

Abstract
Antimicrobial peptides interact specifically with the membrane of a pathogen and kill the pathogen by releasing its cellular contents. Protegrin-1 (PG-1), a β-hairpin antimicrobial peptide, is known to exist as a transmembrane monomer in a 1,2-dilauroylphosphatidylcholine (DLPC) bilayer and shows concentration-dependent oligomerization in a 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) bilayer. To examine its structure, dynamics, orientation, and interaction in membranes, we performed comparative molecular dynamics simulations of PG-1 monomer and dimer in DLPC and POPC bilayers for a total of 840 ns. The PG-1 monomer exhibits larger tilting in DLPC than in POPC due to a hydrophobic mismatch. PG-1 tilting is dependent on its rotation angle. The specific orientation of PG-1 in membranes is governed by the interactions of its aromatic residues with lipid headgroups. The calculated 15N and 13CO chemical shifts of Val16 in DLPC reveal that there are different sets of tilt and rotation angles that satisfy the experimental values reasonably, suggesting that more experiments are needed to determine its orientation. The dimer simulations show that the dimer interface is better preserved in POPC than in DLPC because POPC's greater hydrophobic thickness causes reduced flexibility of the C-terminal strands. Both monomer and dimer simulations show membrane thinning around PG-1, largely due to arginine-lipid interactions.
Introduction
Antimicrobial peptides, often referred to as host defense peptides, are usually 12–50 amino acids long and exist in all living organisms. They play a key role in host defense and innate immune response (1). Due to their ability to kill a broad spectrum of pathogens like bacteria, viruses, and fungi, they are often considered as potential antibiotics (2). Antimicrobial peptides adopt different secondary structures; except for those with β-hairpin conformations reinforced by interstrand disulfide bonds, they are mostly unstructured in solution and form α-helices or β-strands upon partitioning into membranes (3). Antimicrobial peptides are rich in cationic residues such as arginine and lysine. It is the amphipathic characteristic of the peptides that enables them to interact with the pathogen membrane and kill the pathogen by releasing its cellular contents (4).
Protegrin-1 (PG-1) is a β-hairpin antimicrobial peptide of 18 amino acids (RGGRL CYCRR RFCVC VGR) that was first discovered and purified from porcine leukocytes (5). The solution NMR structure of PG-1 shows a β-hairpin structure with two antiparallel β-strands connected by a turn, which is stabilized by two interstrand disulfide bonds (Cys6-Cys15 and Cys8-Cys13) (6). There are six arginine residues in PG-1: Arg1, Arg4, and Arg18 at the terminal regions, and Arg9, Arg10, and Arg11 at the turn region. The β-hairpin structure of PG-1 closely resembles a family of antimicrobial peptides that includes horseshoe crab tachyplesins and mammalian defensins (7). Due to the broad range of antimicrobial activity of PG-1, it is considered a potential pharmaceutical agent (8). Its activity largely depends on the lipid composition of the membrane (9).
PG-1 appears to adopt different oligomeric states in different lipid bilayers. In 1,2-dilauroylphosphatidylcholine (DLPC) membranes, it remains a transmembrane monomer and its tilt angle (55 ± 5°) has been estimated based on solid-state NMR (SSNMR) measurement of 15N and 13CO chemical shifts of Val16 (10). In 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) bilayers, PG-1 shows concentration-dependent oligomerization and the minimal structural unit of PG-1 appears to be a dimer (11,12). The membrane-bound dimer structure in the POPC bilayers (Protein Data Bank (PDB) code 1ZY6) was determined by Hong and co-workers using rotational-echo double-resonance SSNMR (12). Two C-terminal strands of the monomers align parallel at the dimer interface. In anionic membranes such as POPE/POPG, PG-1 dimers form β-barrel pores of 8–10 molecules (13). Due to a wealth of biophysical and structural data on PG-1, several computational studies have been carried out to gain a better understanding of PG-1 behavior on the surface and inside of membrane bilayers, as well as to examine the morphology and behavior of PG-1 oligomeric pore in anionic and zwitterionic membranes (14–18). However, the mechanisms of membrane-dependent dimerization and oligomerization of PG-1 are still poorly understood.
To better understand the molecular driving forces underlying peptide association in different membranes, several questions need to be addressed. What is the orientation of the PG-1 monomer inside different types of lipid bilayers? More specifically, how does this orientation depend on PG-1 interactions with membranes? Furthermore, what types of interactions exist between the peptides, and between the peptide and the different environments? These questions are difficult to address by experimental techniques alone because of the limited resolution of such techniques and the experimental difficulties associated with membrane proteins/peptides. To address these questions, we have performed comparative molecular dynamics (MD) simulations of PG-1 monomers and dimers in DLPC and POPC lipid bilayers for a total of 840 ns to investigate the orientations and interactions of PG-1 peptides in membranes at the atomic level.
Methods
Simulations of PG-1 monomer in DLPC and POPC bilayers
The structure of PG-1 monomer was obtained by taking chain A from PDB 1ZY6 (12) using the PDB Reader module in CHARMM-GUI (http://www.charmm-gui.org) (19) with disulfide bonds and an amidated C-terminus. All arginine residues, as well as the N-terminus, were positively charged. The PG-1 monomer was oriented so that its principal axis was parallel to the membrane normal, i.e., the z axis, with the center of the membrane bilayer at z = 0. The PG-1/membrane systems with DLPC and POPC bilayers were then generated using CHARMM-GUI Membrane Builder (20,21) with 68 DLPC and 64 POPC lipid molecules, respectively. Each system was neutralized with 0.2 M KCl. To sample more conformational/configurational space of PG-1 in each bilayer, we generated a total of three independent systems (M1–M3) for each lipid type, using different lipid conformation/packing and ion distribution. The components of each system are given in Table S1 of the Supporting Material.
We performed 450-ps equilibration and 80-ns MD production of each system using the biomolecular simulation program CHARMM (22) with the simulation protocols used in the Membrane Builder (20,21). In this work, the Nose-Hoover method (23) was employed to maintain a constant temperature at 303.15 K, and the Langevin piston algorithm (24) was used for the NPγT (constant pressure, surface tension, and temperature) dynamics with pressure at 1 atm along the z direction and a surface tension of 20.0 dyne/cm (25,26). We have used the P21 periodic boundary condition (27) to allow the lipid molecules to move between the top and bottom leaflets of the bilayer during the simulations. Fig. S1 shows the initial and final structures of the first (M1) system in DLPC and POPC bilayers. After 20 ns of simulation, there was no big fluctuation in the system size or the lipid number in either lipid leaflet; the standard deviations of the system sizes in the xyz directions were <2.9 Å and those of the lipid number in each leaflet were <2.1. Therefore, all the average properties were calculated using the last 60 ns of each run.
Simulations of PG-1 dimers in DLPC and POPC bilayers
There are currently two PG-1 dimer structures available. Although a solution NMR study of PG-1 in micelles suggested an antiparallel dimer structure with both C-terminal strands in the dimer interface (28), the dimer structure of PDB 1ZY6, determined by rotational-echo double-resonance SSNMR (12), shows a parallel dimer structure with both C-terminal strands in the dimer interface. In this study, we only considered the parallel dimer for the simulations in DLPC and POPC bilayers. Therefore, the dimer structure was taken from PDB 1ZY6 with the same protonation state and terminal patches as those used in the monomer simulations. We followed the procedure described above to build three independent PG-1 dimer/membrane systems (D1–D3) with 78 DLPC and 74 POPC lipid molecules. The components of each system are given in Table S1. Equilibration and 60-ns production were performed using the same simulation protocols as in the PG-1 monomer simulations. Fig. S1 shows the structures in the first system in DLPC and POPC bilayers. After 20 ns of simulation, there was no big fluctuation in the system size or in the lipid number in either of the lipid leaflets; the system size standard deviations in the xyz directions were <2.4 Å and those of the lipid number in each leaflet were <2.2. Therefore, all the average properties were calculated using the last 40 ns of each run.
PG-1 orientation
The orientation of PG-1 in the membrane is defined by the tilt angle (τ) and the rotation angle (ρ). The definitions of τ and ρ are the same as in Lee et al. (29). Briefly, τ is defined as the angle between the hairpin axis (a) and the unit vector () along the z axis, i.e., the membrane normal (Fig. S2 A):
| (1) |
To define ρ uniquely, one has to choose both external and internal references. By using the z axis as the external reference and the backbone N atom of Cys6 as the internal reference, ρ can be defined as the angle between the perpendicular vector, , from a to the N atom and the projection vector, , of the z axis onto the plane made by the second and third principal axes (Fig. S2 B):
| (2) |
where and are normalized. Therefore, each τ and ρ pair yields a unique PG-1 orientation in the membrane.
Results
PG-1 monomer in DLPC and POPC bilayers
Monomer orientation
Fig. 1 shows the distributions of tilt (τ) and rotation (ρ) angles of PG-1 in each DLPC and POPC membrane system. The τ-values range from 0.4° to 54.6° in the DLPC membranes, which is similar to a previous MD study (18). The average τ of each system is 31.4 ± 6.9° (M1), 26.7 ± 6.7° (M2), and 16.9 ± 5.7° (M3). The average value in M3-DLPC was largely affected by the small tilt angles in the first half of the simulation. However, it started to increase from 40 ns (Fig. S3 A). During the last 20 ns, the average tilt angle increased to 21.7 ± 4.7°. Hong and co-workers estimated a tilt angle of 55 ± 5° based on a rigid-body geometric search of τ and ρ of solution NMR structures to best reproduce SSNMR 15N and 13CO chemical shifts of Val16 (10). Although PG-1 visited such large tilt angles sporadically during the simulations, the calculated average tilt angles are much smaller than the experimentally estimated tilt angle. Such discrepancy appears to arise from neglecting conformational flexibility and insufficient number of SSNMR observables in the SSNMR structure determination (see below).
Figure 1.
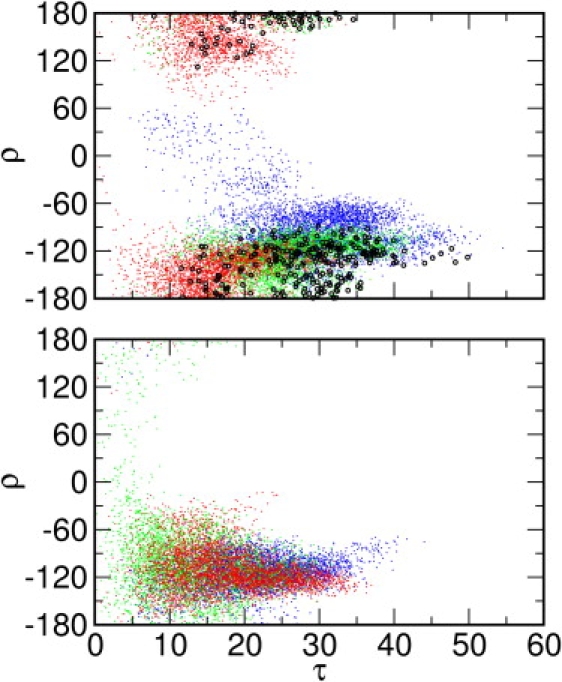
Tilt angle (τ) and rotation angle (ρ) distributions in (Top) DLPC and (Bottom) POPC bilayers. Structures with chemical shift root-mean-square deviations () <4 ppm are highlighted (black circles) in the DLPC bilayer distributions. Systems M1 (blue), M2 (green), and M3 (red) are shown in different colors for clarity.
The accessible tilt-angle range in the POPC membranes is smaller relative to that in DLPC (Fig. 1). The τ-values span from 0.1° to 46.8°, with the average tilt angles at 21.0 ± 6.5° (M1), 15.2 ± 6.6° (M2), and 20.0 ± 6.3° (M3). Recently, we demonstrated that the thermally accessible tilt angles of an isolated transmembrane (TM) helix are determined by both the generic helix precession entropy contribution and the sequence- and length-specific helix-membrane interactions (30). Since the former is the same in both DLPC and POPC membranes, the difference in PG-1 tilting stems from the latter, particularly due to the difference in the membrane hydrophobic thickness. Based on x-ray scattering experiments, the hydrophobic thickness of pure DLPC bilayers is ∼21 Å (31) and that of pure POPC membranes is ∼28 Å (32). The hydrophobic length of PG-1 is ∼30 Å based on the solution NMR structure (6). To maximize the hydrophobic interactions between PG-1 and membranes, PG-1 tilts more in DLPC than in POPC, which is also observed as a major response of single-pass TM helices to a hydrophobic mismatch (30).
There are a couple of interesting features about the rotational preference of PG-1 monomer in both membranes. First, ρ converges to ∼−120° in all the DLPC and POPC systems (Fig. S3 B). Given the rotation angle definition, the negative rotation angle represents a PG-1 structure with two disulfide bonds facing down with respect to the hairpin plane (Fig. S2 B). Second, as shown in the DLPC simulations, there is a strong correlation between τ and ρ: τ becomes larger when ρ → −120°, τ becomes smaller with positive ρ-values, and τ remains in between when ρ is close to 0° or 180°. For example, in the beginning of the M3-DLPC system, τ was considerably smaller than in the two other systems and its ρ hardly visited negative values, but this system eventually evolved to where ρ ≈ −120° and τ was comparable to that of the other systems (Fig. S3 A). Our recent potential of mean force (PMF) calculations of PG-1 as a function of τ and ρ in the EEF1/IMM1 implicit membrane model (29) show that the thermally accessible rotation angles can reach from −70° to −130°, with the minimum free energies at ρ ≈ −90°, whereas the accessible tilt angles range from 0° to 22° and 18° to 34°, depending on the choice of implicit hydrophobic membrane thickness. Both the PMF calculations and the present MD simulations demonstrate that PG-1 monomer strongly prefers a ρ of ∼−90° in membranes. Such a rotational preference appears to arise from the side-chain positions of the aromatic residues. Fig. 2 A shows the molecular structures of three primary rotamers of Tyr7 and Phe12 found in the MD simulations. When τ is large, the χ1 angles of most Tyr and Phe side chains are close to −160° (Fig. S3 A and S4 A). With a conformation of ρ ≈ −90° and χ1 ≈ −160°, the side chain of Tyr7 is located at the hydrophobic/hydrophilic membrane interface and the side chain of Phe12 mostly in the membrane hydrophobic region. On the other hand, in M3-DLPC and M2-POPC, when τ is small, the Tyr7 χ1 angles are ∼−60°, whereas the Phe12 χ1 angles are still ∼−160° (Fig. S3 and Fig. S4). The conformation with χ1 ≈ −60° shows the side chain of Tyr7 positioned mostly in the membrane hydrophobic region (Fig. 2 A), and increasing τ will be energetically unfavorable. Therefore, it becomes clear why larger tilt angles are observed when ρ ≈ −90°, and why PG-1 strongly prefers to have smaller tilt angles otherwise.
Figure 2.
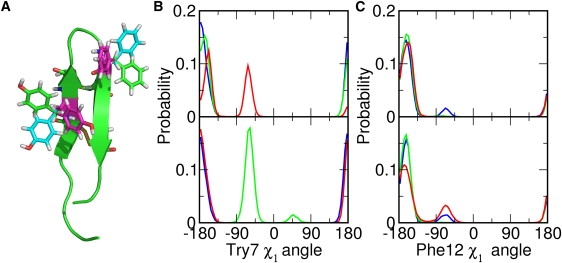
(A) Molecular structure of PG-1 Tyr7 and Phe12 rotamers with χ1= −160° (green), χ1= −60° (cyan), and χ1= 60° (magenta). The PG-1 peptide (green) is shown in cartoon representation. (B) The population of the Tyr7χ1 angle in the DLPC (upper) and POPC (lower) bilayers. (C) The population of Phe12χ1 angle in the DLPC (upper) and POPC (lower) bilayers. Systems M1 (blue), M2 (green), and M3 (red) are shown in different colors for clarity. The molecular structure of PG-1 is produced with PyMOL (41).
Comparison with solid-state NMR observables
The 15N and 13CO chemical shifts of Val16 are available from the SSNMR studies of PG-1 in a DLPC bilayer (10). We calculated the chemical shifts for all the structures in the DLPC trajectories to examine the dependence of the calculated chemical shifts on the orientation and dynamics of PG-1 in the DLPC membranes:
| (3) |
where and are the magnitude and unit vector of the static chemical shift tensor for 15N or 13CO (33). We used the same chemical shift tensors as in the original SSNMR studies (10), i.e., σ11 = 217 ppm, σ22 = 77 ppm, and σ33 = 64 ppm for 15N and σ11 = 244 ppm, σ22 = 178 ppm, and σ33 = 88 ppm for 13CO. For 15N chemical shift calculations, is 17° from the H-N bond on the peptide plane, whereas is tilted 25° from the peptide plane normal. For 13CO chemical shift calculations, is parallel to the CO bond, whereas is perpendicular to the peptide plane normal. We calculated the root mean-square deviation () between the calculated chemical shifts of individual structures and the experimental values as
| (4) |
where and are the calculated 15N and 13CO chemical shifts, and (143 ± 2 ppm) and (216 ± 5 ppm) are the corresponding experimental values (10). Fig. 1, Top shows all the PG-1 conformations of the DLPC systems in the τ and ρ space, and the structures in which < 4 ppm are highlighted. The tilt angles of these structures vary from 7.9° (M3) to 49.8° (M1). Most of these structures have negative rotation angles around −120°. The only four structures with τ close to 50° are found in M1-DLPC. The values of τ and ρ in these structures are 47.7° and −117.1°, 45.1° and −128.8°, 49.8° and −128.1°, and 46.3° and −123.2°, respectively, which correspond roughly to the model proposed by the rigid-body geometrical search (10). Despite the four structures in system M1, most structures with < 4 ppm have tilt angles smaller than that estimated experimentally (55 ± 5°). We also calculated the chemical shifts for the solution NMR structures of PG-1 (PDB 1PG1) (6); the 20 NMR structures are tilted every 1° and rotated every 5° to sample the τ and ρ space. The orientation of structures having < 4 ppm are recorded and plotted in Fig. S5. The tilt angle varies from 24° to 54°. It is interesting that the accessible tilt-angle range is similar to the PG-1 monomer simulations in DLPC bilayers (Fig. 1, Top). However, the rotation angles of these structures are clustered in two different ranges: 115° to 180° and −180° to −155°, whereas the ρ range in the simulations is mostly from −80° to −180°. The difference may arise from the sampling method of the τ and ρ space; the dynamic properties of the peptide in membranes are considered explicitly in the simulations (Fig. 1, Top), but not in the rigid-body orientation search (Fig. S5). This result again illustrates the difficulty in determining the PG-1 orientation solely based on the rigid-body geometrical search. Therefore, it may not be adequate to construct a model simply based on the rigid-body geometrical search, especially when the number of chemical shifts is limited.
The ensemble-average 15N and 13CO chemical shifts are 96.6 ± 22.2 and 219.0 ± 17.6 ppm (M1), 92.1 ± 20.9 and 228.8 ± 11.2 ppm (M2), and 110.3 ± 21.2 and 234.4 ± 11.5 ppm (M3). Although there are individual conformations that closely reproduce the experimental chemical shifts, large fluctuations of the ensemble-average chemical shifts, as well as the discrepancy between the average values and the experimental values, indicate that reliable calculations of SSNMR properties may require much longer simulations, especially when the orientation of the initial structure is not determined by experiments. It is clear that further experimental and computational works are needed to better understand the influence of PG-1 dynamics on the SSNMR observables.
Monomer conformations
PG-1 monomer forms a β-hairpin structure reinforced by two disulfide bonds (Cys6-Cys15 and Cys8-Cys13) in both solution and lipid environments (6). The SSNMR measurement suggests that six potential backbone H-bonds can be formed inside the monomer (34). The amino acids involved in H-bond formation are Leu5-Val16, Tyr7-Val14, and Arg9-Phe12 (Fig. 3 A). The H-bonds play an important role in maintaining the secondary structure of the peptide, which is crucial to the antimicrobial activity (35). In this analysis, a hydrogen bond (D−H⋯A) is defined by an H⋯A distance <2.8 Å and a D−H⋯A angle >120°. To monitor the variations of H-bonding patterns, we calculated the H-bond fraction , where and are the numbers of instantaneous and total possible H-bonds, respectively. The H-bond fraction in each system remains ∼0.8, except in the case of M1-POPC and M2-POPC (Fig. S6 A). The loss of H-bonds takes place mostly in the turn and the terminal region (Table S2), possibly due to higher flexibility in these regions.
Figure 3.
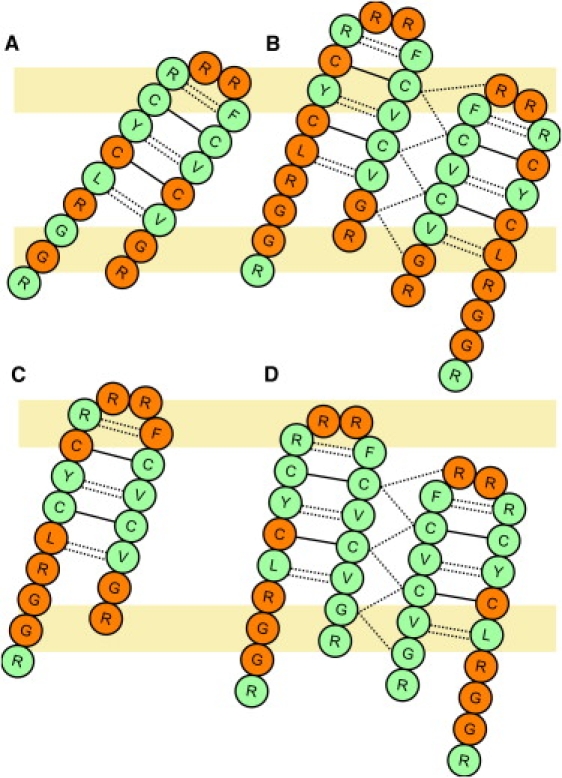
Schematic illustrations of the H-bonding pattern of PG-1 monomer and dimer in DLPC (A and B) and POPC (C and D) bilayers. The residues that form H-bonds with the lipid headgroups are shown in orange and those that do not are in green. Dotted lines represent inter- and intramolecular H-bonds and solid lines represent disulfide bonds. The putative lipid headgroup regions are illustrated by light orange boxes.
Table S2 summarizes the occupancies of the backbone H-bonds inside PG-1 monomer. Each trajectory is sampled every 6 ps to give a reasonable resolution of H-bond gain and loss. H-bonds with occupancy >0.15 are considered significant and listed in Table S2. Generally, the occupancies of H-bonds in the middle of the β-hairpin strands are higher than those at the ends. The two disulfide bonds maintain these H-bonds in the middle of the strands (Fig. 3 A). Notably, in DLPC bilayers, the amide hydrogen of Arg18 readily forms H-bonds with the carbonyl oxygen of Gly3 in the N-terminal strand. The occupancies of Arg18-Gly3 H-bonding are 0.8 (M1) and 0.9 (M3) in the DLPC bilayers. In the POPC bilayers, the H-bond between the backbone of Arg18 and Gly3 only appears in system M3 with the occupancy of 0.5. It can be inferred that in DLPC bilayers, the C-terminal strand of the peptide is bent toward the N-terminal strand in the PG-1 monomer. The same bending of the C-terminal strand is also observed in previous PG-1 simulations in a DLPC bilayer (18).
Peptide-environment interactions
The primary interactions between PG-1 and the environment are electrostatic, including H-bonding. We characterized H-bonds between the backbone amide hydrogen atoms and the oxygen atoms in the lipid headgroups. The H-bonds with occupancy >0.15 are summarized in Table S3. Notably, in DLPC bilayers, Arg4, Cys6, and Cys15, which are in the middle of the β-hairpin, are able to form H-bonds with DLPC headgroups (Fig. 3 A). However, in the POPC bilayers, most H-bonds are formed between residues at the terminal and turn regions of PG-1 and the lipid headgroups (Fig. 3 C). Two factors can contribute to such discrepancy in H-bonding patterns: the hydrophobic thickness of the bilayers and the different tilt angles of the PG-1 monomer in the two bilayers. First of all, since the hydrophobic thickness of the DLPC bilayers is smaller than that of POPC, more H-bond acceptors in the lipid headgroups are accessible to the H-bond donors in the middle of the β-hairpin strands. Moreover, PG-1 tilts more in DLPC bilayers than in POPC bilayers, so that the distance between the donors in the middle of the β-strand and the acceptors in the lipid headgroups is closer, further enabling H-bond formation. Therefore, in the DLPC membrane, PG-1 prefers to remain a monomer because of the H-bonds between the peptide and the lipid molecules. However, in the POPC membrane, the hydrophobic thickness is larger, so that PG-1 tilts less and the donors in the middle of the β-strand cannot stay within H-bonding distance of the H-bond acceptors in the lipid headgroup region. Thus, these free H-bond donors may prefer to form H-bonds with acceptors provided by another PG-1 monomer and form a dimer, suggesting that the free H-bond donors in the middle of the β-strand are crucial for dimer or oligomer formation in POPC.
Other major interactions are found between positively charged arginine guanidinium groups and both lipid headgroups and water molecules. The interaction between the guanidinium group and the phosphate group is ionic. However, the guanidinium group can also form H-bonds with both water and lipid headgroups. The average contact number of the guanidinium group and the environment is calculated with a distance cutoff of 4.5 Å. Fig. 4 shows the average contact numbers of the guanidinium groups. There are ∼20 pairs of interactions for each guanidinium group with both lipid and water molecules, indicating that the guanidinium groups are locating at the hydrophobic/hydrophilic interface, as shown in Fig. S7. By calculating the contact number ratio, ω, between guanidinium-lipid and guanidinium-water interactions, it is observed that Arg4, Arg9, Arg11, and Arg18 interact with more lipid molecules than water molecules (ω > 1). The four arginines appear to play an important role in anchoring the PG-1 peptide inside the lipid bilayers. In particular, the contact ratio of Arg4 with lipid and water molecules differs largely in different lipid bilayers. In the DLPC bilayers, ω is 1.57 (M1), 1.12 (M2), and 1.52 (M3). However, as the hydrophobic thickness of the bilayers increases, ω increases to 4.06 (M1), 2.49 (M2), and 3.52 (M3) in the POPC bilayers, suggesting that the Arg4 guanidinium group is submersed in the lipid bilayers. Unlike the arginines at the terminal and turn regions, the flexibility of the Arg4 side chain is relatively small. As the hydrophobic thickness increases from 21 Å in the DLPC bilayers (31) to 28 Å in the POPC bilayers (32), the guanidinium group gets buried in the region between the lipid headgroups and lipid tails. The strong interaction between the Arg4 guanidinium group and the lipid phosphate groups contributes to the membrane thinning effect around PG-1.
Figure 4.
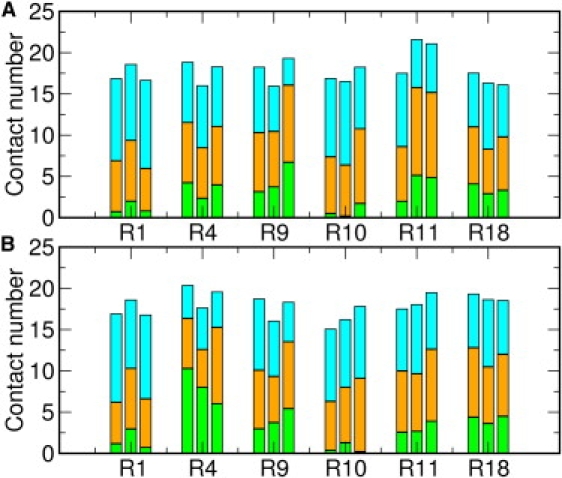
Average contact numbers of guanidinium-water (cyan), guanidinium-lipid headgroups (orange), and guanidinium-lipid tails (green) in (A) DLPC and (B) POPC bilayers. Heavy atoms within 4.5 Å are counted as interacting pairs. The results for systems M1 (left), M2 (middle), and M3 (right) are shown for each arginine residue.
Membrane thinning effects in monomer systems
Membrane thinning was measured upon the concentration-dependent aggregation of the PG-1 peptide on the surface of the DPhPC (1,2-diphytanoylphosphatidylcholine) bilayers using lamellar x-ray diffraction (36). The average thinning is suggested to be 1.5 Å when the PG-1 monomer concentration is below the insertion threshold. However, there is no direct experimental measurement for membrane thinning around PG-1 inside the lipid bilayers. Previous SSNMR studies have suggested a local membrane thinning around a PG-1 monomer in a DLPC bilayer based on a hydrophobic mismatch between the PG-1 model with a tilt of 55 ± 5° and the DLPC hydrophobic thickness (10). To investigate local membrane thinning by PG-1 monomer, the average membrane hydrophobic thickness is defined as the average distance between C2 atoms (the carbon that connects the two aliphatic chains to the carbon leading to the phosphate) in the top and bottom leaflets. Local membrane thinning is then defined as the difference between the thickness of the lipid molecules that have any heavy atoms within 4 Å from PG-1 and the thickness of the rest of the lipid molecules (bulk lipid bilayers). The average thinning is 3.4 ± 1.2 Å (M1), 3.3 ± 1.3 Å (M2), and 4.0 ± 1.1 Å (M3) in the DLPC systems and 3.7 ± 1.5 Å (M1), 4.1 ± 1.4 Å (M2), and 4.5 ± 1.2 Å (M3) in the POPC systems. Similar membrane thinning was observed in previous MD studies (15,16,18). Local membrane thinning appears to be caused by strong interactions between the positively charged arginine guanidinium groups and the negatively charged phosphate groups in the lipid molecules. The lipid/water contact ratios of guanidinium groups in Arg4, Arg9, Arg11, and Arg18 show that these residues have higher contacts with lipid molecules, indicating that the guanidinium groups are closer to the membrane hydrophobic core. The attractive interactions between the guanidinium groups and the phosphate groups pull the lipid headgroups into the membrane hydrophobic region and cause membrane thinning around PG-1. Moreover, the local membrane thinning appears to be slightly larger in POPC than in DLPC. The difference arises from the buried depth of the guanidinium group of Arg4. In the POPC bilayers, PG-1 has a smaller tilt angle. The guanidinium group of Arg4, which is close to the center of the β-strand, gets buried deeper in the hydrophobic tails of the lipids. The phosphate groups interact with the Arg4 guanidinium group and bend into the hydrophobic region, causing a slightly larger local membrane thinning in POPC bilayers. These analyses also clearly demonstrate that membrane thinning around PG-1 does not arise directly from a hydrophobic mismatch between PG-1 and the membrane hydrophobic thickness. Rather, the thinning results from the relative position change of guanidinium groups in the lipid bilayers.
PG-1 dimer in DLPC and POPC bilayers
Dimer stability in the bilayers
PG-1 adopts different oligomeric states in different environments. It appears as a monomer in aqueous, dimethyl sulfoxide solution, and in DLPC bilayers (6,37,38). However, PG-1 dimerizes in dodecylphosphocholine micelles and POPC bilayers (28,38). Based on the SSNMR distance measurement, Hong and co-workers suggest an NCCN parallel packing of the membrane-bound dimer in POPC bilayers (11,12,34), i.e., the C-terminal strands of the monomers are at the dimer interface. Fig. 3 shows the topological arrangement of the NCCN dimer and the proposed H-bonds. Six potential H-bonds can be formed between the monomers. Fig. 5 shows the H-bond fractions between the monomers. The fractions in the DLPC bilayers fluctuate between 0.4 and 0.8, with four H-bonds remaining intact on average. The fluctuation is also observed in the POPC-containing systems; however, the average fraction is 0.8, indicating that all H-bonds but one were maintained during the simulations. It can be inferred that the dimer interface is more stable in POPC than in DLPC. In addition, severe H-bond loss appears to take place inside the monomers in each DLPC dimer system during the simulation (Fig. S6 B). However, the loss of intramolecular H-bonds is much smaller in the POPC dimer systems, which also suggests that PG-1 dimer is more stable in the POPC bilayers.
Figure 5.
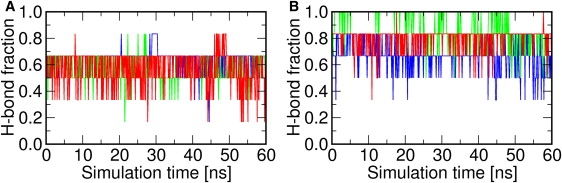
Fractions of intermolecular H-bonds of PG-1 dimer in (A) DLPC and (B) POPC bilayers. Systems D1 (blue), D2 (green), and D3 (red) are presented in different colors for clarity.
To further examine this difference in stability, we investigated other possible intermolecular backbone H-bonds. The results are summarized in Table S4. As mentioned above, there are fewer intermolecular H-bonds in the DLPC systems. The missing H-bonds are those formed between Gly17(A) and Cys15(B) and between Gly17(A) and Gly17(B) at the terminal region of the dimer, where the letter superscripts represent monomers A and B. The H-bond occupancy between the peptide backbone and the lipid molecules, shown in Table S5, reveals that in the DLPC bilayers, Gly17 and Arg18 of each monomer tend to form H-bonds with lipid headgroups because of the smaller hydrophobic thickness. As a result, the C-terminus of each monomer in the dimer is quite flexible in DLPC bilayers. Instead of forming H-bonds between the monomers, these terminal residues prefer to H-bond with lipid phosphate groups. On the other hand, in the POPC bilayers, the C-terminal region of each PG-1 monomer is immobilized due to the larger hydrophobic thickness. Therefore, the PG-1 monomers in the POPC bilayers have a better-preserved dimer interface.
Orientation of the PG-1 dimer in DLPC and POPC bilayers
The cytotoxicity of PG-1 is believed to be the result of oligomeric pore formation (13). According to the 19F and 1H spin diffusion experiment, PG-1 dimer is the minimum structural unit of the pore (11). The membrane-bound PG-1 dimer model was proposed using SSNMR technique in POPC bilayers (12). However, the orientation of the dimer with respect to the membrane remains unsolved. In other words, it is not known how PG-1 dimers assemble to form an oligomeric pore. Motivated by this question, we calculated τ and ρ of each monomer in the PG-1 dimer in DLPC and POPC bilayers (Fig. S8). When combining the trajectories in all the DLPC systems, τ varies from 0° to 48.9°. In the POPC bilayers, the variation of τ is smaller, with a range from 0° to 34.9°. The average τ of each monomer in DLPC is 23.5 ± 6.3°/9.9 ± 5.6° (D1), 15.9 ± 4.7°/27.9 ± 5.3° (D2), and 26.4 ± 6.0°/6.3 ± 3.3° (D3). In the POPC bilayers, the average τ-values are 18.0 ± 3.9°/8.3 ± 3.7° (D1), 7.5 ± 3.5°/16.0 ± 5.5° (D2), and 14.4 ± 3.5°/13.9 ± 3.7° (D3). As in the case of monomer systems, the large τ in DLPC bilayers arises from the smaller hydrophobic thickness.
It is interesting that the ρ of each monomer in the dimer differs from ρ in the unassociated monomer in the DLPC and POPC bilayers. Instead of having a negative rotation angle, each monomer happens to adopt ρ with positive values, i.e., the disulfide bonds are pointing upward due to PG-1 dimerization (Fig. S6). The two monomers in the dimer have their disulfide bonds pointing in different directions with the NCCN parallel dimer packing. As discussed above, a monomer with rotation angles of ρ < 0 has a higher propensity to tilt. The positive ρ angle, on the other hand, restrains each monomer from having a large tilt angle so that the dimer interface can be preserved. As shown in Fig. S8, the rotation angles of the monomers converge to 90° in the DLPC systems and 0° or 180° in the POPC systems. Such convergence is not observed for monomer A in systems D1-POPC and D3-POPC, which appears to be the result of the small tilt angle of the monomer in these two systems. Following the definition of ρ, ρ ≈ 0° or 180° represents a sidewise-tilting orientation of the peptide. We speculate that the sidewise tilting may also present in the dimer unit of PG-1 oligomeric pore in membrane, i.e., that each strand in the β-barrel pore may have a specific tilt angle, as seen in other transmembrane β-barrel pores (39).
Dimer-environment interactions
The H-bond interactions between the PG-1 backbone and lipid headgroups are summarized in Table S5. H-bonding patterns are similar to those observed in the monomer systems. In the DLPC bilayers, Gly17 and Arg18 form H-bonds with the lipid headgroups. However, such H-bonding patterns are not observed in the POPC systems (Fig. 3, B and D). This is largely due to the hydrophobic thickness of the bilayers, as well as the peptide tilting, as discussed previously.
Moreover, 12 guanidinium groups in the dimer reveal strong electrostatic interactions with the lipid headgroups. The contact numbers of the guanidinium group and the environment are measured using the same criterion used in the monomer case. Fig. S9 shows the normalized contact number for each arginine guanidinium group. The guanidinium groups are solvated by water and lipid molecules. The average contact number is ∼20, which is comparable to that in the PG-1 monomer systems. The contact ratio, ω, between guanidinium-lipid and guanidinium-water interactions shows that, similar to the contact ratio in the PG-1 monomer systems, Arg4, Arg9, and Arg18 have closer contacts with the lipid molecules (ω > 1). These arginines may be essential in stabilizing the PG-1 dimer in the membranes. Among these arginines, the contact ratios of Arg4 and Arg18 guanidinium groups increase as a result of increasing membrane thickness. These guanidinium groups are close to the middle of the dimer β-strands. As the bilayer thickness increases, these guanidinium groups are submerged in the hydrophobic region of the bilayers and pull the lipid headgroups toward the bilayer hydrophobic core, which results in membrane thinning around the dimer.
Membrane thinning effects in the dimer systems
Membrane thinning around the PG-1 dimer was calculated for all the dimer systems using the same definition as in the PG-1 monomer systems. Local membrane thinning in the PG-1 dimer system is 2.7 ± 1.0 Å (D1), 3.9 ± 1.0 Å (D2), and 4.3 ± 1.1 Å (D3) in the DLPC bilayers and 6.2 ± 1.0 Å (D1), 4.3 ± 1.2 Å (D2), and 5.3 ± 1.3 Å (D3) in the POPC bilayers. Greater thinning is observed in the POPC bilayers than in the DLPC bilayers. Besides the difference in hydrophobic thickness, such a difference in thinning also arises from the NCCN dimer packing. In the NCCN packing model, the center of mass of each monomer is at different positions along the membrane normal (Fig. 3, B and D). With such an arrangement, arginines at the terminal region and turn region of different monomers are closer to the membrane hydrophobic core, causing the surrounding lipid headgroups to bend toward the bilayer hydrophobic core due to the electrostatic interactions between these guanidinium groups and lipid phosphate groups. As a result, there is greater membrane thinning around the PG-1 dimer in the POPC bilayers.
Discussion
We performed a total of 840 ns of comparative MD simulations of PG-1 monomers and dimers in explicit DLPC and POPC lipid bilayers. The trajectories were analyzed to investigate the peptide orientation in the membranes, as well as the peptide interactions in/with the membranes. The major findings of our simulations are summarized and discussed below.
We observed that the tilt angle of PG-1 is dependent on both the membrane hydrophobic thickness and the rotation angle of the peptide (Figs. 1, Fig. S3, and Fig. S8). In the DLPC bilayers with smaller hydrophobic thickness, PG-1 exhibits a larger tilt angle to minimize the mismatch between the hydrophobic residues and the membrane hydrophobic region. On the other hand, a negative rotation angle (ρ ≈ −120°) is preferred for large PG-1 tilting due to favorable interactions between the lipid molecules and the side chains of Tyr7 and Phe12 (Figs. 2 and Fig. S2, Fig. S3, and Fig. S4). Our finding is also consistent with previous two-dimensional PMF calculations of PG-1 as a function of τ and ρ in the EEF1/IMM1 implicit membrane model (29). In the PG-1 dimer simulations, a sidewise tilting pattern of the monomers in the POPC bilayers is observed. We speculate that the β-strands may exist in such orientation in the proposed PG-1 β-barrel pore (12,40). Moreover, by comparing the 15N and 13CO chemical shifts of Val16 for all PG-1 monomer structures in the DLPC bilayers with the experimental values (10), we found that there were several structures that agreed well with the experimental chemical shifts, but that only four of them had τ and ρ similar to the model previously proposed based on the rigid-body geometrical search (10). Further experimental and computational works are needed to better understand the influence of PG-1 dynamics in membranes on the SSNMR observables.
In addition, H-bonding analyses reveal that dimerization of PG-1 in POPC is related to larger membrane hydrophobic thickness (Figs. 3 and 5). In the DLPC bilayers with smaller hydrophobic thickness, the H-bonds are formed between the acceptors in the lipid headgroups and the backbone amide hydrogen close to the middle β-strands, resulting in a structurally favorable monomer. As the hydrophobic thickness increases, the accessible H-bond acceptors in the lipid headgroups decrease. Instead of forming H-bonds with the acceptors in the lipid headgroups, these donors prefer to form H-bonds with another PG-1 monomer. Although the detailed energetics of PG-1 dimerization (with different PG-1 dimer interfaces) in membranes with different hydrophobic thickness need more extensive PMF calculations, the MD simulations presented here clearly indicate that the PG-1 dimer has a higher degree of association in POPC bilayers than in DLPC bilayers, as observed in the experiments.
Furthermore, by calculating the average contact numbers and ratios for arginine guanidinium groups, we demonstrate that the guanidinium-lipid interactions are crucial to stabilizing PG-1 in lipid bilayers as well as to the local membrane thinning effects (Fig. S7 and Fig. S9). We have also shown that the more significant membrane thinning is related to the relative position of the guanidinium groups with regard to the lipid bilayers. The PG-1 dimer systems in the POPC bilayers have the largest membrane thinning effects. It is feasible that greater thinning may occur in the PG-1 oligomeric pore in anionic membrane such as POPE/POPG bilayers due to the larger hydrophobic thickness, as well as the negatively charged lipid headgroups.
Supporting Material
Five tables and nine figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01035-2.
Supporting Material
Acknowledgments
We are grateful to Taehoon Kim and Kevin Song for helpful comments on the manuscript.
This work was supported by institutional funding from the University of Kansas Alfred P. Sloan Research Fellowship (to W.I.). This research was also supported in part by the National Institutes of Health Centers of Biomedical Research Excellence grant RR-017708 and the National Science Foundation (NSF OCI-0503992) through TeraGrid resources provided by Purdue University.
References
- 1.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 2.Gordon Y.J., Romanowski E.G., McDermott A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 2005;30:505–515. doi: 10.1080/02713680590968637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Z., Wang G.S. APD: the Antimicrobial Peptide Database. Nucleic Acids Res. 2004;32:D590–D592. doi: 10.1093/nar/gkh025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Panchal R.G., Smart M.L., Bowser D.N., Williams D.A., Petrou S. Pore-forming proteins and their application in biotechnology. Curr. Pharm. Biotechnol. 2002;3:99–115. doi: 10.2174/1389201023378418. [DOI] [PubMed] [Google Scholar]
- 5.Kokryakov V.N., Harwig S.S.L., Panyutich E.A., Shevchenko A.A., Aleshina G.M. Protegrins: leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993;327:231–236. doi: 10.1016/0014-5793(93)80175-t. [DOI] [PubMed] [Google Scholar]
- 6.Fahrner R.L., Dieckmann T., Harwig S.S., Lehrer R.I., Eisenberg D. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996;3:543–550. doi: 10.1016/s1074-5521(96)90145-3. [DOI] [PubMed] [Google Scholar]
- 7.Iwanaga S., Muta T., Shigenaga T., Miura Y., Seki N. Role of hemocyte-derived granular components in invertebrate defense. Ann. N. Y. Acad. Sci. 1994;15:102–116. doi: 10.1111/j.1749-6632.1994.tb33566.x. [DOI] [PubMed] [Google Scholar]
- 8.Miyasaki K.T., Lehrer R.I. β-sheet antibiotic peptides as potential dental therapeutics. Int. J. Antimicrob. Agents. 1998;9:269–280. doi: 10.1016/s0924-8579(98)00006-5. [DOI] [PubMed] [Google Scholar]
- 9.Gidalevitz D., Ishitsuka Y.J., Muresan A.S., Konovalov O., Waring A.J. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA. 2003;100:6302–6307. doi: 10.1073/pnas.0934731100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaguchi S., Hong T., Waring A., Lehrer R.I., Hong M. Solid-state NMR investigations of peptide-lipid interaction and orientation of a ss-sheet antimicrobial peptide, protegrin. Biochemistry. 2002;41:9852–9862. doi: 10.1021/bi0257991. [DOI] [PubMed] [Google Scholar]
- 11.Buffy J.J., Waring A.J., Hong M. Determination of peptide oligomerization in lipid bilayers using F-19 spin diffusion NMR. J. Am. Chem. Soc. 2005;127:4477–4483. doi: 10.1021/ja043621r. [DOI] [PubMed] [Google Scholar]
- 12.Mani R., Tang M., Wu X., Buffy J.J., Waring A.J. Membrane-bound dimer structure of a β-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry. 2006;45:8341–8349. doi: 10.1021/bi060305b. [DOI] [PubMed] [Google Scholar]
- 13.Mani R., Cady S.D., Tang M., Waring A.J., Lehrert R.I. Membrane-dependent oligomeric structure and pore formation of β-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA. 2006;103:16242–16247. doi: 10.1073/pnas.0605079103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang H., Ma B., Nussinov R. Conformational study of the protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Struct. Biol. 2007;7:21. doi: 10.1186/1472-6807-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langham A.A., Ahmad A.S., Kaznessis Y.N. On the nature of antimicrobial activity: a model for protegrin-1 pores. J. Am. Chem. Soc. 2008;130:4338–4346. doi: 10.1021/ja0780380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jang H., Ma B., Lal R., Nussinov R. Models of toxic β-sheet channels of protegrin-1 suggest a common subunit organization motif shared with toxic Alzheimer β-amyloid ion channels. Biophys. J. 2008;95:4631–4642. doi: 10.1529/biophysj.108.134551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang H., Ma B., Woolf T.B., Nussinov R. Interaction of protegrin-1 with lipid bilayers: membrane thinning effect. Biophys. J. 2006;91:2848–2859. doi: 10.1529/biophysj.106.084046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khandelia H., Kaznessis Y.N. Structure of the antimicrobial β-hairpin peptide protegrin-1 in a DLPC lipid bilayer investigated by molecular dynamics simulation. Biochim. Biophys. Acta. 2007;1768:509–520. doi: 10.1016/j.bbamem.2006.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jo S., Kim T., Iyer V.G., Im W. Software news and updates - CHARNIM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 20.Jo S., Kim T., Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jo S., Lim J.B., Klauda J.B., Im W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S. Charmm: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 23.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 24.Feller S.E., Zhang Y.H., Pastor R.W., Brooks B.R. Constant-pressure molecular-dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 25.Klauda J.B., Kucerka N., Brooks B.R., Pastor R.W., Nagle J.F. Simulation-based methods for interpreting x-ray data from lipid bilayers. Biophys. J. 2006;90:2796–2807. doi: 10.1529/biophysj.105.075697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skibinsky A., Venable R.M., Pastor R.W. A molecular dynamics study of the response of lipid bilayers and monolayers to trehalose. Biophys. J. 2005;89:4111–4121. doi: 10.1529/biophysj.105.065953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolan E.A., Venable R.M., Pastor R.W., Brooks B.R. Simulations of membranes and other interfacial systems using P2(1) and Pc periodic boundary conditions. Biophys. J. 2002;82:2317–2325. doi: 10.1016/S0006-3495(02)75577-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roumestand C., Louis V., Aumelas A., Grassy G., Calas B. Oligomerization of protegrin-1 in the presence of DPC micelles. A proton high-resolution NMR study. FEBS Lett. 1998;421:263–267. doi: 10.1016/s0014-5793(97)01579-2. [DOI] [PubMed] [Google Scholar]
- 29.Lee J., Ham S., Im W. β-hairpin restraint potentials for calculations of potentials of mean force as a function of β-hairpin tilt, rotation, and distance. J. Comput. Chem. 2009;30:1334–1343. doi: 10.1002/jcc.21154. [DOI] [PubMed] [Google Scholar]
- 30.Lee J., Im W. Transmembrane helix tilting: insights from calculating the potential of mean force. Phys. Rev. Lett. 2008;100:018103. doi: 10.1103/PhysRevLett.100.018103. [DOI] [PubMed] [Google Scholar]
- 31.Kucerka N., Liu Y., Chu N., Petrache H.I., Tristram-Nagle S. Structure of fully hydrated fluid phase DMPC and DLPC lipid bilayers using X-ray scattering from oriented multilamellar arrays and from unilamellar vesicles. Biophys. J. 2005;88:2626–2637. doi: 10.1529/biophysj.104.056606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gawrisch K., Gaede H.C., Mihailescu M., White S.H. Hydration of POPC bilayers studied by H-1-PFG-MAS-NOESY and neutron diffraction. Eur. Biophys. J. 2007;36:281–291. doi: 10.1007/s00249-007-0142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J., Chen J., Brooks C.L., Im W. Application of solid-state NMR restraint potentials in membrane protein modeling. J. Magn. Reson. 2008;193:68–76. doi: 10.1016/j.jmr.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang M., Waring A.J., Hong M. Intermolecular packing and alignment in an ordered β-hairpin antimicrobial peptide aggregate from 2D solid-state NMR. J. Am. Chem. Soc. 2005;127:13919–13927. doi: 10.1021/ja0526665. [DOI] [PubMed] [Google Scholar]
- 35.Lai J.R., Huck B.R., Weisblum B., Gellman S.H. Design of non-cysteine-containing antimicrobial β-hairpins: structure-activity relationship studies with linear protegrin-1 analogues. Biochemistry. 2002;41:12835–12842. doi: 10.1021/bi026127d. [DOI] [PubMed] [Google Scholar]
- 36.Heller W.T., Waring A.J., Lehrer R.I., Harroun T.A., Weiss T.M. Membrane thinning effect of the β-sheet antimicrobial protegrin. Biochemistry. 2000;39:139–145. doi: 10.1021/bi991892m. [DOI] [PubMed] [Google Scholar]
- 37.Aumelas A., Mangoni M., Roumestand C., Chiche L., Despaux E. Synthesis and solution structure of the antimicrobial peptide protegrin-1. Eur. J. Biochem. 1996;237:575–583. doi: 10.1111/j.1432-1033.1996.0575p.x. [DOI] [PubMed] [Google Scholar]
- 38.Buffy J.J., Waring A.J., Lehrer R.I., Hong M. Immobilization and aggregation of the antimicrobial peptide protegrin-1 in lipid bilayers investigated by solid-state NMR. Biochemistry. 2003;42:13725–13734. doi: 10.1021/bi035187w. [DOI] [PubMed] [Google Scholar]
- 39.Hiller S., Garces R.G., Malia T.J., Orekhov V.Y., Colombini M. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang M., Waring A.J., Hong M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 2007;129:11438–11446. doi: 10.1021/ja072511s. [DOI] [PubMed] [Google Scholar]
- 41.DeLano, W. L. 2002. The PyMOL Molecular Graphics System. Palo Alto, CA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.