

Abstract
Receptor mediated endocytosis (RME) plays a major role in the disposition of therapeutic protein drugs in the body. It is suspected to be a major source of nonlinear pharmacokinetic behavior observed in clinical pharmacokinetic data. So far, mostly empirical or semi-mechanistic approaches have been used to represent RME. A thorough understanding of the impact of the properties of the drug and of the receptor system on the resulting nonlinear disposition is still missing, as is how to best represent RME in pharmacokinetic models. In this article, we present a detailed mechanistic model of RME that explicitly takes into account receptor binding and trafficking inside the cell and that is used to derive reduced models of RME which retain a mechanistic interpretation. We find that RME can be described by an extended Michaelis–Menten model that accounts for both the distribution and the elimination aspect of RME. If the amount of drug in the receptor system is negligible a standard Michaelis–Menten model is capable of describing the elimination by RME. Notably, a receptor system can efficiently eliminate drug from the extracellular space even if the total number of receptors is small. We find that drug elimination by RME can result in substantial nonlinear pharmacokinetics. The extent of nonlinearity is higher for drug/receptor systems with higher receptor availability at the membrane, or faster internalization and degradation of extracellular drug. Our approach is exemplified for the epidermal growth factor receptor system.
Electronic supplementary material
The online version of this article (doi:10.1007/s10928-009-9120-1) contains supplementary material, which is available to authorized users.
Keywords: Recepter mediated endocytosis, Nonlinear pharmacokinetics, Michaelis–Menten, Therapeutic proteins, Biopharmaceuticals, Epidermal growth factor receptor, Nonlinear dispostition, Receptor trafficking, Antibodies
Introduction
In recent years, therapeutic proteins have been a major focus of research and development activities in the pharmaceutical industry [1]. Currently, approximately 100 therapeutic proteins have been approved for human use, most of them being biotechnology-derived drug products and many more are under development. Important classes of therapeutic proteins are monoclonal antibodies, growth factors, and cytokines. Generally, therapeutic proteins provide highly attractive but sometimes exceptional behavior in the body [2]: their significant therapeutic potential results from their ability to bind—with high affinity—to specific targets such as receptors or cell surface proteins. For many protein drugs receptor mediated endocytosis (RME) is an important route of cellular uptake and disposition [3]. RME is the process of binding of an endogenous or exogenous ligand to a receptor and subsequent internalization of the resulting complex forming an endosome. Within the cell, the complex may be recycled to the cell surface or intracellularly be cleaved [4, 5]. Receptor-mediated uptake plays a major role in the elimination of protein drugs from the body [3] and is suspected to be a major source for the nonlinear pharmacokinetic (PK) behavior that is observed in clinical data for numerous protein drugs [6].
When aiming at analyzing preclinical/clinical pharmacokinetic data of protein drug trials, typically empirical 1-, 2- or 3-compartmental models including linear and/or nonlinear disposition processes have been developed. Michaelis–Menten terms have often been used to analyze experimental data in order to account for the observed nonlinearity [7–11]. These models have been selected based on, e.g., established statistical criteria (such as maximum likelihood), the precision of estimates of model parameters, and in few cases on model evaluation techniques [12–15]. However, being empirical in nature, these models do not provide a mechanistic understanding of how the different processes of receptor trafficking contribute to the overall pharmacokinetic profile, which is expected to guide, e.g., lead optimization or the design of more efficient dosing regimens. Equally important, there is no theoretical background as to when use the different existing empirical models for nonlinearity.
Less often, models have been developed that also include mechanistic terms to account for nonlinear phenomena, most prominently in terms of target-mediated drug disposition (TMDD) models [16–18]. TMDD explicitly accounts for binding to a target and potential degradation of the resulting complex. Although originally developed to describe effects of extensive drug target binding in tissues, TMDD has more recently gained interest as a model for saturable elimination mechanisms for specific peptide and protein drugs, including RME [6, 18, 19]. TMDD is a general approach for situations where the interaction of a drug with its target is considered to be relevant and might affect the concentration-time profiles. However, it does not explicitly take into account the particular features of receptor trafficking inside cells, such as recycling and sorting, i.e., the process by which receptors and ligands are either targeted for intracellular degradation or recycled to the surface for successive rounds of trafficking [20].
There is a considerable amount of literature about detailed mechanistic descriptions of receptor trafficking systems in the systems biology literature (see, e.g., [5, 21] and references therein). Based on these receptor trafficking systems, our approach is to build a general detailed mechanistic model of RME that takes into account the most relevant kinetic processes of drug binding and receptor trafficking inside the cell. Detailed models derived from the underlying biochemical reaction network have the advantage of a mechanistic interpretation of the kinetic processes and estimated parameters. In [22], a cell-level model of the cytokine granulocyte colony-stimulating factor (G-CSF) and its receptor was incorporated into a pharmacokinetic/pharmacodynamic model to allow for analyzing the life span and potency of the ligand in vivo. However, often these advantages come along with the disadvantage of containing more parameters which, e.g., in population PK analysis of clinal trials may result in poorer performance in the model selection process, since models containing more parameters are usually penalized by the corresponding model selection criteria.
The objective of this article is to develop a framework for RME that is specifically tailored to the needs in PK analysis of clinical trials by bridging the points of view in pharmacokinetics and systems biology. The aims are (i) to develop a detailed model that takes into account the most relevant processes in relation to receptor trafficking; (ii) to derive reduced models of RME which retain a mechanistic interpretation and are defined in terms of a few parameters only, (iii) to offer guidance as to when use them, and (iv) to analyze the impact of the different processes on the extent of nonlinearity. While our approach applies to many receptor systems in general, we will use the epidermal growth factor receptor (EGFR) signalling pathway to illustrate the approach. The EGFR system has been intensively studied over the past 20 years and is one of the most important pathways for cell growth and proliferation as well as angiogenesis and metastasis [23]. The EGFR system comprises a tyrosine kinase receptor, which is activated by a variety of ligands such as the epidermal growth factor (EGF) or the transforming growth factor-α (TGF-α) [24–26]. Mathematical modelling of the EGFR system has proven to be useful for both, measurement of rate constants [27] as well as to elucidate the effects of receptor trafficking as an input to downstream signalling cascades [21, 28]. From a therapeutic point of view, the EGFR system has shown to be a promising target in cancer therapy [29, 30]. Several agents, including therapeutic proteins such as monoclonal antibodies (mAbs), have been developed to specifically target the EGFR with some already approved for drug treatment [31–33].
Theoretical
Throughout the article, the term ’ligand’ refers to both a physiological ligand as well as an exogenous drug ligand.
Detailed model of RME (Model A)
We propose the following detailed model of RME of a ligand as schematically represented in Fig. 1: the ligand Lex is present in the extracellular space. The ligand reversibly binds to free receptor Rm at the cell membrane with association rate constant kon to form the ligand–receptor complex RLm that dissociates with rate constant koff. The complex is internalized with the rate constant kinterRL forming an endosome. The internalized ligand–receptor complex RLi is either recycled to the membrane with the rate constant krecyRL, degraded with the rate constant kdegRL to RLdeg, or dissociates with the rate constant kbreak. The dissociation results in the subsequent degradation of the ligand Ldeg and the availability of the free receptor Ri inside the cell. Free intracellular receptor Ri is recycled to the membrane with the rate constant krecyR and free membrane receptor Rm is internalized with the rate constant kinterR. Inside the cell, the receptor Ri is produced with the rate ksynth and degraded with the rate constant kdegR.
Fig. 1.

Schematic representation of the detailed model of receptor mediated endocytosis. See text for description
Based on the law of mass action, the rates of change for the various molecular species are given by the following system of ordinary differential equations (ODEs):
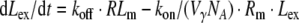 |
1 |
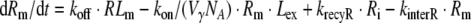 |
2 |
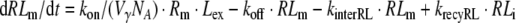 |
3 |
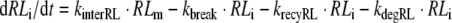 |
4 |
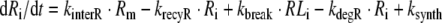 |
5 |
where NA is Avogadro’s number and Vγ is the volume of extracellular space per cell. In the above equations, all variables are expressed in number of molecules. All parameters are first-order rate constants in units [1/time] except for ksynth, which is a zero-order rate constant in units [molecules/time], and kon which is a second-order rate constant in units [1/(concentration × time)]. The factor 1/(VγNA) ensures conversion of units from molar concentration to number of molecules. With respect to the receptor, the above equations comprise the following three overall processes (cf. Fig. 1): (1) synthesis and degradation; (2) distribution of the different receptor species within and between the cytoplasm and the cell membrane; and (3) ligand–receptor interaction. With respect to the ligand, its disposition processes consist of the three overall processes: (i) binding to the receptor; (ii) internalization of the ligand–receptor complex; and (iii) intracellular degradation.
Reduced models of RME
One objective of this study is to derive and analyze reduced models of RME that capture the impact of receptor dynamics on the distribution and elimination of a ligand and that still allow for a mechanistic interpretation. While during short time intervals the transient redistribution processes between the different receptor species Rm, RLm, RLi and Ri may be of interest, these are usually assumed to be negligible on time scales of interest in pharmacokinetics. Therefore, our approach to reduce the detailed RME model will be based on the assumption that the receptor species Rm, RLm, RLi and Ri are in quasi-steady state. In order to finally derive reduced models of RME, it is necessary to make an additional assumption on the time-scale of receptor synthesis and degradation. We distinguish the following two scenarios: (1) the time scale of receptor synthesis and degradation is slow in comparison to the time scale of ligand disposition. In this case, we formally set ksynth = kdegR = kdegRL = 0. As a consequence, the total number of receptors in the system remains constant. Or, (2) the time scale of receptor synthesis and degradation is fast, i.e., comparable to the redistribution processes of the different receptor species. Both scenarios will be used in the following to establish a link between the reduced and the detailed model.
Reduced model of saturable distribution into the receptor system and linear degradation (Model B)
The idea in deriving a reduced model of RME is to use the quasi-steady state assumption for the receptor system (RS). This transforms the differential equations (2)–(5) into algebraic equations for Rm, RLm, RLi, Ri. For a given number of extracellular ligand molecules Lex, these algebraic equations can be solved explicitly. This allows us to compute the total number of ligand molecules in the receptor system LRS = RLm + RLi as a function of the extracellular number of ligands Lex. Based on LRS, the quasi-steady state number of intracellular ligand–receptor complexes RLi can be computed, which determines the extent of elimination.
Model B (see Fig. 2) describes the evolution of the total number of ligands Ltot = Lex + LRS in form of the following ODE:
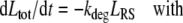 |
6 |
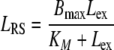 |
7 |
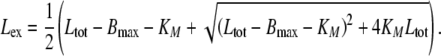 |
8 |
The equations comprise three parameters: the maximal ligand binding capacity Bmax of the receptor system (in units molecules), the number of extracellular ligand molecules corresponding to a half-maximal binding capacity KM (in units molecules), and the degradation rate kdeg (in units 1/time). In this reduced model the combination of saturable distribution and linear degradation results in the overall saturable elimination of the ligand.
Fig. 2.

Models of receptor mediated endocytosis of different resolution: Detailed model (Model A), reduced model of saturable distribution into the receptor system with linear degradation (Model B), and reduced model of saturable degradation (Model C). See text for details
For the two scenarios of slow or fast receptor synthesis and degradation, the functional relation between the parameters Bmax, KM and kdeg and the parameters of the detailed model of RME can be established. In the case of slow receptor synthesis and degradation, it is
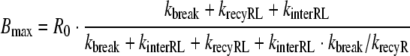 |
9 |
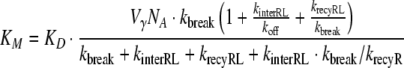 |
10 |
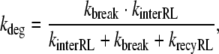 |
11 |
where R0 is the total number of receptors and KD = koff/kon denotes the dissociation constant of the ligand–receptor complex. In the case of fast receptor synthesis and degradation, the relation between the parameters is
 |
12 |
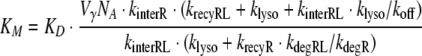 |
13 |
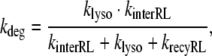 |
14 |
with klyso = kbreak + kdegRL.
Reduced model of saturable degradation (Model C)
The proposed Model C (see Fig. 2) is a further reduction of Model B. It is based on the additional assumption that the amount of ligand distributed into the receptor system is negligible in comparison to the total amount of ligand molecules, i.e., Ltot = Lex + LRS ≈ Lex. More formally, Model C can be derived from Model B under the assumption
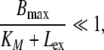 |
15 |
which implies LRS ≪ 1 and thus Ltot ≈ Lex from Eq. 7. Substituting Lex by Ltot in Eq. 7 and LRS into Eq. 6 yields the ODE for the total number of ligand molecules:
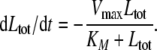 |
16 |
The model comprises two parameters: the maximal elimination rate of ligand molecules Vmax (in units molecules/time) and the number of ligand molecules KM, at which the elimination rate is half-maximal. Exploiting the relation
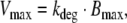 |
17 |
we obtain the functional relations between Vmax and the parameters of the detailed model of RME (Model A). In the case of slow receptor synthesis and degradation, the functional relationship is given by
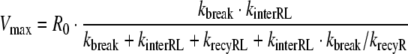 |
18 |
and KM is defined as in Eq. 10. In the case of fast receptor synthesis and degradation, it is
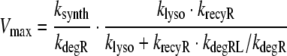 |
19 |
and KM is defined as in Eq. 13.
Integration of RME into compartmental PK models
In order to facilitate the transfer of reduced models of RME into compartmental PK models underlying PK data analysis and for use in the example of therapeutic protein receptor interaction, we explicitly state the system of ODEs for a two-compartment PK model. The model comprises a central compartment (volume V1 (in units volume) and ligand concentration C1 (in units mass/volume)) from which linear elimination CLlin (in units volume/time) takes place and a peripheral compartment (volume V2 and total ligand concentration C2), where saturable elimination via receptor mediated endocytosis CLRS takes place (see Fig. 3). In the peripheral compartment, we further distinguish between the concentration CRS within the receptor system and the extracellular concentration Cex. The inter-compartmental transfer flows are denoted by q12 and q21 (in units volume/time).
Fig. 3.

Two two-compartment models with linear clearance from the central compartment and RME based on Model B (left) and Model C (right) in the peripheral compartment
As in this article we are interested in how to represent RME in PK models, the below mentioned system of ODEs based on the reduced Models B and C represent the proposed structural PK model that can be used for parameter estimation in PK data analysis of nonclinical and clinical trials. The parameter values are determined by performing a fit of the model to the specific in vivo data. Alternatively, the model might be used to scale-up in vitro derived RME parameter values to the in vivo situation (see also Discussion).
If Model B is used to describe the elimination by RME, the system of ODEs is
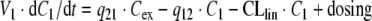 |
20 |
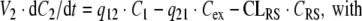 |
21 |
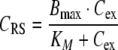 |
22 |
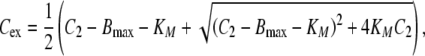 |
23 |
where dosing denotes a mass inflow (in units mass/time) of, e.g., an i.v. infusion over a given time. The parameter Bmax denotes the total maximal ligand binding capacity in mass per volume or mol per volume, KM denotes the concentration at which the binding capacity is half-maximal, CLlin and CLRS denote the total elimination capacities (in units volume/time) in the central and peripheral compartment, respectively. In terms of parameter estimation, the PK model contains eight parameters: V1, V2, q12, q21, CLlin, CLRS, Bmax and KM, plus additional variables relating to dosing.
If Model C is used to describe the elimination by RME, the system of ODEs is
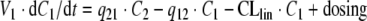 |
24 |
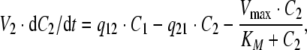 |
25 |
where Vmax denotes the total maximal elimination (in units mass/time), and all remaining parameters are defined as above. In terms of parameter estimation, the PK model contains seven parameters: V1, V2, q12, q21, CLlin, Vmax and KM, in addition to the parameters relating to dosing.
Nonlinear PK caused by RME
In this section, we investigate the extent of nonlinearity in the context of the Michaelis–Menten model defined in Eqs. 24 and 25. We aim to examine the effect of drug and cell properties on the nonlinearity of the pharmacokinetics, e.g., different drug affinities to the receptor (different kon and koff values) or different rates of internalization and recycling of the drug in different cells.
In the chosen setting of the two-compartment PK model (cf. Eqs. 24 and 25, the total clearance CLtot is given by
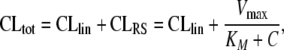 |
26 |
where C denotes the relevant ligand concentration in the RME compartment (e.g., C2 in Eq. 25). While the linear clearance is constant, the clearance attributed to RME varies between Vmax/KM for small ligand concentrations and 0 for high ligand concentrations. Therefore, we consider the quotient Vmax/KM as a measure of the extent of nonlinearity, i.e., the increase in total clearance for small ligand concentrations.
In order to jointly analyze the slow and the fast receptor synthesis and degradation scenario, we set
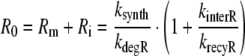 |
27 |
and replace the quotient ksynth/kdegR in Eq. 19 by R0/(1 + kinterR/krecyR) according to Eq. 27. Moreover, we extend the definition of klyso to the slow scenario by setting klyso = kbreak in this case (note: for the fast scenario klyso = kbreak + kdegRL). Then, the extent of nonlinearity for both, the fast and the slow scenario, is given by
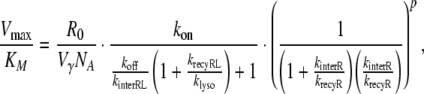 |
28 |
where p = 0 for the slow scenario and p = 1 for the fast scenario. The above equation allows us to study in detail the influence of the various parameters on the extent of nonlinearity.
It can be inferred from Table 1 that ligand-specific, receptor system-specific as well as mixed parameters influence the extent of nonlinearity of the PK: nonlinearity increases for higher affinity drugs (kon) and cell types, which have a higher receptor concentration at the surface of the cell membrane (R0, krecyR) and faster degradation processes (klyso). In contrast, higher values of koff, krecyRL and higher kinterR, kdegR will decrease the extent of nonlinearity by resulting in a lower number of intracellular ligand receptor complexes, free receptor molecules, or a smaller number of receptor molecules at the cell surface membrane.
Table 1.
Contribution of the different parameters to the extent of nonlinearity
| Increase of parameter | Resulting change in extent of nonlinearity |
|---|---|
| R0 | ↑ (RS) |
| krecyR | ↑ (RS) |
| kon | ↑ (L) |
| klyso | ↑ (RS & L) |
| kinterRL | ↑ (RS & L) |
| koff | ↓ (L) |
| kinterR | ↓ (RS) |
| krecyRL | ↓ (RS & L) |
With increasing value of the corresponding parameter the extent of nonlinearity will increase (↑) or decrease (↓). For each parameter, it is indicated by (RS) or (L) whether it is related to the receptor system or the ligand, respectively
In order to more clearly highlight the contribution of the dissociation constant KD, we also give the following alternative representation of Eq. 28:
 |
29 |
As can be inferred from the above relation, the extent of nonlinearity can be very different for ligands with the same dissociation constant KD, but different absolute values of koff. The difference depends on the relative magnitude of the two terms in the first denominator in Eq. 29, i.e., 1/koff to 1/kinterRL · (1 + krecyRL/klyso).
Methods
In order to simulate Models A, B and C, we numerically solved the corresponding system of ODE’s with Matlabs built-in ode15s integrator (The Mathworks, Inc., Natick/MA, USA, version 7.4). Parameter values for the reduced Models B and C were derived from those of Model A using the established relations (12)–(14), and (19) and (13), respectively. Subsequently, numbers of molecules where converted into concentrations (nM).
The models were compared based on the simulated extracellular drug concentration. The specific details of the simulation studies are given in the respective Result section to allow for an easier comparison.
EGFR system with endogenous/physiological ligand
The application of our approach is illustrated using the EGFR system as an example. The properties of the EGF/EGFR system will be analyzed using experimentally measured parameters for the degradation of the epidermal growth factor, binding to the epidermal growth factor receptor and subsequent internalization [20, 34]. The rate constants of the corresponding reactions are listed in Table 2.
Table 2.
Parameter values for the EGF/EGFR system
| Parameter | Numerical value |
|---|---|
| kon | 5.82 1/(nM h) |
| koff | 14.4 1/h |
| R(SS)m | 2 × 105 molecules |
| krecyR | 3.84 1/h |
| kinterR | 4.2 1/h |
| kdegR | 0.96 1/h |
| krecyRL | 1.2 1/h |
| kinterRL | 15 1/h |
| kdegRL | 1.2 1/h |
| Vγ | 4 × 10−10 1/cell |
All parameter values have been extracted from Hendriks et al. [20, 34] and Shankaran et al. [28]. See also section “RME for the EGF/EGFR system”
Hendriks et al. [20, 34] explored EGF as ligand to measure rate constants of the EGFR system. Since receptor is degraded as a consequence of ligand degradation, we choose the scenario of fast receptor synthesis and degradation for all investigations, i.e., Eqs. 12–14 and 19. However, not all rate constants of the herein proposed detailed model of RME were explicitly measured in [20, 34]. Since EGF is predominantly degraded from the EGF-receptor complex [5] rather than from the free form, we set kbreak = 0 resulting in klyso = kdegRL ≠ 0. Since the parameter ksynth was not available in literature, we used the steady state assumption for the receptor system prior to any ligand administration and the experimentally measured steady state number of membrane receptor R(SS)m [28] to determine ksynth using the relation ksynth = kdegR · R(SS)i with R(SS)i = R(SS)m · kinterR/krecyR. The initial number of receptors are Rm(0) = R(SS)m, Ri(0) = R(SS)i, and RLm(0) = RLi(0) = 0; the initial concentration of extracellular ligand is Lex(0) = 40 nM.
EGFR system with exogenous/therapeutic protein ligand
The analysis of drug-EGFR interaction are performed using data from the monoclonal antibody zalutumumab (2F8), as published by Lammerts van Bueren et al. [11]. Zalutumumab is a human IgG1 EGFR antibody that potently inhibits tumor growth in xenograft models and has shown encouraging antitumor results in a phase I/II clinical trial [35, 36]. We transformed the originally published system of difference equations [11, Supplement] into the corresponding continuous system of ordinary differential equations1 (ODEs):
 |
30 |
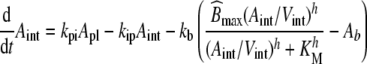 |
31 |
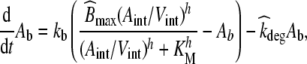 |
32 |
where Apl, Aint and Ab represent the amount of therapeutic protein in the plasma, interstitial and binding compartment, respectively; Vint the interstitial volume, kpi and kip the rate constants for transfer between the plasma and interstitial compartment, kb the rate constant for binding to and dissociation from EGFR, and kel the elimination rate constant. Furthermore, 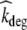 denotes the rate constant for elimination by EGFR internalization and degradation,
denotes the rate constant for elimination by EGFR internalization and degradation,  the maximal binding capacity of the therapeutic protein to EGFR, KM the concentration corresponding to
the maximal binding capacity of the therapeutic protein to EGFR, KM the concentration corresponding to 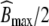 , and h the Hill factor. The initial amount of drug Apl(0) and the parameters are listed in Table 3. The reported value of KM = 5 μg/ml did not allow us to reproduce the results in [11, Fig.1A]. Only a value of KM = 0.5 μg/ml exactly reproduced the in silico data, hence we choose the corrected value for subsequent analyses. Amounts are converted to concentrations by dividing by the corresponding volume.
, and h the Hill factor. The initial amount of drug Apl(0) and the parameters are listed in Table 3. The reported value of KM = 5 μg/ml did not allow us to reproduce the results in [11, Fig.1A]. Only a value of KM = 0.5 μg/ml exactly reproduced the in silico data, hence we choose the corrected value for subsequent analyses. Amounts are converted to concentrations by dividing by the corresponding volume.
Table 3.
Parameter values used by Lammerts van Bueren et al. [11]
| Parameter | Numerical value |
|---|---|
| Vpl | 35 ml/kg |
| Vint | 70 ml/kg |
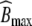 |
2 mg/kg |
| kip | 0.043 1/h |
| kpi | 0.043 1/h |
| kb | 0.069 1/h |
| kel | 0.0055 1/h |
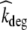 |
0.005 1/h |
| KM | 0.5 μg/ml |
| Apl(0) | 2 and 20 mg/kg |
| h | 1.0 |
KM has been corrected, see text for details. Vpl represents the plasma volume
Transforming the system of ODEs (30)–(32) from units [mg/kg] to [mg/ml] by dividing by the corresponding volumes yields equations for Cpl = Apl/Vpl, Cint = Aint/Vint, Cb = Ab/Vint, in terms of the following scaled parameters q12 = Vpl · kpi, q21 = Vint · kip, 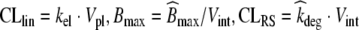 . The model (30)–(32) scaled to units [mg/ml] can be directly compared to our PK model (20)–(23) with C1 = Cpl, Cex = Cint and CRS = Cb, parameterized with the scaled parameters above. We remark that alternatively, our compartmental PK models could have been stated in units [mg/kg].
. The model (30)–(32) scaled to units [mg/ml] can be directly compared to our PK model (20)–(23) with C1 = Cpl, Cex = Cint and CRS = Cb, parameterized with the scaled parameters above. We remark that alternatively, our compartmental PK models could have been stated in units [mg/kg].
Results
RME for the EGF/EGFR system: an example for ligand–receptor interaction
For all subsequent in silico studies, the parameter values are stated as given in section “EGFR system with endogenous/physiological ligand”, unless stated otherwise.
Influence of receptor system properties on RME
We illustrate the approximation features of the two reduced models for predicting concentration-time profiles of the ligand in comparison to the detailed model based on the EGF/EGFR system. The initial concentration is Cex(0) = 40 nM. In Fig. 4(left), the predictions of the extracellular EGF concentration Cex is shown for the three Models A, B and C. All models result in very similar concentration-time profiles: Almost instantaneously, the amount of ligand in the RS is in equilibrium. Due to the high concentration of ligand in comparison to the concentration of receptor, the RS is saturated and the ligand is eliminated at a constant rate. Between approximately 40-60 h, the system undergoes a transition from saturated to non-saturated elimination, which is manifested in the linear decline in the final phase (in the semi-logarithmic representation). For the EGF/EGFR system, the detailed model of RME is well approximated by Model B and also by Model C, the latter taking into account only the apparent saturable elimination. Based on the predictions of Model B, we computed the amount of ligand LRS in the receptor system. In accordance with Eq. 15, LRS is negligible in comparison to extracellular EGF concentration (cf. Fig. 5, solid line) while Cex > 0.01 nM.
Fig. 4.

Concentration-time profile of the extracellular ligand concentration for the Model A (circles on blue solid line), Model B (squares on blue dashed line) and Model C (diamonds on red dashed line). Left: Parameter values used according to Table 2. Right: As in left figure, but decreasing kdegRL 10-fold
Fig. 5.

Evolution of the ratio Bmax/(KM + Cex) for the two scenarios shown in Fig. 4 left (solid line) and right (dashed line)
In order to study the impact of LRS on the approximation quality of Model C, we artificially decrease kdegRL by a factor of 10. All other parameters of the detailed Model A, including the initial EGF concentration, are identical. Parameters of Model B and C have been recalculated according to Eqs. 12–14 and Eqs. 19 and 13, respectively, resulting in particular in an increased maximal binding capacity Bmax. The predictions of the concentration-time profile of the extracellular EGF concentration Cex based on the three Models A, B and C are shown in Fig. 4(right). While Models A and B give almost identical results, the prediction based on Model C differs significantly. Model C over-predicts the extent of elimination by RME. As shown in Fig. 5 the over-prediction corresponds to periods in time where the assumption (15) is violated: While Bmax/(KM + Cex) is small for both settings up to time 60 h, it starts to increase thereafter, in particular for the setting corresponding to Fig. 4(right).
Influence of different cell types on RME
The detailed model A allows us to analyze the influence of processes on the overall disposition of ligand in the extracellular space such as, e.g., the ligand receptor internalization rate constant kinterRL. Alterations in kinterRL have been observed experimentally [37, 38] and could be the result of a mutation of the EGF receptor. In view of Eq. 28 we would expect a decrease in the overall elimination capacity with decreasing internalization rate constant kinterRL. Figure 6(left) shows the impact of an altered kinterRL on the concentration-time course of EGF with Cex(0) = 40 nM. As can be seen, cells with a reduced internalization rate constant kinterRL/4 and kinterRL/16 show a much lower apparent elimination than the reference cells with the rate constant kinterRL. The difference in the apparent elimination does not only depend on the absolute magnitude of change of kinterRL, but more precisely on the magnitude of change of 1/kinterRL · (1 + krecyRL/klyso) in relation to 1/koff, as can been inferred from Eq. 29. Changes in kinterRL will have less impact, if 1/koff is large. This can be seen in Fig. 6(right), which shows the same situation as in the left figure, but with koff decreased by a factor of 100 (we also decreased kon by the same factor in order to keep KD constant).
Fig. 6.

Illustration of the dependence of RME on the rate of internalization using the detailed model of RME (Model A). Parameter values according to Table 2. Left: concentration-time profiles of the extracellular ligand EGF (Lex) for three different internalization rate constants of the ligand–receptor complex: kinterRL (solid line), kinterRL/4 (dashed line), kinterRL/16 (dotted line). Right: same as before, but with decreased association and dissociation rate constants: kon/100 and koff/100, respectively. Note that KD is identical in the left and right graphics
RME in the monoclonal antibody/EGFR system: an example for therapeutic protein–receptor interaction
In this section we will illustrate how our unified theoretical approach to RME allows for resolving seemingly contradictory statements about the performance of empirical models of RME. In [11], Lammerts van Bueren et al. reported about a preclinical study involving a mAb against EGFR in monkeys and their subsequent data analysis. They developed a two-compartment pharmacokinetic model comprising a first-order elimination of the mAb from plasma, a binding compartment (representing EGFR-expressing cells) that equilibrates with the interstitial compartment, and a saturable internalization and degradation of bound mAb. For a detailed description of the model and the corresponding parameters see section “EGFR system with exogenous/therapeutic protein ligand”. Lammerts van Bueren et al. concluded that the observed nonlinear decrease of mAb concentrations in cynomolgus monkeys could not be explained by a saturable elimination in terms of a Michaelis–Menten model and proposed an alternative model, which described the data well. In a different study, the Michaelis–Menten model was reported to successfully describe in vivo data for a monoclonal antibody [10].
The model proposed in [11] is comparable to the two-compartment model introduced in the section “Integration of RME into compartmental PK models”, Eqs. 20–23. In order to understand the inferences made by Lammerts van Bueren et al. [11], we simulated their model defined in Eqs. 30–32 and compared the results to the correspondingly parameterized Models B and C (see Fig. 7, left). Since the experimental data presented in [11] were not available and since model simulations and data were reported to be in good agreement, we used the Lammerts van Bueren model as a surrogate for the experimental data. As in [11], we choose a high and low initial mAb input of 2 and 20 mg/kg. While the predicted mAb plasma concentrations based on Model B are identical to the prediction based on the Lammerts van Bueren et al. model, predictions based on Model C deviate significantly. A closer inspection reveals that the assumption Bmax/(KM + Cex(0)) ≪ 1 is violated for the low dose of 2 mg/kg. Consequently, the amount of mAb inside the RS cannot be neglected and we would expect to see deviations between predictions based on Models B and C. Hence, the use of a Michaelis–Menten based nonlinear elimination in the interstitial compartment, which neglects the drug distributed into the receptor system, leads to an over-prediction of drug elimination by RME (see Fig. 7, left).
Fig. 7.

Comparison of model predictions for zalutumumab (2F8) based on the Lammerts van Bueren et al. model (circles on solid line) and the herein proposed compartment models (20)–(23) (squares on solid line) and (24)-(25) (diamonds on dotted line). Left: parameterization as given in Table 3. Right: maximal receptor capacity Bmax decreased to one 20th of the original capacity
The difference between the predictions based on Model B and C should disappear, if the maximal binding capacity is sufficiently decreased. This is shown in Fig. 7(right), where the binding capacity Bmax has been decreased to one 20th of its original value.
In summary, the inference made in [11] that a Michaelis–Menten term is not adequate for modeling the nonlinearity present in the data is valid for the specific conditions of their experimental design. However, this cannot be generalized to a statement about the validity of the Michael-Menten approximation of RME, as can be seen from Fig. 7(right) and also from the results presented in section “RME for the EGF/EGFR system”.
Discussion
Drugs that demonstrate nonlinear pharmacokinetic behavior at therapeutic concentrations often cause difficulties in designing dosage regimens and determining relations between drug concentrations and effects. The theoretical bases and potential causes of nonlinear/dose-dependent pharmacokinetics are many-fold and have been extensively reviewed (see [17] and reference therein). Therapeutic proteins bind with high affinity to specific targets. For many protein drugs elimination by RME plays a major role in their elimination from the body [3]. RME is suspected to be a major source for the nonlinear pharmacokinetic behavior that is observed in pre-/clinical data of numerous protein drugs [6]. In this article we theoretically investigated the process of RME on the pharmacokinetics of therapeutic proteins.
The detailed Model A (see Fig. 1) represents RME for an endogenous compound in terms of a system of biochemical reactions (1)–(5), including the binding of the ligand to the receptor, subsequent internalization of the complex and eventually degradation as well as receptor recycling, degradation and synthesis. Two reduced models have been derived under the assumption that the redistribution processes between the receptor species Rm, RLm, Ri and RLi are in quasi-steady-state. For the EGFR system, this assumption has been shown experimentally [27]. For other receptor systems, the steady state assumption seems reasonable since intracellular processes are typically much faster than the time scale of interest in pharmacokinetic studies.
With respect to the pharmacokinetics of therapeutic proteins, two aspects of RME are of particular importance:
Distribution as a consequence of the drug binding to the receptor and subsequent internalization of the complex; and
Elimination as a consequence of endocytosis.
Unfortunately both processes typically cannot be differentiated experimentally in pharmacokinetics. Model B explicitly takes into account the amount of drug LRS distributed in the receptor system and the elimination by intracellular degradation, e.g., lysosomes. While the elimination is a linear process in terms of LRS, the distribution into the receptor system itself is a saturable process, specified in terms of Bmax and KM. Model C is derived from model B by assuming in addition that LRS is negligible in comparison to the extracellular amount Lex. In view of the above two sub-processes, this is equivalent to the assumption that the distributional aspect of RME can be neglected. Notably, even if the distributional aspect is negligible, the receptor system could still very efficiently transport ligand molecules into the cell, where they are subsequently degraded. This can be explained from Eq. 17. It states that the maximal elimination rate Vmax is the product of the maximal ligand binding capacity Bmax and the degradation rate constant kdeg. The maximal elimination rate Vmax may still be large due to a large kdeg, even if Bmax is small. The latter implies a negligible amount of ligand LRS within the receptor system. The receptor system acts as a mechanism that transports ligand molecules into the cell to eventually degrade them. Whether or not the receptor system also serves as a distribution phase is independent from the elimination aspect. This yields the following guidance for the usage of the two reduced models:
Model B: Elimination and distribution of ligand into the receptor system are important processes to be considered.
Model C: The distribution of ligand into the receptor system can be neglected, only the elimination process is important, which in this case is non-linear.
Based on Model B and the computable criterion (15) it can easily be checked whether the condition for the applicability of Model C are fulfilled. This has been demonstrated for the EGF/EGFR system in section “RME for the EGF/EGFR system”, see Figs. 4 and 5.
The reduced models are derived under the quasi-steady state assumption that the receptor redistribution processes are much faster than the ligand pharmacokinetics. This assumption is of the same type as the assumption underlying the Michaelis–Menten model of enzyme reactions, where it is assumed that the complex formation, dissociation and catalytic transformation are much faster than the transformation of substrate into product. In order to finally derive reduced models, we have to make an additional assumption on the time-scale of receptor synthesis and degradation. There are three different scenarios: receptor synthesis and degradation is (i) as fast as receptor redistribution (or faster); (ii) slower than the time scale of ligand pharmacokinetics; or (iii) at an intermediate time scale, i.e., comparable or faster than ligand PK but slower than receptor redistribution. The first two scenarios correspond to our fast and slow scenario. Under these assumptions it is possible to either treat receptor synthesis and degradation the same way as the redistribution processes (in the fast scenario) or neglect it and treat the total amount of receptor as a constant (in the slow scenario), since in the latter it would not impact the total number of receptors on the time scale of interest. In the third scenario, however, receptor synthesis and degradation would need to be taken into account in terms of an additional ODE. Unless further assumptions are made, this would require to consider the full system of Eqs. 1–5—which is not suitable for PK parameter estimation in clinical trials.
The elimination process of RME is specified in terms of the parameters Vmax and KM. Noteworthy, the maximal elimination rate Vmax is independent of the processes of complex formation (kon) and dissociation (koff) of the receptor-ligand complex. However, the parameters kon and koff influence the amount of extracellular ligand molecules KM, at which the elimination rate is half-maximal.
In Fig. 6, we studied the impact of different internalization rate constants kinterRL on RME. An altered kinterRL could, e.g., result from a mutation in the EGF receptor, as it has been observed experimentally [37]. Our analysis in section “Nonlinear PK caused by RME” shows that the ligand elimination rate is affected by various processes inside the cell. For example, the elimination rate decreases with decreasing complex internalization rate constant, but the difference is much less pronounced for a ligand with decreased association and dissociation rate constants kon and koff—even though the dissociation constant KD is the same in both scenarios (see Fig. 6, left vs. right). From the detailed Model A, this phenomenon is understandable: given a ligand that forms a complex with rate constant kon, once the ligand–receptor complex is formed at the membrane, its fate is a balance between dissociation (specified in terms of koff) and internalization (specified in terms of kinterRL). If, e.g., koff/kinterRL ≪ 1 then the complex will predominantly be internalized. Based on KD alone, this property of receptor systems can not be observed. The ratio koff/kinterRL has recently been introduced as one of two key parameters to characterize different cell surface receptor systems (termed the consumption parameter) [28]. In general, our analysis shows that reduced ligand elimination from the extracellular space can be due to altered processes inside the cell other than the velocity of internalization of the complex. The influences of the processes can be deduced from Eq. 28 and is summarized in Table 1. The nonlinearity increases with parameters that accelerate’ the processes of receptor availability at the surface (R0, krecyR) or that accelerate’ the transport and intracellular degradation of extracellular ligand (kon, kinterRL, klyso). Counteracting processes (related to the parameters koff, kinterR, krecyRL) decrease the extent of nonlinearity.
Target-mediated drug disposition (TMDD) models explicitly account for binding to a target and potential degradation of the resulting complex [16–18]. Although originally developed to describe effects of extensive drug target binding in tissues, TMDD has more recently also gained interest as a model of saturable elimination mechanisms for specific peptide and protein drugs, including RME [6, 18, 19]. Between TMDD and the herein presented approach, there are a number of distinct differences. First, the TMDD approach considers pharmacological target binding as the key process controlling the complex nonlinear processes. Particular features of receptor trafficking inside the cell are not taken into account. Second, whenever a drug molecule is degraded in the TMDD setting, both, a drug and a receptor molecule are degraded. In the herein presented approach, degradation of the drug does not necessarily imply degradation of the receptor, since the receptor can be recycled. This is, e.g., an important characteristics for the ligand TNF-α. Third, in [16], a reduced model of TMDD is presented based on a equilibrium assumption. In this reduced TMDD model, the unbound extracellular drug concentration is a function of the total concentration, the total receptor concentration Rtot and the equilibrium dissociation constant KD [16, Eq. 11]. In our reduced model B, in contrast, the extracellular drug concentration is a function of the total concentration, the maximal receptor binding capacity Bmax and the quasi-steady state parameter KM (cf. Eq. 8). As a consequence, the models make qualitatively different predictions. For instance, KM does not only depend on the ratio of koff and kon (i.e., KD), but also on the actual magnitude of the two parameters, in addition to the dependence on receptor systems parameters. This implies that two drugs with the same KD but different koff values might be impacted by RME very differently. This has been illustrated in Fig. 6 (compare left and right graphics) and discussed above.
If the reduced models of RME are used as part of structural PK models to estimate parameters in the course of clinical data analysis, the question arises whether or not the identified RME parameters Bmax, KM, kdeg and Vmax allow for a mechanistic interpretation, e.g., whether Bmax can be interpreted as the maximal RME ligand binding capacity. This question is tightly linked to the question of identifiability of model parameters, sometimes referred to as the inverse problem. Identifiability has been studied in detail in the context of compartmental models (see, e.g., [39, Chap. 5–9]). In general, the identifiability of model parameters depends on the structural model (number of compartments, compartment to which the RME process is linked, existence of additional routes of elimination, etc.), prior knowledge of model parameters and the quality of the experimental design [39, Chap. 5]. To illustrate this, we used the detailed model to generate a set of simulated data, to which we fitted the reduced Models B and C (data not shown). We found that for a well-designed experiment (i) the estimated parameters of the reduced RME models obey the expected relations (12)–(14) and (19); (ii) that Model C will not result in a good fit, if the condition Bmax/(KM + Lex) ≪ 1 is violated. This was the case for the in vitro data shown in Fig. 4(left), as well as for the in vivo data shown in Fig. 7(left), where already the authors in [11] reported that they were not able to fit a Michaelis–Menten based PK model to the experimental data. However, if the experimental design is not adequate, then we would expect—in accordance with the parameter identifiability problem [39, Chap. 5–9]—that the above conditions (i) and/or (ii) are violated. This was the case for the in vitro data shown in Fig. 4(right), where both Model B and Model C could be fitted to the generated data based on Model A, although the condition Bmax/(KM + Lex) ≪ 1 was violated (resulting in deviation of the estimated parameters from the expected parameters of 6−20% for Model B and 500% for Model C). Since the criteria in Eq. 15 has not been met, the violation of relations in Eqs. 19 and 13 for Model C is in accordance with our expectations. Furthermore, for the situation corresponding to Fig. 4(right), the expected relation Vmax = kdeg · Bmax (see Eq. 17) was violated, while it was satisfied for the situation corresponding to Fig. 7(left). These results eventually motivate the following recommendation:
- Consistency check: Use both reduced Models B and C to fit the data and check the two conditions (15) and (17):
A violation of the conditions might indicate an insufficient experimental design, and/or insufficient convergence of the fitting algorithm (local minimum).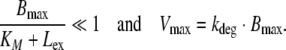
33
Different empirical models have been proposed and used to model the nonlinear pharmacokinetics of therapeutic proteins [7–15]. While, e.g., a Michaelis–Menten based RME model as part of a PK model allowed for describing data in one PK data analysis (e.g., [10]), it failed to do so in another (e.g., [11]). Due to lack of a sound theoretical basis to understand the different performances of empirical models, this certainly was an unsatisfactory situation. The herein presented analysis gives a thorough background of RME and a clear rationale as to when the proposed reduced models are applicable. In addition, the functional relations between the parameters of the detailed Model A and the reduced Models B and C might also serve as a first step to scale in vitro observations on RME to in vivo predictions of either target mediated disposition or Michaelis–Menten elimination, dependent upon the expression level and turnover of the target.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are grateful to the reviewers for their valuable comments on the manuscript.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
The originally published equations in [11, Supplement] are identical to a certain discretization of the system of ODEs (30)–(32). The advantage of stating the system as continuous ODEs is that subsequently any numerical scheme can be used to solve them, in particular high accuracy ODE solver with adaptive step size control.
Electronic supplementary material
The online version of this article (doi:10.1007/s10928-009-9120-1) contains supplementary material, which is available to authorized users.
Implementations of the models in Berkeley Madonna and Matlab are available online as supplementary material.
References
- 1.Meibohm B (2006) Pharmacokinetics and pharmacodynamics of biotech drugs. Wiley-VCH Verlag, Weinheim
- 2.Kuester K, Kloft C (2006) Pharmacokinetics of monoclonal antibodies. In Meibohm B (ed) Pharmacokinetics and pharmacodynamics of biotech drugs, chapter 3. Wiley-VCH Verlag, Weinheim, pp 45–91
- 3.Mahmood I, Green MD (2005) Pharmacokinetic and pharmacodynamic considerations in the development of therapeutic proteins. Clin Pharmacokinet 44:331–347 [DOI] [PubMed]
- 4.Russell-Jones GJ (2001) The potential use of receptor-mediated endocytosis for oral drug delivery. Adv Drug Deliver Rev 46:59–73 [DOI] [PubMed]
- 5.Sorkin A, Von Zastrow M (2002) Signal transduction and endocytosis: close encounters of many kinds. Nat Rev Mol Cell Biol 3:600–614 [DOI] [PubMed]
- 6.Tang L, Persky A, Hochhaus G, Meibohm B (2004) Pharmacokinetic aspects of biotechnology products. J Pharm Sci 93:2184-2204 [DOI] [PubMed]
- 7.Dirks NL, Nolting A, Kovar A, Meibohm B (2008) Population pharmacokinetics of cetuximab in patients with squamous cell carcinoma of the head and neck. J Clin Pharmacol 48:267–278 [DOI] [PubMed]
- 8.Kuester K, Kovar A, Lüpfert C, Brockhaus B, Kloft C (2008) Population pharmacokinetic data analysis of three phase I studies of matuzumab, a humanised anti-EGFR monoclonal antibody in clinical cancer development. Br J Cancer 98:900–906 [DOI] [PMC free article] [PubMed]
- 9.Mould DR, Sweeney KRD (2007) The pharmacokinetics and pharmacodynamics of monoclonal antibodies—mechanistic modeling applied to drug development. Curr Opin Drug Discov Dev 10:84–96 [PubMed]
- 10.Kloft C, Graefe E-U, Tanswell P, Scott AM, Hofheinz R, Amelsberg A, Karlsson MO (2004) Population pharmacokinetics of sibrotuzumab, a novel therapeutic monoclonal antibody, in cancer patients. Invest New Drugs 22:39–52 [DOI] [PubMed]
- 11.Lammerts van Bueren JJ, Bleeker WK, Bøgh HO, Houtkamp M, Schuurman J, van de Winkel JGJ, Parren PWHI (2006) Effect of target dynamics on pharmacokinetics of a novel therapeutic antibody against the epidermal growth factor receptor: implications for the mechanisms of action. Cancer Res 66:7630–7638 [DOI] [PubMed]
- 12.Ette EI, Williams PJ, Ho Kim Y, Lane JR, Liu M-J, Capparelli EV (2003) Model appropriateness and population pharmacokinetic modeling. J Clin Pharmacol 43:610–623 [PubMed]
- 13.Sheiner LB, Beal SL (1983) Evaluation of methods for estimating population pharmacokinetic parameters. III. Monoexponential model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm 11:303–319 [DOI] [PubMed]
- 14.Sheiner BL, Beal SL (1981) Evaluation of methods for estimating population pharmacokinetic parameters. II. Biexponential model and experimental pharmacokinetic data. J Pharmacokinet Biopharm 9:635–651 [DOI] [PubMed]
- 15.Sheiner LB, Beal SL (1980) Evaluation of methods for estimating population pharmacokinetics parameters. I. Michaelis–Menten model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm 8:553–571 [DOI] [PubMed]
- 16.Mager DE, Krzyzanski W (2005) Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm Res 22:1589–1596 [DOI] [PubMed]
- 17.Mager DE, Jusko WJ (2001) General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn 28:507–532 [DOI] [PubMed]
- 18.Mager DE (2006) Target-mediated drug disposition and dynamics. Biochem Pharmacol 72:1–10 [DOI] [PubMed]
- 19.Lobo ED, Hansen RJ, Balthasar JP (2004) Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 93:2645–2668 [DOI] [PubMed]
- 20.Hendriks BS, Orr G, Wells A, Wiley HS, Lauffenburger DA (2005) Parsing ERK activation reveals quantitatively equivalent contributions from epidermal growth factor receptor and HER2 in human mammary epithelial cells. J Biol Chem 280:6157–6169 [DOI] [PubMed]
- 21.Wiley HS, Shvartsman SY, Lauffenburger DA (2003) Computational modeling of the EGF-receptor system: a paradigm for systems biology. Trends Cell Biol 13:43–50 [DOI] [PubMed]
- 22.Sarkar CA and Lauffenburger DA (2003) Cell-level pharmacokinetic models of granulocyte colony-stimulating factor: implications for ligand lifetime and potency in vivo. Mol Pharmacol 63:147–158 [DOI] [PubMed]
- 23.De Luca A, Carotenuto A, Rachiglio A, Gallo M, Maiello MR, Aldinucci D, Pinto A, Normanno N (2008) The role of the EGFR signaling in tumor microenvironment. J Cell Physiol 214:559–567 [DOI] [PubMed]
- 24.Watanabe T, Shintani A, Nakata M, Shing Y, Folkman J, Igarashi K, Sasada R (1994) Recombinant human betacellulin. Molecular structure, biological activities, and receptor interaction. J Biol Chem 269:9966–9973 [PubMed]
- 25.Wells A (1999) EGF receptor. Int J Biochem Cell Biol 31:637–643 [DOI] [PubMed]
- 26.Harari PM (2004) Epidermal growth factor receptor inhibition strategies in oncology. Endocr Relat Cancer 11:689–708 [DOI] [PubMed]
- 27.Wiley HS, Cunningham DD (1981) A steady state model for analyzing the cellular binding, internalization and degradation of polypeptide ligands. Cell 25:433–440 [DOI] [PubMed]
- 28.Shankaran H, Resat H, Wiley HS (2007) Cell surface receptors for signal transduction and ligand transport: a design principles study. PLoS Comput Biol 3:e101 [DOI] [PMC free article] [PubMed]
- 29.Baselga J (2001) The EGFR as a target for anticancer therapy—focus on cetuximab. Eur J Cancer 37(Suppl 4):S16–S22 [DOI] [PubMed]
- 30.Baselga J (2002) Why the epidermal growth factor receptor? The rationale for cancer therapy. Oncologist 7(Suppl 4):2–8 [DOI] [PubMed]
- 31.Baselga J (2000) New therapeutic agents targeting the epidermal growth factor receptor. J Clin Oncol 18:54S–59S [PubMed]
- 32.Goel S, Mani S, Perez-Soler R (2002) Tyrosine kinase inhibitors: a clinical perspective. Curr Oncol Report 4:9–19 [DOI] [PubMed]
- 33.Raymond E, Faivre S, Armand JP (2000) Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs 60(1):15–23; discussion 41–2 [DOI] [PubMed]
- 34.Hendriks BS, Opresko LK, Wiley HS, Lauffenburger DA (2003) Coregulation of epidermal growth factor receptor/human epidermal growth factor receptor 2 (HER2) levels and locations: quantitative analysis of HER2 overexpression effects. Cancer Res 63:1130–1137 [PubMed]
- 35.Bleeker WK, Lammerts van Bueren JJ, van Ojik HH, Gerritsen AF, Pluyter M, Houtkamp M, Halk E, Goldstein J, Schuurman J, van Dijk MA, van de Winkel JGJ, Parren PWHI (2004) Dual mode of action of a human anti-epidermal growth factor receptor monoclonal antibody for cancer therapy. J Immunol 173:4699–4707 [DOI] [PubMed]
- 36.Bastholt L, Specht L, Jensen K, Brun E, Loft A, Petersen J, Kastberg H, Eriksen JG (2007) Phase I/II clinical and pharmacokinetic study evaluating a fully human monoclonal antibody against EGFr (HuMax-EGFr) in patients with advanced squamous cell carcinoma of the head and neck. Radiother Oncol 85:24–28 [DOI] [PubMed]
- 37.Wells A, Welsh JB, Lazar CS, Steven Wiley H, Gill GN, Rosenfeld MG (1990) Ligand-induced transformation by a noninternalizing epidermal growth factor receptor. Science 247:962–964 [DOI] [PubMed]
- 38.Reddy CC, Wells A, Lauffenburger DA (1994) Proliferative response of fibroblasts expressing internalization-deficient epidermal growth factor (EGF) receptors is altered via differential EGF depletion effect. Biotechnol Prog 10:377–384 [DOI] [PubMed]
- 39.Godfrey K (1983) Compartmental models and their application. Academic Press, London
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.