GenBankTM/EBI Data Bank), with cysteines boxed and numbered. The bottom part lists the results of four different web-based disulfide bridge-prediction algorithms calculating the probability of S-S bonding in the N2-Bus. Shown are predicted connectivities (cysteine numbers in red) and the respective contour length of N2-Bus, Lc, for each bonding prediction, assuming 0.36 nm per amino acid. (B) Results of Ellman's test to detect free thiols in recombinant wild-type and mutant (C7S; C100S) human N2-Bus constructs in solution. (Left) Example of a color change of wild-type N2-Bus in Ellman's reagent, in the presence versus absence of reducing agent, NaBH4. (Right) Relative decrease in transmission at 412 nm measured for wild-type and mutant N2-Bus constructs under reducing and nonreducing conditions; a decrease in transmission indicates that thiols became free in the presence of NaBH4 (scale on right).
In attempt to validate this result experimentally, we used Ellman's test to detect free thiols in recombinantly expressed human wild-type or mutated (C7S or C100S) N2-Bus constructs purified under nonreducing conditions. The constructs in solution were studied for absorbance at 412 nm before and after exposure to the reducing agent, NaBH4. Using the wild-type N2-Bus construct (concentration, cProtein = 0.17 g L−1), the solution turned yellow in the presence of NaBH4 (Fig. 2 B, left) and transmission dropped by ∼20% (extinction coefficient of the NaBH4-containing solution, Ereduced = 0.193). A simple calculation using Eq. 2 showed that on addition of NaBH4, n = 6 thiols/molecule became free, suggesting that all sulfhydryl groups in the wild-type N2-Bus are capable of S-S bonding under oxidizing conditions. The predictions of the DiANNA webserver were thus confirmed, although the experiment does not distinguish inter- from intramolecular disulfide bridges. Using the mutants C7S (cProtein = 0.037 g L−1) and C100S (cProtein = 0.048 g L−1), transmission in the presence of NaBH4 was reduced by 11.4% (Ereduced = 0.025) and 12.8% (Ereduced = 0.036), respectively (Fig. 2 B, right). Calculating the number of freed thiols in the mutants using Eq. 2, we obtained n = 4 for both C7S and C100S. These results confirm that both C7 and C100 of the N2-Bus participate in S-S bonding. Since the reduced mutant constructs showed two free thiols less than the wild-type N2-Bus, even though only one cysteine was mutated in each construct, C7 and C100 appear to be involved in intramolecular S-S bonding.
S-S bonding mechanically stabilizes the N2-Bus
To directly test at the single-molecule level whether disulfide bridge(s) form under oxidizing conditions in the N2-Bus, we studied the mechanical properties of this titin segment using AFM force-extension measurements (22,26). To that end, we generated two different recombinant polyprotein constructs containing the human N2-Bus flanked on either side by several Ig domains. One construct had the form (I91)3-N2-Bus-(I91)3, and the other had the form (I24-I25)-N2-Bus-(I26-I27) (Fig. 3). Representative AFM force traces recorded in 200 mM PBS in the absence of reducing agent (“−DTT”) are shown in Fig. 3, A and B, and D and E, respectively. We analyzed only those traces in which a “fingerprint” of successive Ig-unfolding events indicated that the whole N2-Bus had been stretched (see Materials and Methods). By fitting the force-extension data up to the first Ig-domain unfolding peak, we were able to parameterize the elastic properties of the N2-Bus using the WLC model (Eq. 1). Of interest, the fits revealed two different populations of contour length (Lc) for the N2-Bus. One population was centered at 110–120 nm, and the other at 190–210 nm (Fig. 3, C and F). An Lc value of 205–210 nm would be expected if the N2-Bus were a completely unfolded polypeptide (572 residues in N2-Bus, each extending maximally by 0.36 nm). Therefore, a substantial number of N2-Bus molecules were likely to be in an unfolded (unbonded) state. However, the fact that the contour length frequently reached much less than ∼205 nm suggests that in this population, the N2-Bus was stabilized by internal interactions that are unbreakable by AFM, most likely S-S bond(s) forming intramolecularly.
Figure 3.
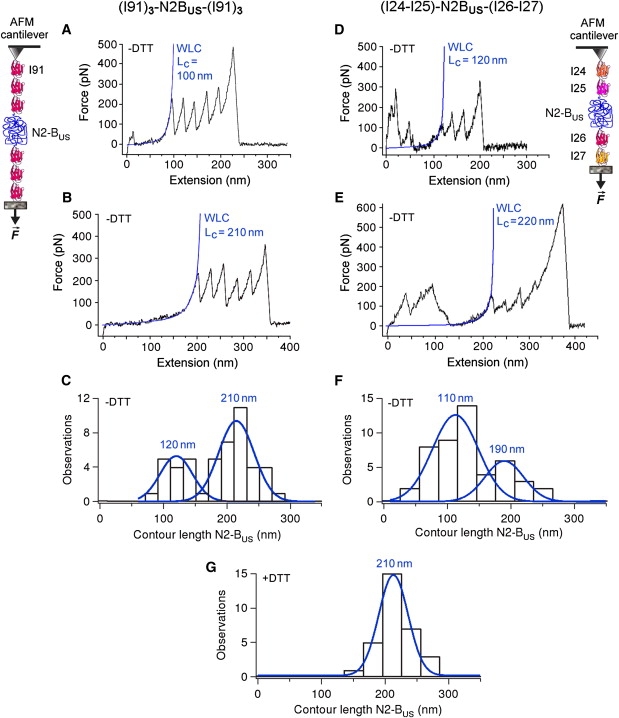
AFM force-extension experiments probing the presence of S-S bonds in the N2-Bus. Polyproteins (I91)3-N2-Bus-(I91)3 (left panels) and (I24)-(I25)-N2-Bus-(I26)-(I27) (right panels) were recombinantly expressed and stretched by AFM. Only those recordings that showed at least four regularly spaced Ig-unfolding peaks for (I91)3-N2-Bus-(I91)3 or three regularly spaced Ig-unfolding peaks for (I24)-(I25)-N2-Bus-(I26)-(I27) were analyzed by WLC fitting, because only then could we be confident that the whole N2-Bus had been stretched. (A–E) Exemplary recordings in 200 mM PBS buffer lacking reducing agent (−DTT), and WLC fit of N2-Bus force-extension behavior up to the first Ig-unfolding peak. Calculated contour length (Lc) values for the N2-Bus are indicated in blue text. (C and F) Histograms showing bimodal contour-length distribution in the absence of reducing agent (−DTT). Blue lines and values are single Gaussian fits and mean Lc. (G) Histogram of contour-length distribution for the N2-Bus in 200 mM PBS buffer supplemented with 10 mM DTT (+DTT). Blue line and value indicate single Gaussian fit and mean Lc. Note the absence of shorter contour lengths.
The shorter mean Lc value detected under these experimental conditions (110–120 nm) is identical to the value one can infer from S-S bonding between cysteines C100 and C367 (DiANNA webserver), leaving 305 residues for extension, such that Lc = 305 × 0.36 nm = 110 nm, or from S-S bonding between cysteines C7 and C264 (CysPRED server), leaving 315 residues for extension, such that Lc = 315 × 0.36 nm ≈ 115 nm (see Fig. 2 A, bottom). These bonding configurations therefore appear to be particularly common. However, we cannot exclude the possibility that a disulfide bridge was formed between other cysteines in the N2-Bus, albeit much less frequently. We sometimes measured a contour length as low as ∼50 nm, a value predicted (DiANNA, PreCys) if S-S bonds occur between C7 and C445 (leaving 134 residues for extension, such that Lc = 134 × 0.36 nm ≈ 50 nm). Also a rare bonding between C264 and C408 is possible (DiANNA prediction; leaving 428 residues for extension, such that Lc = 428 × 0.36 nm ≈ 155 nm), as Lc values near 155 nm were occasionally found (Fig. 3, C and F). Even if the high flexibility of the N2-Bus (persistence length = <1 nm; data not shown, but consistent with Li et al. 22) allowed the formation of more than one S-S bond within the same molecule, the AFM analysis would still detect only one Lc value. Thus, the AFM results support the above notion that up to three disulfide bridges can be present in the N2-Bus under oxidizing conditions. Most frequently, however, this segment contains less than three S-S bonds, and presumably, sometimes only one.
Experimental evidence for redox state-dependent alterations in the mechanical stability of the N2-Bus was obtained when the AFM measurements were performed using the N2-Bus-containing polyproteins in the presence of 10 mM DTT (Fig. 3 G). Under these conditions, the population of short Lc values disappeared and the N2-Bus had a single mean contour length of ∼210 nm. These findings indicate that there was a reductant-induced breakage of one or more intramolecular S-S bonds that previously stabilized the N2-Bus under oxidizing conditions.
WLC modeling predicts redox state-dependent alterations in titin-based passive tension
How would a shortened Lc of the N2-Bus affect the titin spring force? We simulated the force per titin versus I-band extension relationship as that of three WLCs in series (titin Ig segments, PEVK domain, and N2-Bus (5,16,22); see Eq. 3) using custom-written LabView procedures. The cardiac titin isoforms, N2BA and N2B, are expressed in the sarcomere at a ratio that depends on the developmental stage or disease status (2). In normal adult human hearts, the N2B isoform is 3000 kDa (25), whereas N2BA occurs in different isoforms with an average molecular mass of ∼3300 kDa (3,5). The additional mass in N2BA arises because more Ig domains and PEVK residues are spliced into the I-band segment compared to N2B (23). The longer spring makes N2BA more compliant than N2B (21,43). Simulating the force-extension behavior of either isoform using two different Lc values for the N2-Bus (110 nm and 205 nm) while leaving all other WLC parameters unaltered resulted in almost unchanged N2BA curves, but greatly different N2B curves (Fig. 4 A). Using Lc = 110 nm (“oxidizing conditions”), the force of N2B titin was increased by up to 2.2 times compared to a simulation using Lc = 205 nm (“reducing conditions”). When the N2BA:N2B expression ratio in normal human myocardium (35:65% (3,5)) was taken into account, titin-based passive tension was up to 30% higher with Lc = 110 nm than with Lc = 205 nm (Fig. 4 B). Differences between the curves became obvious at SL > 2.1 μm and increased with further stretch, as the N2-Bus elongates significantly at higher physiological SLs (21,43). In summary, WLC modeling predicted that alterations in the redox state could substantially affect titin-based stiffness in myocytes due to altered mechanical stability of the N2-Bus.
Figure 4.
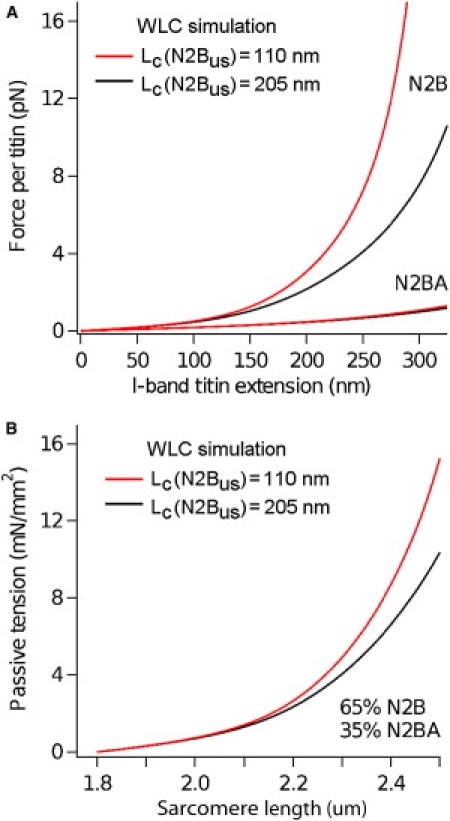
WLC simulations of the effect of a less-extensible N2-Bus on titin-based spring force. (A) Predicted differences in the force/titin versus I-band titin extension curves using Lc values for the N2-Bus of either 110 nm (red curves) or 205 nm (black curves). The simulation was performed separately for the two human cardiac titin isoforms, N2B (3000 kDa) and N2BA (3300 kDa). (B) Predicted difference in the passive tension versus SL relationship of human cardiac titin, using a contour length for the N2-Bus of either 110 nm (red curve) or 205 nm (black curve). This simulation also considered the 35:65 expression ratio of N2BA:N2B titin isoforms in normal human left ventricle.
Reducing agent lowers the passive stiffness of isolated human cardiomyofibrils
We tested whether the above prediction would hold true in titin's natural environment, the sarcomere, by measuring the passive stiffness of isolated human cardiomyofibrils obtained from normal donor hearts in response to 1 Hz stretch-release cycles (Fig. 5 A). The myofibrils were prepared in a buffer lacking a reducing agent. Myofibrillar stiffness was then estimated as the mean peak-to-valley amplitude of the oscillatory force response before and after addition of a recombinant reducing agent, human Trx (in the presence of Trx reductase and NADPH). Trx is an endogenous antioxidant whose downregulation associated with oxidative stress is found in many pathological states, including cardiac hypertrophy and failure (44–46). Recombinant Trx lowered the myofibrillar passive stiffness significantly (typically within a few minutes) by a relative amount ranging between ∼12% and ∼26% (Fig. 5 B). The magnitude of this effect compares well with that predicted from WLC modeling using Lc values for the N2-Bus of 110 and 205 nm, respectively (Fig. 4 B). It is likely that the disulfide bond(s) in the N2-Bus were cleaved by Trx, suggesting the mechanism observed in the AFM experiments also works in situ. In this context, the N2-Bus has been reported to extend maximally to a contour length of ∼105 nm in stretched isolated cardiomyofibrils in the absence of reducing agent—only about half the ∼205 nm expected for the fully unfolded polypeptide (14,21). In contrast, N2-Bus extension was found to reach ∼200 nm at 3.3 μm SL in enzymatically digested, skinned cardiomyocytes incubated in a buffer containing 1 mM DTT (43). Taken together, these results suggest that under oxidizing conditions, internal S-S bonding can limit the extensibility of the N2-Bus in situ, giving rise to increased titin-based stiffness in cardiac muscle cells.
Figure 5.
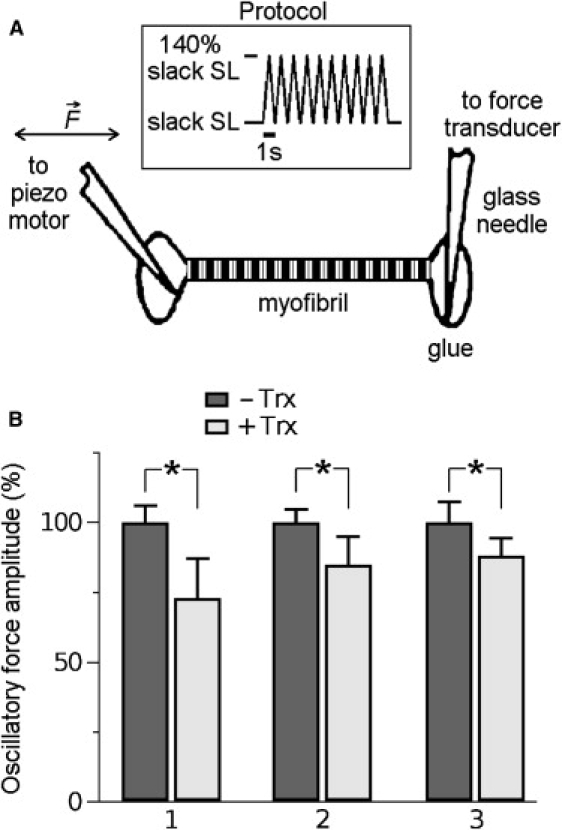
Effect of reducing agent on the passive stiffness of cardiomyofibrils isolated from human donor hearts. (A) Experimental setup and mechanics protocol. (B) Change in the passive stiffness of three different myofibrils studied in relaxing buffer supplemented with 50 nM Trx reductase and 2 mM NADPH, before (−Trx) and after (+Trx) application of human Trx (8 μM). Bars show average oscillatory force amplitudes (mean ± SE) in bursts of 10 stretch-release cycles; data were expressed relative to the mean amplitude before addition of Trx. Time between (−Trx) and (+Trx) recordings is ∼5 min. Asterisks indicate statistically significant differences (p < 0.05 in Student's t-test).
Summary and Conclusions
Contrary to what we expected when we initiated this study, single-molecule AFM force spectroscopy of recombinant polyprotein constructs revealed no evidence of internal disulfide bonding in titin's differentially spliced Ig domains. Instead, our work suggests that one or more S-S bridge(s) can mechanically stabilize the human N2-Bus under conditions that favor the oxidized state. The N2-Bus, a cardiac-specific titin domain, has been speculated to be a permanently unfolded random coil (22,47), a notion that may not hold up in light of the findings presented here. On the other hand, a low degree of secondary structuring may allow the free thiols in the N2-Bus to bond more easily under oxidizing conditions than, e.g., the thiols in the folded titin Ig domain, I1 (18), which in AFM measurements showed only a very low propensity for S-S bonding (19). In this study, disulfide bridges in the N2-Bus were confirmed under oxidizing conditions in vitro. This novel property of the N2-Bus has implications for titin function and adds a twist to our understanding of passive stiffness regulation and dysregulation in the heart.
Normally, disulfide bridges are unlikely to form inside muscle cells because there is a reducing environment, due mainly to the presence of glutathione (48) and other reductants, such as Trx. However, when cardiomyocytes experience oxidative stress, disulfide bridges formed in the N2-Bus will decrease titin extensibility and increase titin-based passive tension, as observed here in isolated human cardiomyofibrils. Titin stiffening could thus contribute to the alterations in global myocardial mechanics associated with oxidant stress, which often accompanies aging or heart failure (49–51). Promoting oxidative mechanisms will thus alter the two major elements known to determine myocardial passive stiffness: the extracellular matrix, which is known to remodel in response to oxidative stress (52,53), and titin. S-S bonding in titin's N2-Bus may add to other aging- or disease-related mechanisms that modify titin-based stiffness, such as titin-isoform transition and alterations in titin phosphorylation (17). Moreover, internal S-S bonding in the N2-Bus could interfere with various other important functions of the N2-B domain, including ligand binding (2), mechanosensing (54), or phosphorylation-dependent stiffness adjustment (16). We conclude that the S-S bonding property of the N2-Bus is likely to be of pathophysiological importance.
Acknowledgments
We thank Dr. Arne Holmgren for providing the recombinant human Trx. We also thank Dr. Hongbin Li, Judith Krysiak, and Stefanie Klede for help with recombinant construct expression.
S.G.-M. received a postdoctoral fellowship from the Generalitat de Catalunya through the NANO and Beatriu de Pinós programs, and financial support from the Fundación Caja Madrid. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 629; Li 690/6-3 to W.A.L.) and the National Institutes of Health (grants HL66030 and HL61228 to J.M.F.).
References
- 1.Granzier H., Labeit S. Structure-function relations of the giant elastic protein titin in striated and smooth muscle cells. Muscle Nerve. 2007;36:740–755. doi: 10.1002/mus.20886. [DOI] [PubMed] [Google Scholar]
- 2.Linke W.A. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc. Res. 2008;77:637–648. doi: 10.1016/j.cardiores.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 3.Neagoe C., Kulke M., del Monte F., Gwathmey J.K., de Tombe P.P. Titin isoform switch in ischemic human heart disease. Circulation. 2002;106:1333–1341. doi: 10.1161/01.cir.0000029803.93022.93. [DOI] [PubMed] [Google Scholar]
- 4.Nagueh S.F., Shah G., Wu Y., Torre-Amione G., King N.M. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155–162. doi: 10.1161/01.CIR.0000135591.37759.AF. [DOI] [PubMed] [Google Scholar]
- 5.Makarenko I., Opitz C.A., Leake M.C., Neagoe C., Kulke M. Passive stiffness changes caused by upregulation of compliant titin isoforms in human DCM hearts. Circ. Res. 2004;95:708–716. doi: 10.1161/01.RES.0000143901.37063.2f. [DOI] [PubMed] [Google Scholar]
- 6.Lahmers S., Wu Y., Call D.R., Labeit S., Granzier H. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ. Res. 2004;94:505–513. doi: 10.1161/01.RES.0000115522.52554.86. [DOI] [PubMed] [Google Scholar]
- 7.Opitz C.A., Leake M.C., Makarenko I., Benes V., Linke W.A. Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ. Res. 2004;94:967–975. doi: 10.1161/01.RES.0000124301.48193.E1. [DOI] [PubMed] [Google Scholar]
- 8.Warren C.M., Krzesinski P.R., Campbell K.S., Moss R.L., Greaser M.L. Titin isoform changes in rat myocardium during development. Mech. Dev. 2004;121:1301–1312. doi: 10.1016/j.mod.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Krüger M., Kohl T., Linke W.A. Developmental changes in passive stiffness and myofilament Ca2+ sensitivity due to titin and troponin-I isoform switching are not critically triggered by birth. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H496–H506. doi: 10.1152/ajpheart.00114.2006. [DOI] [PubMed] [Google Scholar]
- 10.Krüger M., Sachse C., Zimmermann W.H., Eschenhagen T., Klede S. Thyroid hormone regulates developmental titin isoform transitions via the phosphatidylinositol-3-kinase/AKT pathway. Circ. Res. 2008;102:439–447. doi: 10.1161/CIRCRESAHA.107.162719. [DOI] [PubMed] [Google Scholar]
- 11.Yamasaki R., Wu Y., McNabb M., Greaser M., Labeit S. Protein kinase-A phosphorylates titin's cardiac-specific N2B-domain and reduces passive tension in rat cardiac myocytes. Circ. Res. 2002;90:1181–1188. doi: 10.1161/01.res.0000021115.24712.99. [DOI] [PubMed] [Google Scholar]
- 12.Fukuda N., Wu Y., Nair P., Granzier H.L. Phosphorylation of titin modulates passive stiffness of cardiac muscle in a titin isoform-dependent manner. J. Gen. Physiol. 2005;125:257–271. doi: 10.1085/jgp.200409177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krüger M., Linke W.A. Protein kinase-A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J. Muscle Res. Cell Motil. 2006;27:435–444. doi: 10.1007/s10974-006-9090-5. [DOI] [PubMed] [Google Scholar]
- 14.Leake M.C., Grützner A., Krüger M., Linke W.A. Mechanical properties of cardiac titin's N2B-region by single-molecule atomic force spectroscopy. J. Struct. Biol. 2006;155:263–272. doi: 10.1016/j.jsb.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 15.van Heerebeek L., Borbély A., Niessen H.W., Bronzwaer J.G., van der Velden J. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–1973. doi: 10.1161/CIRCULATIONAHA.105.587519. [DOI] [PubMed] [Google Scholar]
- 16.Krüger M., Kötter S., Grützner A., Lang P., Andresen C. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 2009;104:87–94. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 17.Krüger M., Linke W.A. Titin-based mechanical signalling in normal and failing myocardium. J. Mol. Cell. Cardiol. 2009;46:490–498. doi: 10.1016/j.yjmcc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Mayans O., Wuerges J., Canela S., Gautel M., Wilmanns M. Structural evidence for a possible role of reversible disulphide bridge formation in the elasticity of the muscle protein titin. Structure. 2001;9:331–340. doi: 10.1016/s0969-2126(01)00591-3. [DOI] [PubMed] [Google Scholar]
- 19.Li H., Fernandez J.M. Mechanical design of the first proximal Ig domain of human cardiac titin revealed by single molecule force spectroscopy. J. Mol. Biol. 2003;334:75–86. doi: 10.1016/j.jmb.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 20.Witt C.C., Olivieri N., Centner T., Kolmerer B., Millevoi S. A survey of the primary structure and the interspecies conservation of I-band titin's elastic elements in vertebrates. J. Struct. Biol. 1998;122:206–215. doi: 10.1006/jsbi.1998.3993. [DOI] [PubMed] [Google Scholar]
- 21.Linke W.A., Rudy D.E., Centner T., Gautel M., Witt C. I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J. Cell Biol. 1999;146:631–644. doi: 10.1083/jcb.146.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H.B., Linke W.A., Oberhauser A.F., Carrion-Vazquez M., Kerkvliet J.G. Reverse engineering of the giant muscle protein titin. Nature. 2002;418:998–1002. doi: 10.1038/nature00938. [DOI] [PubMed] [Google Scholar]
- 23.Bang M.L., Centner T., Fornoff F., Geach A.J., Gotthardt M. The complete gene sequence of titin expression of an unusual approximately 700-kDa titin isoform and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 2001;89:1065–1072. doi: 10.1161/hh2301.100981. [DOI] [PubMed] [Google Scholar]
- 24.Carrion-Vazquez M., Oberhauser A.F., Fisher T.E., Marszalek P.E., Li H. Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog. Biophys. Mol. Biol. 2000;74:63–91. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 25.Labeit S., Kolmerer B. Titins, giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 26.Linke W.A., Grützner A. Pulling single molecules of titin by AFM—recent advances and physiological implications. Pflugers Arch. 2008;456:101–115. doi: 10.1007/s00424-007-0389-x. [DOI] [PubMed] [Google Scholar]
- 27.Rief M., Gautel M., Oesterhelt F., Fernandez J.M., Gaub H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 28.Puchner E.M., Franzen G., Gautel M., Gaub H.E. Comparing proteins by their unfolding pattern. Biophys. J. 2008;95:426–434. doi: 10.1529/biophysj.108.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferre F., Clote P. DiANNA: a web server for disulfide connectivity prediction. Nucleic Acids Res. 2005;33(Web Server issue):W230–W232. doi: 10.1093/nar/gki412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fariselli P., Riccobelli P., Casadio R. The role of evolutionary information in predicting the disulfide bonding state of cysteines in proteins. Proteins. 1999;36:340–346. [PubMed] [Google Scholar]
- 31.Chen B., Tsai C., Chan C., Kao C. Disulfide connectivity prediction with 70% accuracy using two-level methods. Proteins. 2006;64:246–252. doi: 10.1002/prot.20972. [DOI] [PubMed] [Google Scholar]
- 32.Ceroni A., Passerini A., Vullo A., Frasconi P. DISULFIND: a disulfide bonding state and cysteine connectivity prediction server. Nucleic Acids Res. 2006;34(Web Server issue):W177–W181. doi: 10.1093/nar/gkl266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellman G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 34.Opitz C.A., Kulke M., Leake M.C., Neagoe C., Hinssen H. Damped elastic recoil of the titin spring in myofibrils of human myocardium. Proc. Natl. Acad. Sci. USA. 2003;100:12688–12693. doi: 10.1073/pnas.2133733100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linke W.A., Popov V.I., Pollack G.H. Passive and active tension in single cardiac myofibrils. Biophys. J. 1994;67:782–792. doi: 10.1016/S0006-3495(94)80538-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rief M., Gautel M., Schemmel A., Gaub H.E. The mechanical stability of immunoglobulin and fibronectin III domains in the muscle protein titin measured by atomic force microscopy. Biophys. J. 1998;75:3008–3014. doi: 10.1016/S0006-3495(98)77741-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe K., Muhle-Goll C., Kellermayer M.S., Labeit S., Granzier H. Different molecular mechanics displayed by titin's constitutively and differentially expressed tandem Ig segments. J. Struct. Biol. 2002;137:248–258. doi: 10.1006/jsbi.2002.4458. [DOI] [PubMed] [Google Scholar]
- 38.Grama L., Nagy A., Scholl C., Huber T., Kellermayer M.S. Local variability in the mechanics of titin's tandem Ig segments. Croat. Chem. Acta. 2005;78:405–411. [Google Scholar]
- 39.Carl P., Kwok C.H., Manderson G., Speicher D.W., Discher D.E. Forced unfolding modulated by disulfide bonds in the Ig domains of a cell adhesion molecule. Proc. Natl. Acad. Sci. USA. 2001;98:1565–1570. doi: 10.1073/pnas.031409698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiita A.P., Ainavarapu S.R., Huang H.H., Fernandez J.M. Force-dependent chemical kinetics of disulfide bond reduction observed with single molecule techniques. Proc. Natl. Acad. Sci. USA. 2006;103:7222–7227. doi: 10.1073/pnas.0511035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ainavarapu S.R., Brujic J., Huang H.H., Wiita A.P., Lu H. Contour length and refolding rate of a small protein controlled by engineered disulfide bonds. Biophys. J. 2007;92:225–233. doi: 10.1529/biophysj.106.091561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao M., Wilmanns M., Schulten K. Steered molecular dynamics studies of titin I1 domain unfolding. Biophys. J. 2002;83:3435–3445. doi: 10.1016/S0006-3495(02)75343-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trombitas K., Freiburg A., Centner T., Labeit S., Granzier H. Molecular dissection of N2B cardiac titin's extensibility. Biophys. J. 1999;77:3189–3196. doi: 10.1016/S0006-3495(99)77149-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.World C.J., Yamawaki H., Berk B.C. Thioredoxin in the cardiovascular system. J. Mol. Med. 2006;84:997–1003. doi: 10.1007/s00109-006-0109-6. [DOI] [PubMed] [Google Scholar]
- 45.Berndt C., Lillig C.H., Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1227–H1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 46.Maulik N., Das D.K. Emerging potential of thioredoxin and thioredoxin interacting proteins in various disease conditions. Biochim. Biophys. Acta. 2008;1780:1368–1382. doi: 10.1016/j.bbagen.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe K., Nair P., Labeit D., Kellermayer M.S., Greaser M. Molecular mechanics of cardiac titin's PEVK and N2B spring elements. J. Biol. Chem. 2002;277:11549–11558. doi: 10.1074/jbc.M200356200. [DOI] [PubMed] [Google Scholar]
- 48.Rutten E.P., Engelen M.P., Schols A.M., Deutz N.E. Skeletal muscle glutamate metabolism in health and disease: state of the art. Curr. Opin. Clin. Nutr. Metab. Care. 2005;8:41–51. doi: 10.1097/00075197-200501000-00007. [DOI] [PubMed] [Google Scholar]
- 49.McMurray J., Chopra M., Abdullah I., Smith W.E., Dargie H.J. Evidence of oxidative stress in chronic heart failure in humans. Eur. Heart J. 1995;14:1493–1498. doi: 10.1093/eurheartj/14.11.1493. [DOI] [PubMed] [Google Scholar]
- 50.Singal P.K., Khaper N., Palace V., Kumar D. The role of oxidative stress in the genesis of heart disease. Cardiovasc. Res. 1998;40:426–432. doi: 10.1016/s0008-6363(98)00244-2. [DOI] [PubMed] [Google Scholar]
- 51.Giordano F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang G.X., Kimura S., Nishiyama A., Shokoji T., Rahman M. Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovasc. Res. 2005;65:230–238. doi: 10.1016/j.cardiores.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 53.Henderson B.C., Tyagi S.C. Oxidative mechanism and homeostasis of proteinase/antiproteinase in congestive heart failure. J. Mol. Cell. Cardiol. 2006;41:959–962. doi: 10.1016/j.yjmcc.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 54.Sheikh F., Raskin A., Chu P.H., Lange S., Domenighetti A.A. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J. Clin. Invest. 2008;118:3870–3880. doi: 10.1172/JCI34472. [DOI] [PMC free article] [PubMed] [Google Scholar]