

Abstract
The hyperpolarization-activated cyclic nucleotide-modulated channel gene family (HCN1-4) encodes the membrane depolarizing current that underlies pacemaking. Although the topology of HCN resembles Kv channels, much less is known about their structure-function correlation. Previously, we identified several pore residues in the S5-P linker and P-loop that are externally accessible and/or influence HCN gating, and proposed an evolutionarily conserved pore-to-gate mechanism. Here we sought dynamic evidence by assessing the functional consequences of Cys-scanning substitutions in the unexplored P-S6 linker (residues 352–359), the HCN1-R background (that is, resistant to sulfhydryl-reactive agents). None of A352C, Q353C, A354C, P355C, V356C, S357C, M358C, or S359C produced functional currents; the loss-of-function of Q353C, A354C, S357C, and M358C could be rescued by the reducing agent dithiothreitol. Q353C, A354C, and S357C, but not M358C and HCN1-R, were sensitive to Cd2+ blockade (IC50 = 3–12 μM vs. >1 mM). External application of the positively charged covalent sulfhydryl modifier MTSET irreversibly reduced I−140mV of Q353C and A354C to 27.9 ± 3.4% and 58.2 ± 13.1% of the control, respectively, and caused significant steady-state activation shifts (∆V1/2 = –21.1 ± 1.6 for Q353C and −10.0 ± 2.9 mV for A354C). Interestingly, MTSET reactivity was also state dependent. MTSET, however, affected neither S357C nor M358C, indicating site specificity. Collectively, we have identified novel P-S6 residues whose extracellular accessibility was sterically and state dependent and have provided the first functional evidence consistent with a dynamic HCN pore-to-gate model.
Keywords: Outer pore, Pacemaker channels, HCN
The hyperpolarization-activated cyclic nucleotide-modulated channel gene family (HCN1-4) encodes the membrane depolarizing current that underlies pacemaking (Biel et al. 2002; Brown et al. 1977; Bucossi et al. 1997; Chen et al. 2001; Creighton 1992; DiFrancesco 1981a, b, 1993, 2006; Lieu et al. 2008; Ludwig et al. 1998; Siu et al. 2006). Functionally, HCN channels nonselectively permeate Na+ and K+ (with a ratio of 1:4 vs. ≤1:100 K+ channels) and are activated upon hyperpolarization rather than depolarization of classical voltage-gated K+ (Kv) channels (Azene et al. 2003; Prole and Yellen 2006; Siu et al. 2006). Despite these differences, HCN and Kv channels have highly homologous primary amino acid sequences (Gauss et al. 1998; Giorgetti et al. 2005; Kaupp and Seifert 2001). Structurally, both are tetramers of monomeric subunits consisting of six membrane-spanning segments (S1–S6) (Gauss et al. 1998; Ludwig et al. 1998; Santoro et al. 1998; Xue et al. 2002). The region between S5 and S6, which inserts back into the membrane to form part of the ion-conducting pore, can be further subdivided into three regions (Fig. 1): while the S5-P and P-S6 linkers form the extrapore mouth, the descending and ascending limbs of the P-loop, where the permeation determinant GYG motif is located, constitute the deeper region of the extracellular pore. The cytoplasmic side of the pore is believed to consist of the S6 segments (Prole and Yellen 2006).
Fig. 1.

Schematic representation of HCN1-R. The six transmembrane segments of a monomeric subunit of the HCN1-R are shown. Six endogenous cysteine residues are removed together with the truncation of the –COOH terminus. Five of the six remaining cysteine residues are replaced by a serine, threonine, or isoleucine substitution. C303 in the S5 segment is left in the final HCN1-R construct because of lack of functional current upon mutations. Their approximate locations, as well as the GYG signature motif, are highlighted as shown. The eight amino acid residues in the P-S6 motif of the final HCN1-R construct are individually replaced by cysteine and named accordingly
Although the topology of HCN resembles Kv channels, much less is known about their structure-function correlation. Previously, we identified several pore residues in the S5-P linker (C318 HCN1 numbering), the descending (H334, F339) and ascending (A352, A354) P-loop limbs that are externally accessible and/or influence HCN gating (Au et al. 2008; Azene et al. 2005; Xue and Li 2002). Based on these results, we proposed an evolutionarily conserved pore-to-gate mechanism (Au et al. 2008; Azene et al. 2005). However, the functional role of the P-S6 linker, putatively defined as residues 352–359 here, and its accessibility have not been explored. To obtain further insights into the footprint of the pore, here we investigated the functional consequences of systematically introducing single Cys-scanning substitutions into the P-S6 linker in the background of HCN1-R channels (Bell et al. 2004), in which all endogenous cysteines except C303 have been removed or replaced to render the wild-type (WT) resistant to sulfhydryl-reactive agents such as Class IIB divalent Cd2+ and covalent methanethiosulfonate (MTS) modifying compounds (Karlin and Akabas 1998). Using this approach, we successfully identified several novel P-S6 linker residues whose extracellular accessibility was sterically and state dependent. These results provide the first functional evidence that the HCN pore undergoes dynamic rearrangements consistent with a pore-to-gate coupling mechanism.
Materials and Methods
Molecular Biology
Murine HCN1 and an MTS/Cd2+-resistant HCN1 construct, HCN1-R (Bell et al. 2004), provided by Dr. Steven Siegelbaum, were subcloned into the pGHEM-HE vector (Santoro et al. 1998). The construction of HCN1-R has been described (Bell et al. 2004). In brief, 11 of 12 endogenous cysteine residues of WT HCN1 were removed or substituted: 6 cysteines were removed by truncating the COOH terminus and 4 of the 6 remaining cysteines were conservatively replaced by serine or threonine (i.e., C55S, C318S, C347S, and C374T), with C298 substituted by isoleucine, according to the homologous residue in cyclic nucleotide-gated (CNG) channels. As we and others have reported, substitution of C303 with any of the other 19 amino acids did not produce functional channels (Azene et al. 2003; Bell et al. 2004). Therefore, C303 was not replaced in HCN1-R. All cysteine substitutions investigated in this study were generated in the HCN1-R background using the Stratagene QuickChange site-directed mutagenesis kit (Akabas et al. 1992; Benitah et al. 1996; Karlin and Akabas 1998; Tsushima et al. 1997). The desired mutations were confirmed by DNA sequencing. cRNA was transcribed from NheI-linearized DNA using the Ambion MEGAscript transcription kit.
Oocyte Preparation and Heterologous Expression
All constructs were heterologously expressed and studied in Xenopus oocytes (Xue and Li 2002; Xue et al. 2002). Stage IV–VI oocytes were surgically removed from female Xenopus laevis anesthetized by immersion in 0.3% 3-aminobenzoic acid ethyl ester, followed by digestion with 1 mg/ml collagenase (type IA) in OR-2 solution containing 88 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 5 mM HEPES (adjusted to pH 7.6 with NaOH) for 30–60 min After defolliculation by incubation in OR-2 solution with 10% fetal bovine serum for 15–30 min, isolated oocytes were injected with cRNA (50 ng/oocyte) and incubated in ND96 solution containing 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES (adjusted to pH 7.6 with NaOH) supplemented with 50 μg/ml gentamycin, 5 mM pyruvate, and 0.5 mM theophylline for 1–2 days before experiment.
Preparation of Oocyte Membrane Fractions and Western Blotting
As we previously described, injected and control (uninjected) oocytes were homogenized in ice-cold PBS containing an EDTA-free protease inhibitor cocktail (1 tablet/10 ml; Roche Applied Science) and centrifuged for 10 min at 1000g to remove pigment and nuclear and mitochondrial materials. The supernatant was fractioned into cytosolic and membrane fractions by ultracentrifugation at 120,000g for 1 h at 4°C. The membrane fraction was resuspended in 1× LDS sample buffer and electrophoresed on a 10% polyacrylamide-SDS gel. Membrane proteins were then transferred onto a nitrocellulose membrane, followed by blockade with 5% nonfat dried milk for 1 h. Blocked membranes were washed in Tris-buffered saline-Tween (TBST) and incubated with a purified rabbit polycolonal anti-HCN1 antibody (Alomone Labs, Jerusalem, Israel) at a 1:200 dilution for 4°C overnight, followed by a wash in TBST and an incubation with horseradish peroxidase-conjugated donkey anti-rabbit immunoglobulin (Chemicon, Temecula, CA) diluted to 1:10,000 in 5% nonfat dried milk for 1 h at room temperature. Membranes were washed again three times in TBST, and bound secondary antibodies were detected with an ECL detection kit (Chemicon) by autoradiography.
Electrophysiology
Two-electrode voltage-clamp recordings were performed at room temperature using a Warner OC-725B amplifier 1–2 days after cRNA injection. Measuring electrodes (TW120-6; World Precision Instruments Inc., Sarasota, FL) were pulled using a Sutter micropipette puller P-97 with final tip resistances of 2–4 MΩ when filled with 3 mM KCl. The recording bath solution contained 96 mM KCl, 2 mM NaCl, 2 mM MgCl2, and 10 mM HEPES (adjusted to pH 7.5 with NaOH).
For the steady-state current-voltage (I–V) protocol, whole-cell currents were measured at the end of a 3-s pulse from −140 to 0 mV from a holding potential of −30 mV against the test potentials. The voltage dependence of HCN channel activation was assessed by plotting tail currents measured immediately after a pulse to −140 mV as a function of the preceding 3-s test pulse normalized to the tail current at −140 mV. Data were fitted to the Boltzman function using the Marquardt–Levenberg algorithm in a nonlinear least-squares procedure:
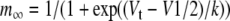 |
where Vt is the test-voltage, V1/2 is the midpoint, and k = RT/zF is the slope factor of steady-state activation (m∞), and R, T, z, and F have their usual meanings.
For tail I–V relationships, currents were recorded immediately after stepping to a family of test voltages ranging from −100 to +40 mV preceded by a 3-s prepulse to either −140 or −20 mV. The difference in tail currents resulting from the two prepulse potentials was plotted against the test potentials and fitted with linear regression to obtain the reversal potential (Erev).
To test for Cd2+ sensitivity, each oocyte expressing an HCN1r pore cysteine mutant was first stimulated by a 2-s voltage pulse from a holding voltage of 0 mV to a test voltage of −140 mV. The steady-state current at −140 mV was used as the initial current magnitude against which currents after Cd2+ exposure were compared. Immediately after the initial test pulse, cells were exposed to varying concentrations of CdCl2 (3 μM to 1 mM). Voltage pulses (from 0 to −140 mV, 2-s duration) were then applied every minute until current block reached an equilibrium level. The fractional block by Cd2+ was plotted against the concentration to generate a binding curve. Data were fitted to the Hill equation using the Marquardt–Levenberg algorithm in a nonlinear least-squares procedure:
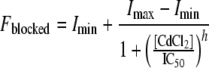 |
where Fblocked is the fractional current block, Imin is the minimum fractional current block, Imax is the maximum fractional current block, IC50 is the (Benitah et al.) that led to 50% fractional block, and h is the Hill coefficient.
2-Trimethylammoniumethylmethane thiosulfonate (MTSET; Toronto Research Chemicals, Toronto, ON, Canada) powder was dissolved in deionized water at 0.2 M shortly before experiments. The stock solution was stored on ice and used within 1 h. The MTSET shock solution was diluted with the bath solution to 2 mM and applied extracellularly to the oocytes after control data were obtained. The time course of MTSET modification was fit with the following single-exponential equation:
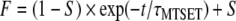 |
where F is the fraction of remaining current measured at −140 mV in the presence of MTSET, t is the cumulative exposure time, τMTSET is the time constant for MTSET modification, and S is the steady-state plateau (Xue and Li 2002).
All data reported are mean ± SE. Statistical significance was determined from all individual data points and fitting parameters using one-way ANOVA and the Tukey HSD post hoc test at the 5% level.
Results
As our first step, we characterized and compared the baseline properties of WT and HCN1-R channels. Figure 2a shows representative current tracings of WT and HCN1-R channels recorded using the electrophysiological protocol given in the inset. Consistent with previous studies (Au et al. 2008; Bell et al. 2004), oocytes injected with HCN1-R cRNA robustly expressed hyperpolarization-activated currents comparable to those of WT (I–140mV = −12.3 ± 2.5 and −15.6 ± 3.3 μA, respectively; p > 0.05). The corresponding current-voltage relationships are given in Fig. 2b. Figure 2c shows their steady-state activation curves. In comparison to WT, HCN1-R channels displayed a modest but insignificant hyperpolarizing shift of their activation midpoint (V1/2 = −80.3 ± 2.5 mV [n = 6] vs. 86.4 ± 1.7 mV [n = 5] of WT; p > 0.05). The slope factor k was also not different (17.2 ± 0.6 and 16.2 ± 0.5, respectively; p > 0.05). These activation gating parameters are summarized in Table 1.
Fig. 2.

a Representative current tracings of WT HCN1 and HCN1-R. Currents were elicited by stepping to a family of 3-s electrical pulses ranging from 0 to –140 mV in 10-mV increments from a holding potential of –30 mV. b Steady-state current–voltage (I–V) relationships (at 3 s). c Steady-state activation curves
Table 1.
Summary of steady-state activation properties of WT, HCN1-R, and various P-S6 mutants
| Channel | V1/2 (mV) | K | N |
|---|---|---|---|
| WT HCN1 | –86.4 ± 1.7 | 16.2 ± 0.5 | 5 |
| HCN1-R | –80.3 ± 2.5 | 17.2 ± 0.6 | 6 |
| Q353Ca | –91.3 ± 1.2 | 14.4 ± 0.5 | 12 |
| A354Ca | –87.4 ± 1.0 | 18.3 ± 0.9 | 15 |
| S357Ca | –75.5 ± 1.4* | 16.1 ± 0.5 | 5 |
| M358Ca | –69.2 ± 0.6* | 11.5 ± 0.5 | 9 |
aMeasurable currents are only produced with 24-h incubation with DTT
* Statistically different (p < 0.05) from WT HCN1 channels
Effect of Dithiothreitol (DTT) on P-S6 Cys-Substituted HCN1 Channels
Unlike WT and HCN1-R, none of the eight Cys-substituted constructs A352C, Q353C, A354C, P355C, V356C, S357C, M358C, and S359C produced functional currents (Fig. 3a). Since HCN channels are tetrameric, and only the cRNA of a single Cys-substituted construct was injected, each expressed channel protein carried four copies of the introduced cysteine. If these introduced sulfhydryls in the pore are in close enough geometric proximity, it is possible that they cross-link and thereby restrict the pore for permeation. To test this possibility, we incubated injected oocytes with the reducing agent DTT (2.5 mM). Interestingly, the loss of function of Q353C, A354C, S357C, and M358C could be rescued by DTT, as evident by the presence of hyperpolarization-activated currents (Fig. 3b). By contrast, A352C, P355C, V356C, and S359C failed to produce measurable currents even after DTT treatment. As we previously reported (Au et al. 2008), DTT had no detectable effect on the steady-state I–V relationship and activation gating of WT and HCN1-R channels (data not shown). The steady-state activation curves of DTT-treated HCN1-R, Q353C, A354C, S357C, and M358C are given in Fig. 3c. While the steady-state activation gating properties of Q353C and A354C were not statistically different from HCN1-R, those of S357C and M358C were depolarized (p < 0.05). All gating parameters are summarized in Table 1.
Fig. 3.

Representative current tracings of the eight P-S6 Cys-substituted constructs (a) before and (b) after DTT. c Steady-state activation curves of Q353C, A354C, S357C, and M358C after DTT treatment
Effect of Cd2+ on Cys-Substituted HCN1 Constructs
Given the high-affinity binding of class II divalents to free sulfhyfryl, we next examined the sensitivity of HCN1R, Q353C, A354C, S357C, and M358C channels for current blockade by Cd2+ (Fig. 4). Representative current tracings recorded in the absence and presence of Cd2+ are given. Consistent with previous reports, HCN1-R channels were completely insensitive to Cd2+ (n = 3) (Au et al. 2008). Similarly, Cd2+ of up to 1000 μM led to no current blockade of M358C channels at −140 mV (n = 3). By contrast, 100 μM Cd2+ sufficed to reduce I–140mV of S357C to 23 ± 5% (n = 3) and even completely blocked Q353C (n = 4) and A354C (n = 4) channels. Steady-state I–V relationships of Q353C, A354C, and S357C recorded in the presence of Cd2+ (at IC50) did not shift the V1/2 of Q353C (∆V1/2 = −4.6 ± 1.4 mV), A354C (−1.2 ± 0.8 mV), and S357C (−0.1 ± 0.2 mV) (p > 0.05), suggesting that the observed current reductions were not secondary to changes in gating properties. Figure 4b shows the dose-response relationships of current inhibition by Cd2+ block for HCN1-R, Q353C, A354C, S357C, and M358C channels. From these binding curves, the IC50 values for Cd2+ block of Q353C, A354C, and S357C were 3.1 ± 1.1 (n = 4), 3.7 ± 1.1 (n = 3), and 12.4 ± 1.0 (n = 3) μM, respectively (Table 2). Interestingly, a residual component for S357C was observed at high concentrations. The relationships for HCN1-R and M358C were flat across the concentration range examined.
Fig. 4.

Effects of Cd2+ on HCN1-R, Q353C, A354C, S357C, and M358C channels. a Representative current tracings measured at −140 mV in the absence and presence of Cd2+ as indicated. b Dose–response relationships
Table 2.
Summary of effects of CdCl2 on Q353C, A354C, and S357C channels
| Channel | IC50 (μM) | Hill coefficient | V1/2 (mV) at [CdCl2] of IC50 | N |
|---|---|---|---|---|
| Q353Ca | 3.1 ± 1.1 | 1.55 | –89.3 ± 1.5 | 3 |
| A354Ca | 3.7 ± 1.1 | 1.15 | –85.7 ± 0.9 | 4 |
| S357Ca | 12.4 ± 1.0 | 1.95 | –73.3 ± 1.2 | 4 |
aMeasurable currents are only produced with 24-h incubation with DTT
Covalent Modification of P-S6 Pore Cys-Substituted Constructs by MTSET
To complement our Cd2+ experiments, we next examined the effect of the permanently charged, membrane-impermeant covalent sulfhydryl modifier MTSET (Figs. 5–7 and Table 3). Since the size of MTSET is known (5.8 × 5.8 × 10 Å), successful modification of the inserted sulfhydryl would enable us to estimate the lower limit of the size of the accessible extrapore region. Similar to the MTS-resistant HCN1-R (Au et al. 2008; Bell et al. 2004), external application of 2.5 mM MTSET to Cd2+-resistant M358C channels did not result in any observable changes in the whole-cell currents, gating parameters, and reversal potentials (Fig. 6; p > 0.05). Interestingly, Cd2+-sensitive S357C channels were also resistant to MTSET modification (Fig. 6; p > 0.05). In stark contrast, MTSET application led to a significant current reduction at steady state at −140 mV of Q353C (IMTSET/IControl = 27.9 ± 3.4%; n = 5) and A354C (58.2 ± 13.1%; n = 8) (Figs. 5 and 7a; p < 0.05).
Fig. 5.

Representative current tracings of Q353C, A354C, S357C, and M358C before and after external application of MTSET
Fig. 7.

a Steady-state I–V relationships of Q353C and A354C before and after MTSET. b Steady-state activation curves. c Tail I–V relationships
Table 3.
Summary of activation and deactivation kinetics of WT and mutant HCN1 channels before and after MTSET modification
| Mutant | τactivation | τdeactivation | τactivation (MTSET) | τdeactivation (MTSET) | IMTSET/IControl |
|---|---|---|---|---|---|
| HCN1R | 371 ± 6 | 65 ± 10 | 390 ± 6 | 64 ± 9 | 100.3 ± 4.9% |
| Q353C | 403 ± 40 | 50 ± 3 | 1212 ± 228 | 236 ± 10 | 27.9 ± 3.4% |
| A354C | 444 ± 12 | 353 ± 23 | 1488 ± 57 | 537 ± 54 | 58.2 ± 13.1% |
| S357C | 661 ± 13 | 318 ± 17 | 729 ± 10 | 341 ± 11 | 97.0 ± 4.7% |
| M358C | 320 ± 6 | 79 ± 6 | 322 ± 2 | 77 ± 7 | 96.0 ± 3.3% |
Fig. 6.

a Steady-state I–V relationships of S357C and M358C before and after MTSET. b Steady-state activation curves. c Tail I–V relationships
Since current reduction induced by MTSET could result from changes in the permeation pathway, gating properties, or both, we next studied the effect of MTSET on steady-state activation to distinguish among these possibilities. Figure 7b showed that MTSET modification similarly and significantly shifted the activation curves of Q353C (V1/2,MTSET = −103.3 ± 2.0 mV vs. V1/2,Control = −82.2 ± 0.9 mV [n = 7]; p < 0.01) and A354C (V1/2,MTSET = −96.3 ± 2.1 mV vs. V1/2,Control = −86.3 ± 1.9 mV [n = 8]; p < 0.05) channels in the hyperpolarizing direction. Importantly, the steady-state activation plateau was reached at −140 mV for both constructs, implying that macroscopic I−140mV reductions observed could primarily result from gating modification. However, an effect on permeation cannot be excluded given that the open probability of HCN channels may not reach 100% even at saturating hyperpolarizing potentials (Flynn et al. 2007; Chen et al. 2007). In general, these phenotypes of Q353C and A354C were similar to our previously reported S5-P C318 whose MTS-induced reduction is primarily attributed to the hyperpolarizing activation shift (Xue and Li 2002). MTSET also slowed the activation and deactivation kinetics (Table 3) of Q353C and A354C but without altering their reversal potentials (Fig. 7c).
State-Dependent MTSET Modification of Q353C and A354C
The Shaker residues M448 and P450, whose analogous residues in HCN1 are Q353 and P355, respectively, have been shown to become highly accessible or even capable of cross-linking in the C-type inactivated state (Liu et al. 1996). Given the demonstrated accessibility of Q353C and the proximity of A354 to P355 (whose Cys substitution led to dysfunctional channels; cf. Fig. 3), we next examined if MTSET modification of Q353C and A354C channels was state dependent. With a stimulation frequency of 0.03 Hz, I−140mV of Q353C reduced progressively upon external MTSET application (Fig. 8a). The modification time constant (τMTSET) was 381.6 ± 24.7 s (n = 3), with steady-state block achieved ~12 min after application. However, when the stimulation frequency was slowed to 0.016 Hz, steady-state modification required ~20 min and τMTSET significantly increased to 892.6 ± 82.1 s (n = 3; p < 0.05). Similarly, MTSET modification of A354C was also state dependent (Fig. 8b). At 0.03 Hz, τMTSET was 364.1 ± 35.7 s and steady-state block was completed at ~8 min. These were changed to 693.3 ± 61.5 s and ~20 min, respectively, when the slower frequency of 0.016 Hz was applied. Interestingly, no apparent differences were observed for both Q353C and A354C channels when normalized I−140mV was plotted against the pulse number (rather than time) (Fig. 8c and d). These data implicate that residues C353 and C354 were reactive to MTSET only during the open state when depolarized. Taken collectively, our observations were consistent with the pore-to-gate coupling hypothesis that the P-S6 extrapore region undergoes dynamic conformational changes during gating.
Fig. 8.

Time course of the development of current inhibition at −140 mV upon external application of 2.5 mM MTSET with a stimulation frequency of 0.03 and 0.016 Hz to a Q353C and b A354C channels. c, d The same experimental data from A and B are plotted as normalized I−140mV against the pulse number (rather than time)
A352C, P355C, V356C, and S359C channels were dysfunctional but synthesized.
To further explore the basis of the loss-of-function phenotype of A352C, P355C, V356C, and S359C channels even after DTT treatment, we performed Western blot analysis (Fig. 9). For WT and HCN1-R channels prepared under control (oxidizing) conditions and after DTT incubation, only single bands at 76 kDa, as expected for the size for the HCN1 monomer, were detected. Without DTT, additional bands at 150 kDa that appeared to be cross-linked dimers were seen for A352C, Q353C, V356C, S357C, and S359C. However, similar dimers were not seen for A354C, P355C, and M358C channels. Interestingly, all dimers disappeared after DTT treatment, and only monomers similar to those of WT and HCN1-R channels were observed. All constructs displayed bands with intensity comparable with those of WT and HCN1-R channels (with the identical amounts of cRNA injected), suggesting that protein synthesis and translational efficiency were not affected by the Cys substitutions. The same pattern was observed in three other experiments performed independently with different batches of Xenopus oocytes.
Fig. 9.

Western blot analysis of the membrane proteins isolated from oocytes injected with cRNA of WT HCN1, HCN1-R, and eight pore Cys-substituted constructs under control and reducing conditions
Discussion
In the present study, we employed Cys-scanning substitutions to investigate the previously unexplored P-S6 linker that is C-terminal to the GYG motif. Based on our results, we conclude that the P-S6 linker residues 352–359 are important determinants of the structure-function properties of HCN1 channels. Among these pore residues, Q353, A354, and S357 are exposed to the aqueous phase as gauged by Cd2+ and/or MTSET sensitivities. The accessibility of Q353 and A354 is state dependent, consistent with an allosteric pore-to-gate coupling model that involves dynamic rearrangements of the extrapore. Using the same approach, we previously identified the endogenous cysteine C318 from the S5-P linker (Xue and Li 2002) and H344 from the descending limb of the P-loop (Au et al. 2008) immediately N-terminal to GYG as extracellularly accessible residues. The collective data sets provide structural and functional insights for deducing a more comprehensive footprint of the HCN pore, as further discussed below.
The Cd2+-sensitive but MTSET-resistant S357C channels were phenotypically distinctive from Q353C and A354C in that the latter were sensitive to both. Furthermore, Cd2+ led to complete blockade of Q353C and A354C but a residual component (~20%) remained for S357C even at high concentrations (cf. Fig. 4). Our findings were also consistent with previously reported cysteine-scanning experiments of the sea urchin sperm (sp) HCN channels, whose F434 residue homologous to HCN1 Q353 when replaced by cysteine similarly enhanced macroscopic current blockage by extracellular Cd2+ (Roncaqlia et al. 2002). A possible explanation for these observations is that the side chain of S357C is indeed exposed to the extracellular milieu but at a location that is more superficial than those of Q353 and A354 (Fig. 10). As such, Cd2+ binding to C357 does not lead to complete occlusion of the pore. Functionally, however, the bound Cd2+ can inhibit conduction by electrostatic repulsion of the incoming permeant ions. Also, as a result of its superficial location in the extrapore, even successful MTSET modification cannot lead to functional changes because (i) the ethyl linker that attaches the MTS moiety to C357 precludes direct pore occlusion and (ii) residue 357 is not involved in dynamic pore rearrangements during gating, unlike Q353C, A354C, and C318C. By contrast, MTS modification of Q353C or A354C occludes the pore, increases the energy required for channel opening (as reflected by a hyperpolarizing shift of V1/2), and slows the gating kinetics (due to the attachment of the bulky MTS moiety to a dynamic pore region).
Fig. 10.

Schematic representation of the HCN pore summarizing our present results and those of our two previously published studies. Gray circles, cysteinyl side chain; open circles, permeant ion; filled circles, endogenous amino acid
Unlike Q353C, A354C, and S357C, however, M358C has a phenotype that is similar to the F339C (N terminal to the GYG motif in the P region) that we previously described (Au et al. 2008). Both M358C and F339C channels displayed loss-of-function that could be rescued by DTT, but the restored currents were sensitive to neither Cd2+ nor MTSET. We cannot exclude the possibility that Cd2+ binding or MTSET modification of M358C does not lead to functional changes due to its distance from the permeation pathway (particularly given the proposed superficial location of S357). Further experiments will be needed to dissect the underlying mechanisms. The lack of measurable functional currents from A352C, P355C, V356C, and S359C channels, however, did not enable us to assess their accessibility.
Our present results were consistent with our previously proposed pore-to-gate coupling model of HCN channels (Azene et al. 2003, 2005; Xue and Li 2002) and provide further structural and functional insights. To date, we have identified that C318 from the S5-P linker (Xue and Li 2002), H344 of the ascending arm of the P-loop (Au et al. 2008), and Q353, A354, and S357 of the P-S6 segment (from the present study) are exposed to the aqueous phase and externally accessible by Cd2+. MTS modification of C318, H344, Q353, and A354, but not S357, alters activation gating, presumably due to their locations in pore regions that undergo dynamic rearrangements. MTS reactivity of C318, however, is not state dependent as we previously demonstrated (Xue and Li 2002).
Other than the extracellularly accessible residues Q353, A354, and S357, our results also support the notion that other P-S6 linker residues studied are also structural and functional determinants of HCN channels. Although channel proteins were expressed at levels comparable to WT and HCN1-R, HCN functions were completely abolished by substituting any of A352, P355, V356, M358, or S359 with a Cys. Specifically, A352C, Q353C, V356C, S357C, and S359C were capable of cross-linking, although our experiments did not allow us to rule out the possibility of at least one HCN subunit cross-linking with endogenous oocyte proteins. Nonetheless, such resultant dimers could be reduced by DTT coincubation. Whereas the loss-of-function of Q353C, A354C, S357C, and M358C could be rescued after dimer reduction, others remained dysfunctional even after DTT treatment. Although the endogenous C303 in HCN1-R that has not been replaced could cross-link with the inserted sulfhryl of the same subunit to abolish channel functions, this possibility can be ruled out because such an internal disulfide bridge would not lead to the formation of dimers. Assuming that the HCN pore is analogous to the crystal structure of KcsA (Doyle et al. 1998; Roux and MacKinnon 1999), the predicted distance (~35 Å) makes it unlikely for C303 from one monomer to cross-link with an introduced P-S6 cysteine from another monomer. Indeed, disulfide bond formation is a dynamic collisional process that requires at least occasional proximity (~3.5 Å) of the free sufhydryls involved with their α-carbons oriented at particular angles.
Based on our results, we conclude that the P-S6 linker residues 352–359 are important determinants of the structure-function properties of HCN1 channels. Among these pore residues, Q353, A354, and S357 are exposed to the aqueous phase. The accessibility of Q353 and A354 is state dependent, consistent with an allosteric pore-to-gate coupling model that involves dynamic rearrangements of the extrapore.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 HL72857 to R.A.L.), Croucher Foundation Fellowship (C.W.S), the University of Hong Kong Seed Funding Programme for Basic Research (HKU 200808159001 to C.W.S), the Hong Kong Research Grant Council General Research Fund (HKU 7747/08 M to C.W.S., H.F.T., and R.A.L.), and the CC Wong Stem Cell Fund (to H.F.T. and R.A.L.).
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Akabas MH, Stauffer DA, Xu M, Karlin A (1992) Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science 258:307–310 [DOI] [PubMed]
- Au KW Siu CW, Lau CP, Tse HF, Li RA (2008) Structural and functional determinants in the S5-P region of HCN-encoded pacemaker channels revealed by cysteine-scanning substitutions. Am J Physiol Cell Physiol 19:136–141 [DOI] [PubMed]
- Azene EM, Xue T, Li RA (2003) Molecular basis of the effect of potassium on heterologously expressed pacemaker (HCN) channels. J Physiol 547:349–356 [DOI] [PMC free article] [PubMed]
- Azene EM, Sang D, Tsang SY, Li RA (2005) Pore-to-gate coupling of HCN channels revealed by a pore variant that contributes to gating but not permeation. Biochem Biophys Res Commun 327:1131–1142 [DOI] [PubMed]
- Bell DC, Yao H, Saenger RC, Riley JH, Siegelbaum SA (2004) Changes in local S4 environment provide a voltage-sensing mechanism for mammalian hyperpolarization-activated HCN channels. J Gen Physiol 123:5–19 [DOI] [PMC free article] [PubMed]
- Benitah JP, Tomaselli GF, Marban E (1996) Adjacent pore-lining residues within sodium channels identified by paired cysteine mutagenesis. Proc Natl Acad Sci USA 93:7392–7396 [DOI] [PMC free article] [PubMed]
- Biel M, Schneider A, Wahl C (2002) Cardiac HCN channels: structure, function, and modulation. Trends Cardiovasc Med 12:206–212 [DOI] [PubMed]
- Brown HF, Giles W, Noble SJ (1977) Membrane currents underlying activity in frog sinus venosus. J Physiol 271:783–816 [DOI] [PMC free article] [PubMed]
- Bucossi G, Nizzari M, Torre V (1997) Single-channel properties of ionic channels gated by cyclic nucleotides. Biophys J 72:1165–1181 [DOI] [PMC free article] [PubMed]
- Chen S, Wang J, Siegelbaum SA (2001) Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol 117:491–504 [DOI] [PMC free article] [PubMed]
- Chen S, Wang J, Zhou L, George MS, Siegelbaum SA (2007) Voltage sensor movement and cAMP binding allosterically regulate an inherently voltage-independent closed-open transition in HCN channels. J Gen Physiol 129:175–188 [DOI] [PMC free article] [PubMed]
- Creighton TE (1992) Protein folding. W.H. Freeman, New York
- DiFrancesco D (1981a) A new interpretation of the pace-maker current in calf Purkinje fibres. J Physiol 314:359–376 [DOI] [PMC free article] [PubMed]
- DiFrancesco D (1981b) A study of the ionic nature of the pace-maker current in calf Purkinje fibres. J Physiol 314:377–393 [DOI] [PMC free article] [PubMed]
- DiFrancesco D (1993) Pacemaker mechanisms in cardiac tissue. Annu Rev Physiol 55:455–472 [DOI] [PubMed]
- DiFrancesco D (2006) Serious workings of the funny current. Prog Biophys Mol Biol 90:13–25 [DOI] [PubMed]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280:69–77 [DOI] [PubMed]
- Flynn GE, Black KD, Islas LD, Sankaran B, Zagotta WN (2007) Structure and rearrangements in the carboxy-terminal region of SpIH channels. Structure 15:671–682 [DOI] [PMC free article] [PubMed]
- Gauss R, Seifert R, Kaupp UB (1998) Molecular identification of a hyperpolarization-activated channel in sea urchin sperm. Nature 393:583–587 [DOI] [PubMed]
- Giorgetti A, Carloni P, Mistrik P, Torre V (2005) A homology model of the pore region of HCN channels. Biophys J 89:932–944 [DOI] [PMC free article] [PubMed]
- Karlin A, Akabas MH (1998) Substituted-cysteine accessibility method. Methods Enzymol 293:123–145 [DOI] [PubMed]
- Kaupp UB, Seifert R (2001) Molecular diversity of pacemaker ion channels. Annu Rev Physiol 63:235–257 [DOI] [PubMed]
- Lieu DK, Chan YC, Lau CP, Tse HF, Siu CW, Li RA (2008) Overexpression of HCN-encoded pacemaker current silences bioartificial pacemakers. Heart Rhythm 5:1310–1317 [DOI] [PubMed]
- Liu Y, Jurman ME, Yellen G (1996) Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron 16:859–867 [DOI] [PubMed]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M (1998) A family of hyperpolarization-activated mammalian cation channels. Nature 393:587–591 [DOI] [PubMed]
- Prole DL, Yellen G (2006) Reversal of HCN channel voltage dependence via bridging of the S4–S5 linker and Post-S6. J Gen Physiol 128:273–282 [DOI] [PMC free article] [PubMed]
- Roncaqlia P, Mistrik P, Torre V (2002) Pore topology of the hyperpolarization-activated cyclic nucleotide-gated channel from sea urchin sperm. Biophys J 83:1953–1964 [DOI] [PMC free article] [PubMed]
- Roux B, MacKinnon R (1999) The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science 285:100–102 [DOI] [PubMed]
- Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR (1998) Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell 93:717–729 [DOI] [PubMed]
- Siu CW, Lieu DK, Li RA (2006) HCN-encoded pacemaker channels: from physiology and biophysics to bioengineering. J Membr Biol 214:115–122 [DOI] [PubMed]
- Tsushima RG, Li RA, Backx PH (1997) P-loop flexibility in Na+ channel pores revealed by single- and double-cysteine replacements. J Gen Physiol 110:59–72 [DOI] [PMC free article] [PubMed]
- Xue T, Li RA (2002) An external determinant in the S5-P linker of the pacemaker (HCN) channel identified by sulfhydryl modification. J Biol Chem 277:46233–46242 [DOI] [PubMed]
- Xue T, Marban E, Li RA (2002) Dominant-negative suppression of HCN1- and HCN2-encoded pacemaker currents by an engineered HCN1 construct: insights into structure-function relationships and multimerization. Circ Res 90:1267–1273 [DOI] [PubMed]