

Abstract
The functional imaging of neuronal circuits of the central nervous system is crucial for phenotype screenings or investigations of defects in neurodegenerative disorders. Current techniques yield either low penetration depth, yield poor resolution, or are restricted by the age of the animals. Here, we present a novel ultramicroscopy protocol for fluorescence imaging and three-dimensional reconstruction in the central nervous system of adult mice. In combination with tracing as a functional assay for axonal transport, retrogradely labeled descending motor neurons were visualized with >4 mm penetration depth. The analysis of the motor cortex shortly before the onset of clinical prion disease revealed that >80% neurons have functional impairments in axonal transport. Our study provides evidence that prion disease is associated with severe axonal transport defects in the cortical motor neurons and suggests a novel mechanism for prion-mediated neurodegeneration.
Introduction
Visualization of large areas in the central nervous system (CNS) is required for a wide range of studies including phenotype screenings, studies of neuronal circuits, or studies of transport processes in neuronal projections. Axonal transport plays a crucial role in neuron viability and differentiation (1) and is impaired at different neurodegenerative disorders, such as amyotrophic lateral sclerosis, Huntington's disease, or Alzheimer's disease (1–3). A powerful method to investigate the transport in neuronal projections is retrograde axonal tracing labeling neurons often located in large CNS areas. The imaging combining deep specimen penetration with high resolution is still not achieved. Confocal, electron microscopy, and objective-coupled planar illumination microscopy (4) keep high resolution at penetration depths of up to 0.15 mm. Histological sectioning necessary for investigations of larger CNS areas produce side effects causing information loss and compromising three-dimensional (3D) reconstruction (5). Magnetic resonance imaging, ultrasound imaging, optical coherence, and optical projection tomography reveal deeper penetration but do not yield cellular resolution (6).
Ultramicroscopy is a method using an orthogonal thin light sheet to visualize macroscopic specimens (7). Two types of optical layouts apply cylindrical lenses to focus an expanded laser beam into a light sheet. The simpler version overlaps the light sheet with the imaging plane of a microscope objective (8). Another version utilizes laser excitation from two opposing sides to reduce shadowing artifacts (9,10). The latter setup combined with procedure to clear brain tissues (11) achieved 2 mm penetration for mouse embryos or 0.41 mm for 35-day-old mice (9). The dependence on green fluorescent protein (GFP) expression and age limitations of the clearing efficacy restricted the method application to young animals.
Prion diseases, e.g., bovine spongiform encephalopathy in cattle and Creutzfeldt-Jakob disease (CJD) in humans, are fatal neurodegenerative disorders of the CNS. The clinical symptoms include motor dysfunctions and dementia (12). Spongiosis, neuronal cell loss, microglial activation, and proliferation of astrocytes are typical neuropathological hallmarks (13). The defects in axonal functions in vivo (14–16) and in vitro (17) were also reported as possible factors for the clinical manifestation of the disease. The role of spinal cord transport pathways in prion pathogenesis was implicated by the study on transgenic mice expressing the truncated prion protein. These mice did not demonstrate any detectable pathological changes in the brain, but did show prion-induced loss of motor neurons in the spinal cord (18). Other reports, however, implied that prion disease is independent of the neuronal transport (19,20).
In this work, we report novel ultramicroscopy setup, which achieves penetration depths of up to 4.2 mm in adult mouse brain and spinal cord specimens. Combining this powerful imaging technique with the tracing as a functional assay for the axonal transport, we show that 78–98% of motor cortex (MC) neurons reveal impaired axonal transport at the onset of clinical prion disease.
Material and Methods
Mouse inoculation and AAV application
All the procedures with laboratory animals were approved by the committee for the Care and Use of Laboratory Animals by the State of Bavaria, Germany. Mice used in the study: C57Bl/6 (wild-type, wt), Elevage Janvier, Le Genest Saint Isle, France; Tga20 transgenic mice, which express 10-fold PrP protein as compared with the wt (21). The animals were challenged with the Rocky Mountain Laboratory (RML) prion strain. The inoculum consisted of a 1% (w/v) brain homogenate from terminally scrapie-sick CD1 mice infected with the RML prion strain.
Animals were inoculated with 1 μl of either 1% (w/v) of RML prions or with 1% brain homogenate from healthy littermates (mock) into right sciatic nerve (i.n.) as previously described (22). In brief, animals were anesthetized with 40 mg/kg Ketanest (Parke-Davis, c/o Pfeizer, Freiburg, Germany) and 40 mg/kg Rompun (Bayer, Leverkusen, Germany). The right sciatic nerve was surgically exposed by dislodging Musculus gluteus superficialis and Musculus biceps femoris, placed onto a metal plate (2 × 5 × 1 mm) and prion inoculate was applied with 33-gauge Hamilton syringe over a period of 5 min. After inoculation, the sciatic nerve was repositioned and the wound was closed with Vicryl resorbable sutures (Johnson & Johnson, Duesseldorf, Germany).
Single-stranded adeno-associated virus (AAV) is widely used as a retrograde delivery system in mice and rats (23–26). It is also used to express AAV-CNTF-GFP or AAV-BDNF-GFP (27). The distribution of transgenic AAV in CNS and transport along neuronal projections was also extensively studied (28–30). Also the internalization as well as transport properties of AAVs are well investigated, which is reviewed elsewhere (31,32). Deletion of terminal resolution site in AAV results in the generation of a double-stranded AAV vector. Modified double-stranded AAVs are also used as a delivery system. They possess essentially the same transport properties, but higher speed of transgene delivery and expression (33). A modified AAV type 2 vector expressing DsRed-Express (AAV-REx) was used to target neurons in the cortico- and rubrospinal tract. Before the onset of clinical scrapie mice were anesthetized with ketanest/rompun, a cut was made on the level of the cervical spinal cord and 1 × 108 genome containing AAV particles were inoculated in a total volume of 1 μl into the cervical spinal cord with a 33-gauge Hamilton syringe over the period of 2 min. After inoculation, the wound was closed with Vicryl resorbable sutures (Johnson and Johnson, Duesseldorf, Germany).
Sample preparation
At the terminal stage of the prion disease, animals were sacrificed using an overdose of Ketanest/Rompun and transcardially perfused with PBS followed with 4% paraformaldehyde in sodium-phosphate buffer (pH 7.2). Brains and spinal cords were removed and postfixed in 4% paraformaldehyde for 1 h at 4°C. Brain and spinal cord samples for ultramicroscopy were washed for 2 h in phosphate-buffered saline (PBS) and subsequently in 30%, 50%, 70%, 80%, 96%, and two times in 100% alcohol 10 h each at room temperature. The specimens were prepared by excising the area of interest. After 1 h incubation in n-hexane (Sigma, Taufkirchen, Germany) the samples were transferred into clearing solution (CS) composed of one part of benzyl alcohol and two parts of benzyl benzoate, both from Sigma (9). To avoid air contact, because it showed to affect the clearing procedure, n-hexane was substituted with CS in 3–4 steps for 20–30 min each. Afterward, the sample was incubated in pure CS for 14–18 h at room temperature.
The samples for confocal microscopy were postfixed in 4% paraformaldehyde for 1 h at 4°C and incubated into 20% sucrose in PBS for cryoprotection for overnight at 4°C. Subsequently, the samples were frozen in Tissue Tek (Sakura, Zoeterwoude, Netherlands), and 20 μm-thick cryosections were made for confocal microscopy analysis.
Ultramicroscopy
The SPIM (selective plane illumination microscopy) or ultramicroscopy setup had an optical excitation path similar to those previously reported (8,34,35). The major differences included a high-power 8W Ar Ion Laser (Innova I-308, Coherent, Santa Clara, CA) and an objective inverter (LSM Tech, Stewartstown, PA) built into a commercial inverted microscope (Axiovert 200, Zeiss, Goettingen, Germany). The laser excitation beam (diameter 4 mm) was expanded 3.3-fold using a telescope (Thorlabs, Auburn, CA). A 10 mm-tall (with focus beam waist of 3–4 μm) light sheet was created by focusing the beam with a cylindrical lens (focal length 5 cm, Newport, Irvine, CA). It was adjusted exactly along the focus plane of a microscope objective by tilting the input angle of the excitation beam and positioning it exactly into the center of cylindrical lens with a mirror (Thor Labs, Auburn, CA). To focus the light sheet exactly on the specimen, the cylindrical lens was moved using a translation stage (Standa, Vilnius, Lithuania).
Epiplan 10X with 0.2 numerical aperture (NA) and LD Achroplan 20× with 0.4 NA objectives (Zeiss, Goettingen, Germany) were used in this work. They were placed 14 mm and 6 mm from a home-built sample chamber, respectively. The sample chamber was made of 36 × 25 mm rectangular 0.4 mm-thick cover glasses (Paul Marienfeld GmbH, Lauda-Koenigshofen, Germany) glued together with fast-drying acrylamide glue (Pattex, Henkel, Duesseldorf, Germany). After sample fixation and clearing (see “Sample preparation”), the specimen was affixed with acrylamide glue (Pattex, Henkel, Duesseldorf, Germany) to a home-built glass rod. The sample was positioned with a stepper motor-controlled stage located above the sample chamber (Standa, Vilnius, Lithuania). The stage enabled XY and rotational step motion with XY accuracy of 0.1 μm and rotational accuracy of 0.1° and also including a feedback-memory system. The stepper motors were controlled by the software package LabView (National Instruments, Austin, TX).
The emission light was directed from the objectives to the microscope via an objective inverter system allowing a flexible horizontal positioning of the objective at a greater distance and range than in an inverted microscope (LSM Tech, Stewartstown, PA). This setup enabled microscope to be precisely visually focused. The same beam path and filtering optics were used for visual observation and for the camera.
The emission light beam from the objective inverter went through a dichroic mirror (DCLP 555, Chroma, Rockingham, VT) and then through a band pass filter (HQ 607/75, Chroma) to remove any scattered excitation light from the laser. A back-illuminated electron multiplying charge-coupled device (EMCCD) camera with resolution of 512 × 512 pixels, 16 μm/pixel (Cascade II, Photometrics, Tucson AZ) was used in this work.
Laser illumination was synchronized with EMCCD camera exposure, as previously reported (35,36) to minimize illumination time and, consequently, photobleaching of the probe. The exposure time for image acquisition was 500 ms per frame. The synchronization of laser illumination and EMCCD camera exposure was administered by a solid-state electronically tunable acousto-optic filter (AOTF, AA Sa, Orsay, France). Electronic output signal from the EMCCD camera was filtered and temporally readjusted using a function generator (Hameg, Mainhausen, Germany).
Data acquisition and storage were administered by the software package MetaMorph 7.1 (Molecular Devices, Downingtown, PA).
Confocal microscopy
Serial 20 μm-thick cryosections were performed for the whole MC area of prion- and mock-challenged mice. Every third section was taken for anatomical analysis and confocal imaging. A series of five from selected sections were anatomically localized in the same MC region (evaluated region length 260 μm) for all studied samples. Serial confocal Z-stacks were taken with a step of 1 μm on a Leica SP5 laser-scanning confocal microscope (Leica, Mannheim, Germany) with a 20× (NA 0.7) immersion objective. The fluorescence emission of REx resulted from excitation from a 514 nm laser and was detected with 580–620 nm spectral band width on Leica TCS SP5 scanner and LAS AF software (Leica, Mannheim, Germany). Z-stacks were combined and processed with ImageJ software package (Psion Image, National Institutes of Health, Bethesda, MD).
Multiphoton microscopy
Multiphoton experiments were carried out using a specialized two-photon microscope that was customized in collaboration with the manufacturer on a commercial scanning system (TriMScope, LaVision BioTec, Bielefeld, Germany). We used photomultipliers (PMTs; Hamamatsu Photonics Deutschland, Hemsching am Ammersee, Germany) for detection and also a single beam for excitation (37).
Two modules each comprising four detector PMTs in nondescanned arrangement were used for the single-beam mode allowing simultaneous signal acquisition in forward as well as backward direction. Switchable dichroic mirrors and band pass filters were used to separate the signals before each PMT. A Ti:Sapphire laser (Chameleon, Coherent, Santa Clara, CA) and an optical parametric oscillator (OPO; APE, Berlin, Germany) were coupled into the scan head via two separate ports, and both were directed through two independent prechirp paths consisting of two prisms to compensate pulse broadening due to the intermediate optics and the objective lens. To minimize losses, all optical elements in the OPO beam pathway including tube and objective lens were adapted for high transmission and wavelength correction. The wavelength tuning was completely automated by the ImSpector scanner software (Max-Planck Institute for Biophysical Chemistry, Goettingen, Germany) allowing 2-photon excitation and emission spectrometry within short time ranges (37).
Both infra-red laser beams were overlapped on a short-pass dichroic mirror (DCSP 750, Chroma) for reflecting the scanning excitation beams into the objective (20× 0.95 W IR, Olympus Deutschland, Hamburg, Germany) and on the sample, and also to allow the transmission of the fluorescence to the detectors. The OPO was set to 1100 nm to specifically excite the REx. A band pass filter (HQ 620/60M, Chroma) was used to block out auto fluorescence and to specifically detect the REx. 512 × 512 pixel images for 500 μm × 500 μm image field were scanned at 800 lines/second in Z-stack steps of 3 μm.
Image analysis and quantification of neurons and REx-positive cells
Z-projection of each confocal stack was done on ImageJ software package (Psion Image). Obtained Z-projections for each side of M were combined in Adobe Photoshop (Adobe Systems, Munich, Germany) to obtain complete picture of tracer-positive neurons on the given side of given section. Obtained images were combined in one stack for each mouse and then in one file for simultaneous thresholding and quantification.
Ultramicroscopy stacks for the quantification were kept 250 μm thick and anatomically localized to match the anatomical region similar to the one evaluated by confocal microscopy. They were initially contrasted to remove background and leave only tracer-positive objects. Subsequently, all the stacks were combined in one file similar to the confocal one.
REx-positive cells were quantified with ImageJ software package (Psion Image). The mask parameter for counting was adjusted so that the number of AAV-REx-positive objects correlated with the number of PGP9.5-stained neurons in the same MC region of mock-inoculated animals. All statistical analyses were done using Prism software (GraphPad Software, San Diego, CA).
The ultramicroscopy stacks of MC were analyzed with Volocity software package (Improvision, Tübingen, Germany) where it is indicated. The fluorescent objects having the brightness between 40% and 100% were selected, and the objects larger than 200 μm3 were filtered out. The three-dimensional coordinates obtained in this analysis were transferred to MATLAB software (The MathWorks, Munich, Germany) to make 3D reconstruction of AAV-REx positive cells in MC.
As an internal control experiment, Ni2+-NTA agarose beads (QIAGEN, Hilden, Germany) were coated with his6-tagged DsRed protein (35), washed, and briefly centrifuged. The DsRed coated beads were imbedded in 1% low-melting agarose (Roth, Karlsruhe, Germany) and imaged with ultramicroscopy, multiphoton microscopy, and confocal microscopy under the same imaging and sample clearing conditions that were used for the imaging of REx-positive MC neurons. Because the confocal imaging was performed on thin cryosections in this study, the DsRed coated beads were also imaged on the surface of a cover glass. These experiments revealed that the relative fluorescence intensities are similar for the multiphoton and confocal imaging methods, but 1.5-fold higher for the ultramicroscopy (Supporting Material, Fig. S1).
Immunohistochemistry and PET-blots
For immunohistochemistry, 20 μm cryosections were used. The antibodies used were: mouse monoclonal anti-NeuN (working concentration: 33 μg/ml, Millipore, Schwalbach, Germany) and rabbit polyclonal anti-PGP9.5 (working concentration: 16 μg/ml, Abcam, Cambridge, UK). NeuN was visualized with biotin-SP-conjugated AffiniPure rabbit anti-mouse IgG (1:100, Dianova, Hamburg, Germany) combined with Cy5-conjugated streptavidin (1:100, Dianova). PGP9.5 was visualized with goat anti-rabbit IgG (1:100, Dianova) combined with Cy5-conjugated streptavidin (1:100, Dianova).
The paraffin-embedded tissue blot (PET-blot) method was used to visualize the prions in the brain tissues. PET-blot was performed as previously described elsewhere (38,39) with minor changes. Briefly, 5 μm-thick paraffin sections were transferred onto 0.45 μm pore nitrocellulose membranes (Schleicher und Schuell, Dassel, Germany), treated with Proteinase K (Roche Applied Sciences, Mannheim, Germany), and PrPSc was detected using the mouse monoclonal anti-PrP 6H4 antibody (Prionics, Schlieren, Switzerland).
The localization of prions on 20 μm-thick cryosections was done using polyclonal rabbit anti-PrP antibodies produced in our laboratory (R340) with the working concentration of 33 μg/ml. They were visualized with biotinylated goat anti-rabbit IgG (1:100, Dianova) combined with Cy5-conjugated streptavidin (1:100, Dianova). To remove the host PrP, the sections were treated with 100% formic acid for 8 min before antibody treatment. The mock controls were always stained in the same experiment with the sections from prion-infected animals.
Results
Axonal tracing as a functional assay for axonal transport upon prion challenge
Retrograde tracing as an assay for axonal transport was performed with a viral vector system based on the AAV type 2 (25) expressing the fluorescent marker DsRed-Express (REx). After injection into the spinal cord, AAV is retrogradely transported along axons, and REx expression labels the bodies of neurons in the spinal cord and several brain regions, including the red nucleus (RN) and MC, which we analyzed in this study. Prion inoculation into the right sciatic nerve results in the initially asymmetrical PrPSc accumulation in the RN and MC on the side contralateral to the inoculation. At the onset of clinical disease, the PrPSc is bilaterally localized in different CNS centers including spinal cord, RN, and MC (22,40). To investigate whether the axonal transport in cortico- and rubrospinal neurons is involved in prion pathogenesis, we applied AAV-REx to label the RN, MC, and gray matter in the cervical spinal cord cranially to the site of application (Fig. 1 A). Either C57Bl/6 wt or transgenic mice (Tga20) were used for tracer application at 136 and 54 days after prion infection, respectively. This time point is a few days before the onset of clinical disease, which allowed us to associate observed molecular effects with the onset of clinical disease symptoms, such as ataxia and partial paralysis of hind limbs.
Figure 1.
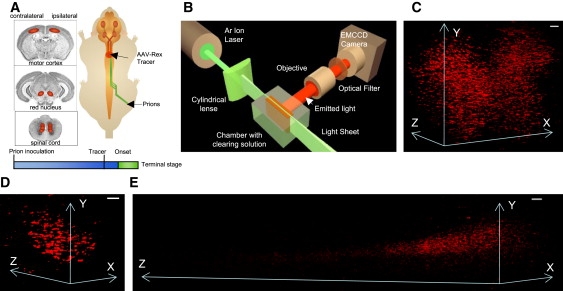
(A) Prions inoculated into the right sciatic nerve spread along axonal projections into the RN and the MC. Application of AAV-REx tracer into the spinal cord resulted in retrograde labeling of motor neurons in CNS centers. The areas analyzed in this study are shown on the images of coronar sections taken from www.hms.harvard.edu/ and http://biology.clc.uc.edu/, respectively. To visualize defects in axonal transport, the tracer was applied shortly before the onset of the disease (onset). (B) For the ultramicroscopy, the specimens placed into the chamber with CS were moved through light sheet obtained with cylindrical lens. Emitted light was collected with 10× objective located at 90° to the light sheet, let through optical filter allowing only REx to efficiently pass and imaged with an EMCCD camera. (C–E) 3D reconstructions of ultramicroscopy optical stacks showing REx-positive cells in spinal cord, 0.3 mm-thick stack, 225-day-old Tga20 mouse (C); RN, 0.2 mm-thick stack, 244-day-old Tga20 mouse (D), and MC, 4.2 mm-thick stack, 242-day-old wt mouse (E). Scale bars, 100 μm.
Improved sample preparation and imaging
We improved CNS imaging with optimized specimen preparation and clearing procedures as well as a superior ultramicroscopy setup. The specimens were prepared to maximally expose the area of interest. They were removed from the CS before reaching the state of complete clearance. Longer incubation times turned out to reduce the fluorescence and, as a consequence, seriously diminish signal/background. Clearing of brain samples was consistent for the animals aged between 180 and 400 days. Our ultramicroscopy setup (Fig. 1 B) relied on the one-sided single-beam version, because the fluorescence intensity across cleared tissues remained constant. High-power lasers; a tightly focused and high-intensity light sheet; a high-sensitivity and low-noise EMCCD camera; and a motorized, direct sample positioning device further contributed for the enhanced performance of this simple, yet high-resolution and deep penetration imaging system.
The improved ultramicroscopy protocol enabled us for the first time to visualize REx-positive neurons with 0.3 mm-deep penetration in the spinal cord (Fig. 1 C and Movie S1, 264-day-old Tga20 mouse). The brain tissue in RN was somewhat denser than in spinal cord; however, 0.2–0.3 mm deep penetration depth could be achieved for mice 200–396 days of age (Fig. 1 D, 225-day-old Tga20 mouse). Moreover, imaging in MC was accomplished with a penetration depth of 4.2 mm in 273-day-old wt mouse. Minor contrasting was required for quick and robust 3D reconstruction of the entire MC (Fig. 1 D and Movie S2). Achievement of such penetration depths establishes ultramicroscopy as a powerful tool for imaging neurons in large CNS areas with cellular resolution.
Axonal tracing and imaging in the RN
Large pyramidal neurons in the RN were located in an area of 0.2–0.35 mm diameter. Ultramicroscopy imaging showed bright and clearly recognizable neurons (Fig. 2 A, 396-day-old wt mouse) with resolution comparable to the confocal imaging (Fig. 2 B, 242-day-old wt mouse). The signal/background of ultramicroscopy images were comparable to the confocal microscopy, but the uniform background (the deviation between maximal and minimal background brightness only 1.2 ± 0.1-fold) allowed us to obtain clear images and to generate a robust 3D reconstruction. Such reconstructions resulted in important information on neuron location observed at different angular views (Fig. 2 C, the same specimen as in Fig. 2 A). In contrast to ultramicroscopy, the background of confocal images was extremely variable, differing by 67.5- ± 7.7-fold, which makes them more difficult to process and analyze. Moreover, the cryosectioning caused artifacts, which also compromised 3D reconstruction. Because the sections were acquired independently, it was impossible to exactly position them to obtain continuous stacks (Fig. S2). Finally, imaging of cryosections showed image quality variations within every single section. Consequently, 3D reconstructions of the confocal image stacks were inundated with so much background that REx-positive neuronal cells in the RN could be visualized only in a few positions upon intense manipulation and thresholding (Fig. 2 D, the same specimen as in Fig. 2, B).
Figure 2.
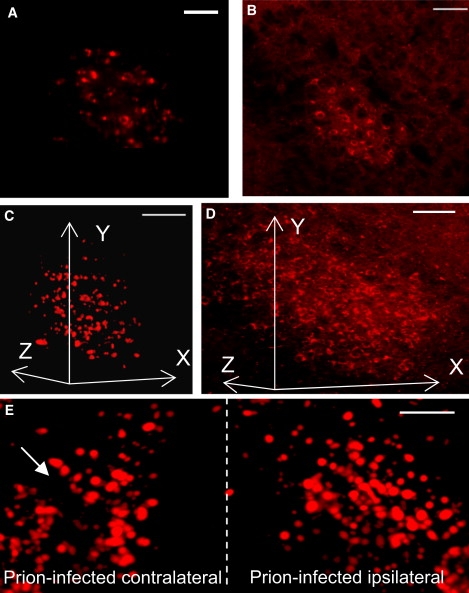
(A) Ultramicroscopy image from 0.2 mm-thick stack from RN of 396-day-old wt mock mouse. The image quality is comparable to the confocal imaging (B). (B) Confocal image of the RN, 70-day-old wt mock mouse. (C) 3D reconstruction of 0.2 mm-thick ultramicroscopy RN stack, the same specimen as in A. The ultramicroscopy enables 3D reconstruction of superior quality and containing lower background as compared to the confocal one (D). (D) 3D reconstruction of six serial confocal stacks from RN. Because of cryosectioning, the reconstruction contains high background. (E) 3D reconstruction of ultramicroscopy stacks from prion-challenged Tga20 mouse. The side of the RN contralateral to prion inoculation contains reduced amount of REx-positive cells (arrows). Scale bars, 100 μm.
The ultramicroscopy imaging revealed decreased numbers of REx-positive neurons in the RN only on the side contralateral to the prion inoculation (Fig. 2 E and Movie S3, 225-day-old Tga20 mouse). After the initial asymmetric distribution of prions, our data demonstrate functional defects in axonal projections of RN neurons contralaterally to the site of prion inoculation.
Ultramicroscopy enables both superior imaging and deep penetration into the MC area
The improved ultramicroscopy setup demonstrated several advantages for the imaging of the MC over the confocal microscopy. A penetration depth of up to 4.2 mm allowed the complete reconstruction of the MC area (Fig. 3 A, 273-day-old wt mouse). The ultramicroscopy images were of better quality as compared to the confocal ones (Fig. 3 B, 136-day-old wt mouse) in the beginning as well in the end of 3.8 mm-thick stack (Fig. 3, C and D, the same specimen as in Fig. 3 A). Signal/background (2.7 ± 0.5) was significantly higher than for the confocal images (1.2 ± 0.1), which was visible especially for prion-challenged animals (Fig. 3, B–D). Ultramicroscopy generated uniform images with a background deviation of 1.3- ± 0.1-fold, which were easy to process and perform in-depth analysis. The differing background in the confocal images (27.5- ± 8.4-fold deviation between maximal and minimal brightness in the background) as well as low signal/background seriously complicated data analysis and interpretation.
Figure 3.
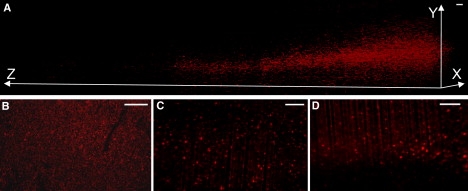
(A) 3D reconstruction of 3.8 mm-thick ultramicroscopy stacks showing REx-positive cells in the MC of 273-day-old wt mouse challenged with prions i.n. (AAV-REx application at 136 days post infection). (B) Confocal image of the MC from 264-day-old prion-infected wt mouse (AAV-REx application at 136 days post infection). (C and D) First (C) and last (D) ultramicroscopy images from MC stack done with prion-infected wt mouse (the same as in A). Graph coordinates indicate the distances in micrometers. Scale bars, 100 μm.
Majority of cortical motor neurons demonstrate impaired axonal transport at the onset of clinincal prion disease
The prion inoculation into the right sciatic nerve leads to prion accumulation in various CNS centers including spinal cord, lateral vestibular nucleus, RN, hindlimb MC, and thalamus (22,40). We observed prion accumulation in MC and RN of wt and Tga20 mice at the terminal stage of the disease after i.n. inoculation (Fig. S3), which correlates with the data on hamster (22) and murine (40) disease models. Upon inoculation in the sciatic nerve, wt and Tga20 animals demonstrated incubation times of 168 ± 5 and 67 ± 2 days post infection (dpi), respectively, until the terminal stages of the disease. The time points of the initial clinical disease onset were 149 dpi for wt and 59 dpi for Tga20 mice. We applied AAV-REx before the disease onset at 136 dpi and 54 dpi for wt and Tga20 mice, respectively (Fig. S4), and demonstrated the accumulation of prions in the MC and RN also at the same time points (Fig. S5). Visualization of REx positive neurons in the MC revealed uniform images within each experimental group (Fig. S6). The dramatic differences were detected between prion-challenged animals and mock controls (Fig. 4 and Fig. S6), which were consistent for both confocal (Fig. 4 A) and ultramicroscopy imaging (Fig. 4 B and Movie S4). Ultramicroscopy yielded images with superior quality ready for in-depth analysis. Upon image processing, we were able to implement automated cell-tracking and cell-positioning protocols to identify the location and number of cells in the MC and other CNS regions (Fig. 4 C).
Figure 4.
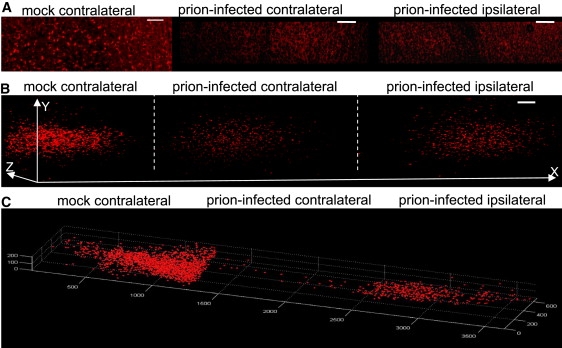
(A) Number of REx-positive neurons is reduced bilaterally in the MC of i.n. prion-infected mice as compared to mock control. Confocal imaging done with 288-day-old mock and 264-day-old prion-challenged wt mice (AAV-REx application at 136 days post infection). Scale bar, 100 μm. (B) 3D reconstructions of 0.2 mm-thick ultramicroscopy stacks also reveal the reduction of traced neurons in the MC upon prion challenge. The imaging was done on 244-day-old mock and 225-day-old i.n. prion-challenged Tga20 mice (AAV-REx application at 54 days post infection). Scale bar, 100 μm. (C) 3D cell position and image analysis of the data from Fig. 3C are shown in the MC of mock and prion-challenged mice (the same specimens as in B).
To quantify the REx positive neurons in mock and prion-challenged animals, we adapted automated label recognition for confocal and ultramicroscopy stacks. We colocalized REx deposits with NeuN-positive neurons in the MC and adapted the deposit properties for REx quantification. (Fig. 5 A). Moreover, the stacks from mock and prion-inoculated animals were combined in the single file, simultaneously edited and subsequently used for the quantification. The software properties for the label recognition were additionally adjusted to the quantity of neurons in the same MC level assessed by the staining with antibodies against PGP9.5, a neuronal marker (Fig. S7).
Figure 5.
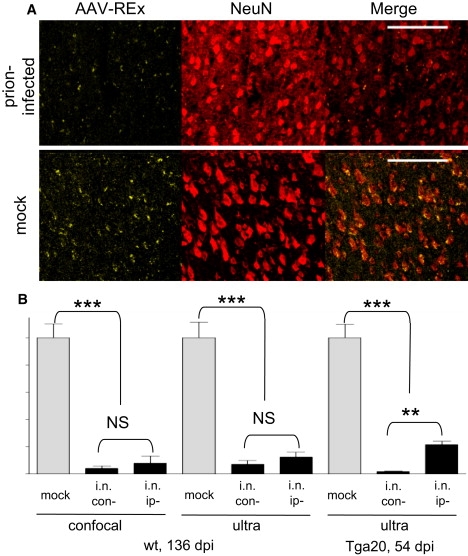
(A) REx deposits colocalize with NeuN-positive neurons in the MC of mock-challenged mouse. The deposit sizes allow distinguishing the REx-positive neurons from background fluorescence. Prion-infected animals show dramatically less labeled neurons. Scale bars, 100 μm. (B) Graph of the REx-positive neuron quantification. Despite the difference in the incubation times (168.8 days post infection, dpi and 67.9 dpi for wt and Tga20 mice, respectively), both mouse lines showed similar reduction of REx-positive neurons. Wt demonstrates 96% ± 4% impaired neurons on the contralateral side and 92% ± 11% on the ipsilateral side as evaluated on confocal images, (n = 4), and 93% ± 7% and 87 ± 6%, respectively, as evaluated on 0.25 mm-thick ultramicroscopy stacks (n = 5). Tga20 shows 98% ± 0.1% and 78% ± 4% impaired neurons, on the contra- and ipsilateral sides, respectively, as evaluated on 0.25 mm-thick ultramicroscopy stacks (n = 3). ∗∗∗p < 0.0001 ∗∗p = 0.0005 (unpaired t-test).
As shown in Fig. 5 B, quantification of tracer-positive neurons in the MC of wt animals by confocal microscopy revealed a decrease of REx-positive neurons for 96% ± 4% and 92% ± 11% on the side contralateral and ipsilateral to the prion challenge (n = 4), respectively. Confocal and ultramicroscopy analyses demonstrated severe impairments in retrograde axonal transport of prion-infected cortical motor neurons as judged by their inability to express REx in the MC neurons. Moreover, a similar 98% ± 0.1% and 78% ± 4% decrease for the contra- and ipsilateral sides, respectively, was also obtained for the Tga20 mice (Fig. 5 B). Such a dramatic decrease of REx-positive neurons cannot be explained simply by the cell loss in the MC. The quantification of NeuN- and PGP9.5-positive neurons in the MC revealed no significant differences between mock controls and prion-challenged animals (Fig. S8). Moreover, the analysis of REx-positive cells in the spinal cord revealed also no differences between mock and prion-inoculated littermates (Fig. S9). Because the spinal cord demonstrates the high PrPSc content (22,40), this experiment rules out the hypothesis that the observed decrease of REx-positive neurons in the MC could be attributed to the general prion influence on the uptake and/or expression directed by the AAV-REx.
Discussion
Investigations of the biological processes in vivo, especially of the nervous tissue containing a lot of interconnected cells with long projections, often requires imaging methods combining deep penetration and high resolution. The areas of interest in the CNS are frequently larger than the maximal penetration depth of 0.15 mm, currently achieved by laser-scanning confocal microscopy or objective-coupled planar illumination microscopy. The first ultramicroscopy protocol combined ≥2mm-deep penetration into the specimen with a cellular resolution for embryos or was restricted to 0.41 mm penetration depth for mice not older than 35 days (9). Consequently, this method could not be used to study animal models of infectious or neurodegenerative diseases.
The ultramicroscopy imaging technique presented in this work enabled the effective visualization and 3D analysis in adult mouse CNS up to 400 days of age. The improved ultramicroscopy protocol allowed remarkable penetration into the specimen beyond 4 mm and thereby establishes this imaging technique as the method of choice for studying such large CNS areas as MC or spinal cord. Along with the deep penetration, this method keeps the resolution on the confocal microscopy level without sectioning side effects. Areas of interest in MC or spinal cord are long (1.5–2 cm), making serial sectioning and analysis extremely laborious. Consequently, simplified approaches are used, like the imaging of every third MC section, which excludes a meaningful 3D reconstruction. A complete reconstruction of the confocal image stacks from the entire MC is extremely difficult to process due to the shifts incurred from each histological section, distracting background auto fluorescence, and simply big dimensions of the center implying a lot of sectioning.
The ultramicroscopy yielded images of high quality, which did not require any expensive and time-consuming image processing for specimen analysis and 3D reconstruction. Simple rescaling algorithms similar to previous reports (41) were enough to reduce background and remove shadowing artifacts. The ultramicroscopy can be accomplished within hours producing stacks suitable for immediate reconstruction and analysis making this technique also a suitable choice for analysis of statistically significant animal groups. Such analysis can be done with free software (ImageJ) or with more expensive and sophisticated software packages (Volocity), which enables further in-depth study, analysis, and quantification.
In contrast to the MC and spinal cord, imaging of the RN could be accomplished with confocal microscopy; however, sectioning side effects also largely compromises 3D reconstruction of the whole area. Here we demonstrate that our ultramicroscopy protocol also represents a valuable technique for the analysis of small and nonuniformly built centers, with resolution comparable to confocal microscopy and suitable for the effective 3D reconstruction of a whole brain center.
An alternative method, intravital deep-tissue multiphoton microscopy (37), revealed high resolution and 0.5 mm optical penetration depth in the MC, but yielded signal/background comparable to confocal imaging (2.0 ± 0.1) with high background deviations of 5.8- ± 1.7-fold (Fig. S10). Moreover, due to the high background, the RN and spinal cord tissues could not be imaged with this technique.
The combination of tracing, as a functional assay for the retrograde axonal transport, and ultramicroscopy, as an imaging technique, were successfully applied to study motor neurons in prion disease. We demonstrated for the first time, to our knowledge, that impairments of the transport processes in rubrospinal and corticospinal motor neurons correlated with the onset of clinical prion disease. Damaged neurons appeared unilaterally in the RN after initial prion distribution of prions upon unilateral challenge (22). The RN is an important movement coordination center in rodents, which partially lost its role in higher developed animal species, like primates or humans (42). The MC, on the other hand, is involved in the voluntary movement of both rodents and humans. The delivery of REx into the cell bodies was blocked in 78–98% of motor neurons due to functional impairments in retrograde transport. We observed similar decrease of REx-positive neurons for two different mouse lines despite dramatic differences in the incubation times. Such effects were observed before the onset of disease symptoms including ataxia and hind limb paralysis. Because the decrease of neuronal cells could not be observed on stainings with the neuronal marker NeuN in MC of prion-infected animals (Fig. 5 A, and Fig. S7 and Fig. S8), the phenomenon cannot be attributed to neuronal loss but rather to the functional impairments in retrograde axonal transport in living neurons. This conclusion is also supported by the fact that there is no difference of REx-positive cells in the gray matter of the spinal cord, the CNS center also infected by the prions. Recent studies in experimental models for prion diseases (43,44) as well as in sporadic CJD patients (15,16) suggest the role of the axonal defects in prion pathogenesis supporting the evidence for the transport impairments demonstrated in this work. These studies, however, were restricted to the description of either prion protein deposits (44) or changes in the axonal structure during the disease (16). The data presented in this work provide the first direct evidence, to our knowledge, proving that prion disease is associated with severe defects in the axonal transport of cortical motor neurons and establishes the transport impairments as a possible mechanism for the prion-mediated neurodegeneration. The role of axonal transport as a common pathogenesis target was demonstrated for various neurodegenerative diseases (1) including amyotrophic lateral sclerosis (45) and Alzheimer's disease (46).
Supporting Material
Ten figures and four movies are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00507-4.
Supporting Material
Acknowledgments
The authors thank Julia Merk and Eva Guhl for excellent technical support; Christoph P. Mauch and Monika Zelman-Femiak for helpful comments about the clearing procedure; Francesco Pampaloni and Ernst Stelzer for advice on methods of sample preparation, microscopy, and imaging; as well as Markus Hirschberg for help with the multiphoton imaging.
This research was funded by the German Research Foundation (DFG) to E.F. (Emmy Noether Program FL387/1-2), to M.K. and E.F. (SFB581, TP-A6), and to G.H. (FZ-82), and by the European Commission 6th Framework Program to T.C. (ZNIP–037783).
Footnotes
V. Ermolayev's present address is Molecular Microscopy Group, Rudolf Virchow Center, DFG Research Center for Experimental Biomedicine, University of Würzburg, Würzburg, Germany.
E. Flechsig's present address is Biotest AG, Dreieich, Germany.
References
- 1.Goldstein L.S. Do disorders of movement cause movement disorders and dementia? Neuron. 2003;40:415–425. doi: 10.1016/s0896-6273(03)00630-5. [DOI] [PubMed] [Google Scholar]
- 2.Gunawardena S., Goldstein L.S. Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch. Neurol. 2005;62:46–51. doi: 10.1001/archneur.62.1.46. [DOI] [PubMed] [Google Scholar]
- 3.Stokin G.B., Goldstein L.S. Axonal transport and Alzheimer's disease. Annu. Rev. Biochem. 2006;75:607–627. doi: 10.1146/annurev.biochem.75.103004.142637. [DOI] [PubMed] [Google Scholar]
- 4.Holekamp T.F., Turaga D., Holy T.E. Fast three-dimensional fluorescence imaging of activity in neural populations by objective-coupled planar illumination microscopy. Neuron. 2008;57:661–672. doi: 10.1016/j.neuron.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Weninger W.J., Mohun T. Phenotyping transgenic embryos: a rapid 3-D screening method based on episcopic fluorescence image capturing. Nat. Genet. 2002;30:59–65. doi: 10.1038/ng785. [DOI] [PubMed] [Google Scholar]
- 6.Helmchen F., Denk W. Deep tissue two-photon microscopy. Nat. Methods. 2005;2:932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- 7.Siedentopf H., Zsigmondy R. Über Sichtbarmachung und Größenbestimmung ultramikroskopischer Teilchen, mit besonderer Anwendung auf Goldrubingläser. Ann. Phys. 1903;10:1–39. [Google Scholar]
- 8.Huisken J., Swoger J., Del Bene F., Wittbrodt J., Stelzer E.H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305:1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- 9.Dodt H.U., Leischner U., Schierloh A., Jahrling N., Mauch C.P. Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nat. Methods. 2007;4:331–336. doi: 10.1038/nmeth1036. [DOI] [PubMed] [Google Scholar]
- 10.Huisken J., Stainier D.Y. Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM) Opt. Lett. 2007;32:2608–2610. doi: 10.1364/ol.32.002608. [DOI] [PubMed] [Google Scholar]
- 11.Spalteholz, W. 1914 Über das Durchsichtigmachen von menschlichen und tierischen Präparaten und seine theoretischen Bedingungen. S. Hierzel, Leipzig.
- 12.Flechsig E., Weissmann C. The role of PrP in health and disease. Curr. Mol. Med. 2004;4:337–353. doi: 10.2174/1566524043360645. [DOI] [PubMed] [Google Scholar]
- 13.Aguzzi A., Heikenwalder M., Polymenidou M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 2007;8:552–561. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- 14.Clinton J., Forsyth C., Royston M.C., Roberts G.W. Synaptic degeneration is the primary neuropathological feature in prion disease: a preliminary study. Neuroreport. 1993;4:65–68. doi: 10.1097/00001756-199301000-00017. [DOI] [PubMed] [Google Scholar]
- 15.Iwasaki Y., Yoshida M., Hashizume Y., Kitamoto T., Sobue G. Pyramidal tract degeneration in sporadic Creutzfeldt-Jakob disease. Neuropathology. 2007;27:434–441. doi: 10.1111/j.1440-1789.2007.00812.x. [DOI] [PubMed] [Google Scholar]
- 16.Kovacs G.G., Budka H. Prion diseases: from protein to cell pathology. Am. J. Pathol. 2008;172:555–565. doi: 10.2353/ajpath.2008.070442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novitskaya V., Makarava N., Sylvester I., Bronstein I.B., Baskakov I.V. Amyloid fibrils of mammalian prion protein induce axonal degeneration in NTERA2-derived terminally differentiated neurons. J. Neurochem. 2007;102:398–407. doi: 10.1111/j.1471-4159.2007.04537.x. [DOI] [PubMed] [Google Scholar]
- 18.Flechsig E., Shmerling D., Hegyi I., Raeber A.J., Fischer M. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron. 2000;27:399–408. doi: 10.1016/s0896-6273(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 19.Hafezparast M., Brandner S., Linehan J., Martin J.E., Collinge J. Prion disease incubation time is not affected in mice heterozygous for a dynein mutation. Biochem. Biophys. Res. Commun. 2005;326:18–22. doi: 10.1016/j.bbrc.2004.10.206. [DOI] [PubMed] [Google Scholar]
- 20.Kunzi V., Glatzel M., Nakano M.Y., Greber U.F., Van Leuven F. Unhampered prion neuroinvasion despite impaired fast axonal transport in transgenic mice overexpressing four-repeat tau. J. Neurosci. 2002;22:7471–7477. doi: 10.1523/JNEUROSCI.22-17-07471.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer M., Rülicke T., Raeber A., Sailer A., Moser M. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 22.Bartz J.C., Kincaid A.E., Bessen R.A. Retrograde transport of transmissible mink encephalopathy within descending motor tracts. J. Virol. 2002;76:5759–5768. doi: 10.1128/JVI.76.11.5759-5768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borrell V., Kaspar B.K., Gage F.H., Callaway E.M. In vivo evidence for radial migration of neurons by long-distance somal translocation in the developing ferret visual cortex. Cereb. Cortex. 2006;16:1571–1583. doi: 10.1093/cercor/bhj094. [DOI] [PubMed] [Google Scholar]
- 24.Dodge J.C., Haidet A.M., Yang W., Passini M.A., Hester M. Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol. Ther. 2008;16:1056–1064. doi: 10.1038/mt.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaspar B.K., Erickson D., Schaffer D., Hinh L., Gage F.H. Targeted retrograde gene delivery for neuronal protection. Mol. Ther. 2002;5:50–56. doi: 10.1006/mthe.2001.0520. [DOI] [PubMed] [Google Scholar]
- 26.Kaspar B.K., Vissel B., Bengoechea T., Crone S., Randolph-Moore L. Adeno-associated virus effectively mediates conditional gene modification in the brain. Proc. Natl. Acad. Sci. USA. 2002;99:2320–2325. doi: 10.1073/pnas.042678699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leaver S.G., Cui Q., Plant G.W., Arulpragasam A., Hisheh S. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006;13:1328–1341. doi: 10.1038/sj.gt.3302791. [DOI] [PubMed] [Google Scholar]
- 28.Chamberlin N.L., Du B., de Lacalle S., Saper C.B. Recombinant adeno-associated virus vector: use for transgene expression and anterograde tract tracing in the CNS. Brain Res. 1998;793:169–175. doi: 10.1016/s0006-8993(98)00169-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rolling F., Shen W.Y., Tabarias H., Constable I., Kanagasingam Y. Evaluation of adeno-associated virus-mediated gene transfer into the rat retina by clinical fluorescence photography. Hum. Gene Ther. 1999;10:641–648. doi: 10.1089/10430349950018715. [DOI] [PubMed] [Google Scholar]
- 30.Ward, H.L., J.V. Gardiner, W.M. Kong, K.G. Murphy, and S.R. Bloom. 2002. The pattern of green fluorescent protein (GFP) expression following intrahypothalamic injection of adeno-associated virus-GFP. Endocrine Abstracts. 3:186.
- 31.Ding W., Zhang L., Yan Z., Engelhardt J.F. Intracellular trafficking of adeno-associated viral vectors. Gene Ther. 2005;12:873–880. doi: 10.1038/sj.gt.3302527. [DOI] [PubMed] [Google Scholar]
- 32.Goncalves M.A. Adeno-associated virus: from defective virus to effective vector. Virol. J. 2005;2:43–60. doi: 10.1186/1743-422X-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z., Ma H.I., Li J., Sun L., Zhang J. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. 2003;10:2105–2111. doi: 10.1038/sj.gt.3302133. [DOI] [PubMed] [Google Scholar]
- 34.Engelbrecht C.J., Stelzer E.H. Resolution enhancement in a light-sheet-based microscope (SPIM) Opt. Lett. 2006;31:1477–1479. doi: 10.1364/ol.31.001477. [DOI] [PubMed] [Google Scholar]
- 35.Steinmeyer R., Noskov A., Krasel C., Weber I., Dees C. Improved fluorescent proteins for single-molecule research in molecular tracking and co-localization. J. Fluoresc. 2005;15:707–721. doi: 10.1007/s10895-005-2978-4. [DOI] [PubMed] [Google Scholar]
- 36.Michel R., Steinmeyer R., Falk M., Harms G.S. A new detection algorithm for image analysis of single, fluorescence-labeled proteins in living cells. Microsc. Res. Tech. 2007;70:763–770. doi: 10.1002/jemt.20485. [DOI] [PubMed] [Google Scholar]
- 37.Friedl P., Wolf K., von Andrian U.H., Harms G. Biological second and third harmonic generation microscopy. Curr. Protoc. Cell Biol. 2007;4:1–21. doi: 10.1002/0471143030.cb0415s34. [DOI] [PubMed] [Google Scholar]
- 38.Lezmi S., Bencsik A., Baron T. PET-blot analysis contributes to BSE strain recognition in C57Bl/6 mice. J. Histochem. Cytochem. 2006;54:1087–1094. doi: 10.1369/jhc.5A6892.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schulz-Schaeffer W.J., Tschoke S., Kranefuss N., Drose W., Hause-Reitner D. The paraffin-embedded tissue blot detects PrP(Sc) early in the incubation time in prion diseases. Am. J. Pathol. 2000;156:51–56. doi: 10.1016/S0002-9440(10)64705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glatzel M., Aguzzi A. PrP(C) expression in the peripheral nervous system is a determinant of prion neuroinvasion. J. Gen. Virol. 2000;81:2813–2821. doi: 10.1099/0022-1317-81-11-2813. [DOI] [PubMed] [Google Scholar]
- 41.Verveer P.J., Swoger J., Pampaloni F., Greger K., Marcello M. High-resolution three-dimensional imaging of large specimens with light sheet-based microscopy. Nat. Methods. 2007;4:311–313. doi: 10.1038/nmeth1017. [DOI] [PubMed] [Google Scholar]
- 42.Kennedy P.R. Corticospinal, rubrospinal and rubro-olivary projections: a unifying hypothesis. Trends Neurosci. 1990;13:474–479. doi: 10.1016/0166-2236(90)90079-p. [DOI] [PubMed] [Google Scholar]
- 43.Lombardi G., Casalone C., D'Angelo A., Gelmetti D., Torcoli G., Barbieri I., Corona C., Fasoli E., Farinazzo A., Fiorini M., Gelati M., Iulini B., Tagliavini F., Ferrari S., Caramelli M., Monaco S., Capucci L., Zanusso G. Intraspecies transmission of BASE induces clinical dullness and amyotrophic changes. PLoS Pathog. 2008;4:e1000075. doi: 10.1371/journal.ppat.1000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medrano, A.Z., S.J. Barmada, E. Biasini, and D.A. Harris. 2008. GFP-tagged mutant prion protein forms intra-axonal aggregates in transgenic mice. Neurobiol Dis. [DOI] [PMC free article] [PubMed]
- 45.De Vos K.J., Grierson A.J., Ackerley S., Miller C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- 46.Stokin G.B., Lillo C., Falzone T.L., Brusch R.G., Rockenstein E. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.