

Abstract
We have developed a multiscale structure prediction technique to study solution- and adsorbed-state ensembles of biomineralization proteins. The algorithm employs a Metropolis Monte Carlo-plus-minimization strategy that varies all torsional and rigid-body protein degrees of freedom. We applied the technique to fold statherin, starting from a fully extended peptide chain in solution, in the presence of hydroxyapatite (HAp) (001), (010), and (100) monoclinic crystals. Blind (unbiased) predictions capture experimentally observed macroscopic and high-resolution structural features and show minimal statherin structural change upon adsorption. The dominant structural difference between solution and adsorbed states is an experimentally observed folding event in statherin's helical binding domain. Whereas predicted statherin conformers vary slightly at three different HAp crystal faces, geometric and chemical similarities of the surfaces allow structurally promiscuous binding. Finally, we compare blind predictions with those obtained from simulation biased to satisfy all previously published solid-state NMR (ssNMR) distance and angle measurements (acquired from HAp-adsorbed statherin). Atomic clashes in these structures suggest a plausible, alternative interpretation of some ssNMR measurements as intermolecular rather than intramolecular. This work demonstrates that a combination of ssNMR and structure prediction could effectively determine high-resolution protein structures at biomineral interfaces.
Introduction
High-resolution structures of surface-adsorbed proteins cannot be determined by contemporary experimental methods (1), presenting a unique challenge for computational structure prediction in biomineralization systems. On one hand, the absence of training sets and/or benchmarks derived from experimental data makes rigorous algorithmic testing and parameterization difficult. On the other hand, this limitation places special emphasis on the computational investigation of protein-surface interactions, as computation is currently the only means of obtaining atomic coordinates for protein-adsorbed states.
Protein-surface simulations that model the protein with entirely atomistic or entirely coarse-grained representations address different problems (2). Atomistic simulations can calculate adsorption energies (3–9), compare face or phase preferences of proteins for materials (6,7), can include explicit solvent (4,5,10), etc., but require too much computer time to predict entire structures a priori. Coarse-grained models can predict bulk phenomena of polymers/proteins interacting with surfaces (11–13) or enumerate all conformations of hypothetical polymers on two-dimensional square lattices (12), but cannot resolve atomic aspects of molecular systems. As in other disciplines of computational biology, a multiscale approach can be used to rapidly search conformation space while maintaining atomic resolution (14,15).
A handful of solid-state NMR (ssNMR) techniques can determine the relative position of atoms at the protein-surface interface (16–26). Of protein-surface systems, the human salivary protein statherin interacting with hydroxyapatite (HAp) has the greatest number of published ssNMR measurements: three protein-surface intermolecular distances (19,21,22), seven protein intramolecular distances, and five protein backbone torsion angles (20,24,25), making the statherin-HAp system the most amenable for comparison with high-resolution protein-surface structure prediction algorithms. Statherin, a 43-residue doubly phosphorylated salivary protein (27), inhibits HAp crystallization and spontaneous calcium phosphate precipitation in vivo (28) and binds HAp in vitro (29). HAp is the primary mineral component in all mammalian skeletal and dental tissues (30). Statherin's structure is thought to affect fimbriae mediated bacterial colonization of dentin, a cause of human periodontal disease (31).
In a previous study, our lab modified the Rosetta structure prediction suite (32–38) to dock proteins to solid surfaces (RosettaSurface) and predicted a set of statherin conformers bound to the (001) monoclinic face of HAp (39). Although that work represented a significant improvement in protein-structure prediction at solid surfaces, simplifications were necessary to make the system tractable. Specifically, statherin had been prefolded in implicit solvent using Rosetta ab initio (20) and refined using four ssNMR intramolecular distance constraints (acquired from statherin bound to HAp); the docking simulation that ensued assumed a fixed backbone and included only a single crystal face of HAp. One critique of that approach is that the number of protein conformations satisfying only four intramolecular distance constraints is expected to be large, and selecting only one conformation neglects the effect of HAp on statherin's backbone conformation. Some of these effects might include surface catalyzed structural transitions and conformational selection by HAp for statherin. Similarly, specificity/promiscuity cannot be thoroughly tested by simulating adsorption to a single HAp face, and it is not known which face(s) is biologically relevant for statherin adsorption. Also, a fixed-backbone simulation requires advanced structural knowledge, an advantage not common in protein-surface studies.
In this study, we present what is, to our knowledge, the first structure-prediction-based algorithm capable of folding a protein in the presence of a solid surface. The algorithm can rapidly generate and energy-minimize protein folds and docked orientations, representing a significant improvement in the conformational space accessible during simulation. Because surfaces are suspected to catalyze structural transitions, this RosettaSurface protocol generates large ensembles in the solution and adsorbed states. With this protocol, we address the following questions: i), How similar are the solution- and adsorbed-state structures? ii), How does the protein structure differ at three different HAp surfaces ((001), (010), and (100))? iii), Is the RosettaSurface algorithm robust enough to capture known structural features without any bias beginning from a fully extended peptide chain in solution? Finally, we add ssNMR constraints to the RosettaSurface simulation. The outcome of constrained simulation helps interpret the RosettaSurface and ssNMR structural data and provides proof of concept for a combined technique to aid in adsorbed-state structure determination.
Materials and Methods
Each execution of the RosettaSurface algorithm developed for this study folds a peptide from a fully extended conformation and results in one energy-minimized candidate solution- and adsorbed-state structure. Large ensembles of 105 candidate solution- and adsorbed-state structures were generated from which the lowest-energy structures, from each state, were chosen for further analysis. See the Supporting Material for a complete description of the new structure prediction methods developed for this study, construction of all materials (peptides, surfaces, etc.), and data analysis methods.
Results
We applied the RosettaSurface protocol to statherin interacting with the HAp (001) monoclinic surface. We also demonstrated the algorithmic portability by adsorbing a control peptide to the HAp (001) surface. In addition, we ran two NMR-biased simulations (adsorbing statherin to HAp's (001) face) to assess the quality of unbiased RosettaSurface predictions and to help infer structure from ssNMR measurements. Finally, we applied the RosettaSurface protocol to two other HAp surfaces ((010) and (100)) to probe statherin specificity.
In all simulations, we generated 105 decoys for both the solution and adsorbed state. Approximately 3.5 × 104 conformers were sampled for each of the 105 decoys generated; therefore, ∼3.5 × 109 conformers were assessed by the RosettaSurface energy function in each “run”. For all runs, the top scoring 0.1% (100 decoys) from each state was selected for further analysis. RosettaSurface generation of every 104 decoys produced structurally similar top-scoring (i.e., converged) decoys. We generated 105 decoys to enrich top-scoring decoys for subsequent statistical analysis.
Because the middle segment of statherin is thought to be unstructured, analysis is often carried out with respect to three individual statherin segments (40,41). Here we adopt a similar convention when applicable: residues 1–15 referred to as N-terminal, 16–29 as middle, and 30–43 as C-terminal.
Statherin in implicit solvent
Fig. 1 details the predicted structure of statherin in solution. Fig. 1 a shows the population of three secondary structure motifs at each residue, averaged over the 100 top-scoring decoys from each of three runs. The predicted structure of the N-terminal segment is mostly helical from residues 4–12, with a “frayed” helical motif from residues 13–15; residues 1 and 2 are mostly unstructured. The C-terminal segment exhibits moderate helical structure, which initiates at Pro-36 and fluctuates between turn- and helix-like hydrogen-bond configurations. The middle segment is more difficult to characterize due to structural dispersion. The Ramachandran plot (Fig. 1 b) shows that Rosetta predicts a mostly polyproline II (PPII) and β-turn structure in the middle segment, with moderate right- and left-handed helical structure. The occurrence of torsion angles indicative of canonical secondary structure (Fig. 1 b) coupled with a lack of persistent local hydrogen-bonded secondary structure (Fig. 1 a) may arise from the abundance of proline residues (seven from residues 20–36). Coordinates for the 10 top-scoring decoys (Fig. 1 c) show regular helical structure in the N-terminal segment, a structurally dispersed middle segment, and partially structured C-terminal segment.
Figure 1.
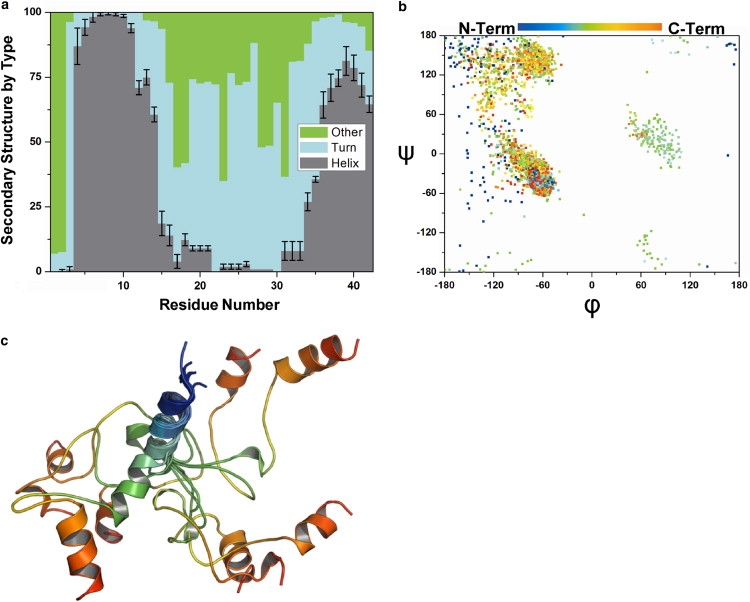
Predicted statherin solution-state structure. (a) Distributions of three secondary structure motifs averaged for three independent simulations (error bars represent standard deviation from the mean, for clarity, shown only for helix). (b) Ramachandran plot for 100 top-scoring structures shows regions of populated (ϕ,ψ) space at each residue (blue to red = N- to C-terminus). (c) Coordinates for 10 top-scoring solution-state decoys superimposed about the N-terminal binding domain.
Previous experimental measurements on solvated statherin support these predictions. Circular dichroism (CD) spectra of a solvated statherin fragment comprising the N-terminal segment display a significant population of helix (17,41). NMR experiments on full-length statherin in the helix-stabilizing solvent 2,2,2-trifluoroethanol (TFE) suggest a PPII type secondary structure for statherin's middle segment (40). CD experiments on a statherin fragment comprising the middle segment found a β-turn structure in TFE and a PPII structure in phosphate buffered saline (41). CD spectra of a statherin fragment comprising the C-terminal segment show turn-like secondary structure (41), whereas NMR experiments on full-length statherin in TFE predict a helical structure from residues 36–43 (40).
Structure and binding of statherin at the HAp (001) surface
Fig. 2 a shows the change in secondary structure upon adsorption (upper panel), average change in side-chain solvent-accessible surface area (Å2) upon adsorption (middle panel), and the frequency that each residue adsorbed to HAp in the 100 top-scoring decoys (lower panel); all are plotted against residue number.
Figure 2.
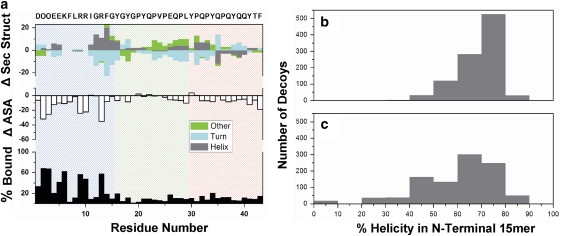
Predicted binding and structural statistics for statherin adsorbed to HAp. (a) Three adsorption phenomena plotted against residue for the 100 top-scoring decoys adsorbed to the (001) surface. The upper panel shows the average change in secondary structure upon adsorption. The middle panel shows the average change in solvent-accessible surface area (Å2) upon adsorption. The lower panel shows the adsorption frequency. Background shading (blue, green, and red) indicates the defined statherin segments (N-terminal, middle, and C-terminal respectively). Statherin amino-acid (one-letter code) sequence plotted along upper x axis. (b) Histogram showing distribution of N-terminal helicity for 1000 top-scoring and (c) 1000 randomly selected decoys.
In the N-terminal binding domain, the dominant structural difference between states occurs between residues 12 and 14 (upper panel, Fig. 2 a). RosettaSurface predicts that Gly-12 is a helical cap in the solution state, a common role of glycine residues at the carboxy terminus of helices (42). The frayed helix motif at residues 12–14 is stabilized upon adsorption (Fig. 3).
Figure 3.
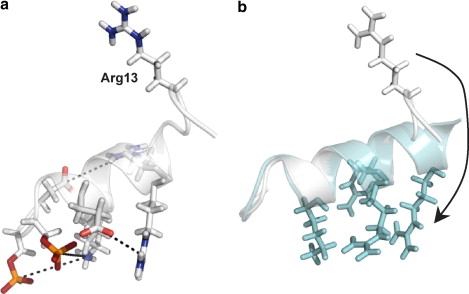
Adsorption induced folding event about statherin's Gly-12 as predicted by RosettaSurface. (a) RosettaSurface predicts that the statherin solution-state N-terminal 15-mer is partially stabilized by an electrostatic network (dashed lines) among its ionic residues (shown in stick representation). (b) Superposition of representative solution- (gray) and adsorbed-state (teal) decoys depicting predicted adsorption induced folding event.
In solution (Fig. 3 a), we find that electrostatics cause the binding residues to orient in a manner that complements the chemistry and geometry of the HAp surface (lattice matching), with the exception of Arg-13. Arg-13 is the only ionic residue in the binding domain without a residue of complementary charge nearby. Therefore, Arg-13 has a higher tendency to position its side chain away from Arg-9 and Arg-10 and thus away from the binding interface. Upon adsorption (Fig. 3 b), however, HAp shields local protein electrostatics and facilities a conformational change extending the helix, placing the positively charged arginine side chain in contact with negatively charged HAp phosphates. NMR measurements support an adsorption-induced shift toward helical ϕ angles at glycine 12 (25).
With the exception of the above-mentioned structural difference between states, predicted solution- and adsorbed-state structures are similar. In our previous study (39), we found that a helical fold for the N-terminal segment facilitates a lattice-matching arrangement of binding residues. Here we find that lattice-matching conformers also dominate the solution-state energy minima, and that a combination of van der Waals forces, electrostatic, and hydrogen-bond interactions cause unfolded states to score poorly relative to folded states at the interface and in solution.
The structural similarity between solution- and adsorbed-states might come from insufficient sampling; however, analysis to this point had been restricted to decoys in the top-scoring 0.1%. We compared the 1000 top-scoring decoys with 1000 randomly selected decoys to see if diverse structures were being sampled at the interface but not being selected by the RosettaSurface energy function. Fig. 2, b and c, show the distribution of helical content for the N-terminal binding domain, in the context of full-length statherin, for the 1000 top-scoring and 1000 randomly selected adsorbed-state decoys respectively. More than 55% percent of the top-scoring 1000 (Fig. 2 b) decoys have greater than 70% helicity in the adsorbed-state N-terminal binding domain, whereas only 30% of the randomly chosen decoys meet this criteria (Fig. 2 c). Also, 2% of the randomly chosen decoys have a completely unfolded (i.e., 0% helicity) adsorbed-state N-terminal binding domain, whereas completely unfolded decoys are absent in the top 1000. Thus, sampling includes both nonhelical and helical states, but helical states are selected by the RosettaSurface energy function.
Fig. 2 a also shows the predicted binding pattern of HAp-adsorbed statherin at each residue (middle and lower panels). The middle panel in Fig. 2 a shows the change in solvent accessibility upon adsorption, and the lower panel shows the adsorption frequency of each residue. In general, high binding frequency at a residue should correspond to a decrease in solvent accessibility relative to the solution state; however, the correlation can vary based on changes in protein fold. RosettaSurface predicts that the acidic residues Sep-2 and Sep-3 and the basic residues Lys-6, Arg-9, Arg-10, and Arg-13 adsorb frequently. Strong electrostatic interactions locate these residues at the surface, and in the case of the basic residues, there is also a large energetic contribution from hydrogen bonding with HAp surface phosphates. Interestingly, Glu-5 adsorbs significantly more often than Glu-4. The fact that neighboring glutamate residues bind the surface disproportionately suggests some orientational specificity. This specificity may arise from statherin's fold coupled with strong binding of its Sep and basic residues. Sep-2, Sep-3, Lys-6, Arg-9, Arg-10, and Arg-13 have been implicated in binding by mutagenesis (41,43), deletion (41), and ssNMR experiments (19). An important experiment might investigate the absence/presence of preferential adsorption for one of the neighboring glutamate residues at positions 4 and 5.
The middle and C-terminal segments (Fig. 2 a) show minimal change in secondary structure (Fig. 2 a, upper panel) and minimal binding (Fig. 2 a, middle and lower panels). The combined effect of strong N-terminal binding, a semirigid proline-rich middle segment, and the energetic cost of desolvating the HAp surface enables only weak interaction of middle and C-terminal segments. Aside from a lone glutamate residue at position 26, there are no charged side chains downstream of Arg-13, and therefore little electrostatic compensation for HAp desolvation. It has been experimentally demonstrated that the middle and C-terminal segments have little or no affinity for HAp (41).
Adsorption of a control peptide
Although unfolded states were significantly populated at the time of adsorption (Fig. 2 c), those states did not significantly populate the top-scoring decoys (Fig. 2 b). As a further test of the algorithm's ability to capture extended states on the surface, we sought a control peptide of known conformation whose ionizable side-chain positioning would require adsorption-induced rearrangement to complement HAp surface electrostatics. One candidate is the 17-residue peptide designed by Marqusee et al. (44). This polyalanine peptide was designed to fold into a helix by placing three glutamate-lysine pairs at i and i + 4 positions in its primary sequence; the peptide was determined to be helical using CD spectroscopy. We simulated the adsorption of this control peptide to the (001) surface of HAp using the same protocol.
RosettaSurface predicts a mostly helical fold for the control peptide in solution, staggering its ion pairs about the circumference of its helix. A significant unfolding event is required if the ionizable side chains are to compliment the surface. Fig. 4 a shows that the control peptide undergoes drastic conformational change upon adsorption accompanied by strong binding of the ionic residues. Adsorption stabilizes the otherwise frayed termini and the central portion unfolds, placing ionizable residues in contact with the HAp surface. The surface induced/selected unfolding event is depicted structurally in Fig. 4 b. This control shows that the RosettaSurface protocol developed for this study can select for gross structural differences between solution and adsorbed states.
Figure 4.
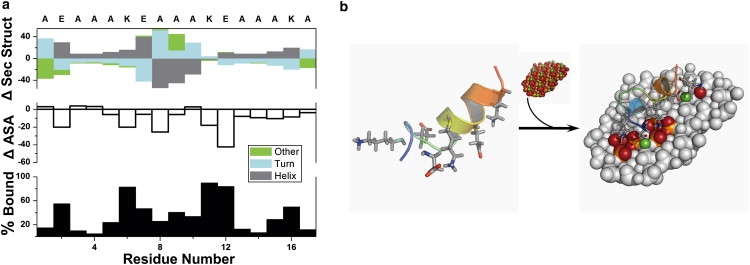
Adsorption induced folding event in a control peptide predicted by RosettaSurface. (a) Three adsorption phenomena plotted against residue number (see Fig. 2 a caption for complete description of these three phenomena) show strong HAp binding via the control peptide's ionic residues that is facilitated by a considerable unfolding event in the peptide's central region. (b) Representative solution-state (left) and HAp-absorbed models for control peptide.
Comparison with NMR data
We compared RosettaSurface decoys with all published high-resolution ssNMR distance and angle measurements (acquired from HAp-adsorbed statherin). The histograms in Supporting Material, Fig. S2, a–c, show this comparison separated into angle, intra-, and intermolecular distance measurements for statherin adsorbed to the HAp (001) surface. RosettaSurface predictions agree with the 15 published ssNMR measurements; however, the following measurements disagreed: the long-range intramolecular distances Pro-23–Pro33 and Pro-23–Tyr-34, and the intermolecular distance between Phe-14 and the nearest HAp phosphorus. These distances vary between top-scoring models in our simulation, and no single measurement is predicted.
To investigate the above-mentioned discrepancies, we added a harmonic potential to the RosettaSurface energy function to bias the simulation toward satisfying all ssNMR distance and angle measurements. The histograms in Fig. S2, a†–c†, show the increased accuracy, with respect to ssNMR measurements, that resulted from biasing the simulation. NMR-biased simulation satisfies the long-range distance measurements between Pro-23 and Pro-33 and Pro-23 and Tyr-34. To determine why these long-range distance measurements disagreed with unbiased RosettaSurface simulation, we analyzed the residue-specific energies for biased and unbiased simulations. In general, decoys generated from biased simulations had poor repulsive van der Waals scores and scored worse than decoys obtained from unbiased simulations. Satisfying the long-range distance, measurements between residues 23 and 33 and 23 and 34 always produced steric clashes among intervening residues. These steric clashes likely excluded decoys satisfying long-range distance measurements from being enriched in the 100 top-scoring decoys from unbiased simulations. Fig. S3 a shows a representative structure highlighting clashes caused by the long-range distance constraint between Pro-23 and Tyr-34.
A surprising consistency between NMR biased and unbiased simulation was the predicted ∼6.5 Å Pro-33–Tyr-38 distance. The experimentally measured distance of ∼5.5 Å was rarely captured despite the potential function bias to meet that distance constraint. The Pro-33–Tyr-38 and Tyr-34–Tyr-38 distance constraints always produced clashes. Clashes were so severe between residues 33 and 38 that even with bias only 31 of the 100 top-scoring decoys satisfied the constraint. However, in 59 of the 100 top-scoring decoys the Pro-33–Tyr-38 distance is between 6 and 7 Å; these decoys achieved good atomic packing without creating steric clashes. Fig. S3 b shows some clashes created when satisfying the Pro-33–Tyr-38 distance measurement.
Inclusion of ssNMR bias led to a significant adsorption-induced folding event in the C-terminal segment (data not shown). As much as 55% of the population shifted from turn to helix between residues 31 and 38 during biased predictions; no such transition results from unbiased predictions (Fig. 2 a, upper panel). Based on the Pro-23–Pro-33, Pro-23–Tyr-34, Pro-33 and Tyr-38, and Tyr-34–Tyr-38 ssNMR measured distances, Goobes et al. (20) concluded that a significant C-terminal folding event accompanied HAp adsorption.
In the absence of a biasing potential, Phe-14 tends to adsorb infrequently relative to the rest of the binding domain. It is difficult to determine whether this departure from ssNMR experiment arises from insufficient sampling and/or a deficiency in the energy function. To see if we could eliminate the energy function as a possibility, we ran a second biased simulation, whereby the protein-surface intermolecular constraints were only enforced to form the initial adsorbed-state complex. Subsequent moves did not bias the relative rigid-body positions of the protein and surface. Therefore, top-scoring decoys will likely display enriched binding of Phe-14 if the energy function favors the sampled adsorbed-state conformations. We found that even though the initial adsorbed-state complex satisfied the HAp–Phe-14 distance constraint, subsequent moves often removed Phe-14 from the surface. Therefore, even when conformations with Phe-14 adsorbed are sampled, the energy function does not favor the adsorption of Phe-14. Although this suggests the energy function is deficient, a second possibility is that Phe-14 adsorbs but the correct statherin conformations were not found despite extensive sampling of adsorbed-state conformations. Another possibility is that the Phe-14 ssNMR measured distance is not relevant to the predictions made here. Phe-14 ssNMR distance measurements were acquired in the context of a statherin molecule truncated at residue 15, leaving a negatively charged carboxyl group adjacent to Phe-14 that was not present in our models.
Statherin adsorption at the (010) and (100) faces of HAp
It is not known to which face(s) of HAp statherin binds. To test whether RosettaSurface could distinguish surfaces relevant to statherin recognition, we applied our protocol to dock statherin to two additional HAp surfaces, (010) and (100). Atomically flat, mixed-charge terminations were used at each surface. Simulation methodology was identical at all three surfaces.
Surprisingly, similar statherin conformers adsorbed to three HAp faces ((001) face in Fig. 2 a and (010) and (100) faces in Fig. S4, a and b, respectively). In all three runs, the adsorbed state is mostly helical, and the N-terminal acidic and basic residues bind the charged HAp surface.
The most significant difference in statherin's conformation at the three surfaces is the more pronounced folding event about Gly-12 at the (001) (Fig. 2 a) surface compared to the (010) and (100) surfaces (Fig. S4, a and b, respectively). One reason folding may differ slightly at each surface is the geometry of the binding moieties replicated across the three surfaces. In Fig. 5 a, the white parallelogram shows the interstice of the phosphate-oxygen triad (IPOT) motif, the motif hypothesized to be responsible for binding statherin's basic residues at the HAp (001) face in our previous fixed-backbone docking study (39). The IPOT motif is a periodic replication of open phosphate clusters, which expose hydrogen-bond-accepting oxygen atoms and afford favorable van der Waals forces and electrostatic interactions. Similar motifs are shown for the (010) and (100) surfaces in Figs. 5, b and c, respectively. Among these motifs, the IPOT on the (001) surface has unique dimensions (a parallelogram with 9.42 Å sides and 16.2 Å diagonal) when compared to the motifs on the (010) and (100) surfaces (both are rectangles, 9.42 by 6.88 Å and an 11.64 Å diagonal). The motif dimension at the (001) surface is more easily complemented by a statherin binding domain in the extended helix fold (∼16 Å in length). This correlation may account for the elevated binding of Arg-13 at the (001) HAp face, whereas there is elevated binding of Asp-1 and/or Glu-4 at the (010) and (100) faces.
Figure 5.
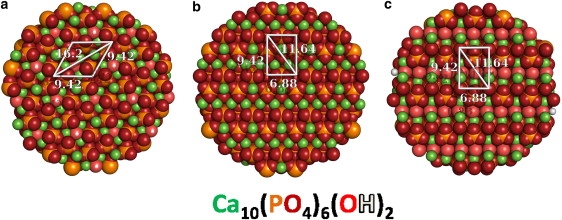
Predicted binding motifs on three HAp surfaces. (a) The IPOT motif (38) of the (001) monoclinic face of HAp and similar motifs on the (b) (010) and (c) (100) monoclinic faces of HAp.
Discussion
Structural differences in the solution and adsorbed states
Protein adsorption has historically been associated with unfolding and/or structural rearrangement (45). This notion may have arisen, to some extent, from the fact that protein-surface interactions are often studied using stock-room proteins such as lysozyme, albumin, and fibrinogen that have not evolved to bind to the surfaces studied in those experiments (46–50). Whereas gross unfolding events may be expected to accompany nonspecific adsorption owing to an increase in entropy, stable, well-defined adsorbed-state folds have been experimentally inferred in several evolved systems including antifreeze proteins binding ice (51–53) and HAp and calcite biomineralization systems (54–57). The findings in this study suggest that the binding domain of statherin has evolved such that the solution and adsorbed states are similar, and hence little conformational rearrangement upon adsorption is necessary. This finding is supported by CD experiments that suggest the N-terminal segment of statherin to be stable and partially helical in solution (17,41) and ssNMR measurements that predict that the adsorbed state is also helical (24,25).
The only significant structural difference between statherin's predicted solution and HAp-bound states is the extension of the helical conformation in the binding domain of the HAp-bound states. This prediction agrees with ssNMR measurements (25) and addresses an apparent conflict between macro and microscopic studies on statherin's solution and adsorbed states. Naganagowda et al. (40) observed a single sharp 31P NMR signal at -2 and Sep-3, and Shaw et al. (25) measured ϕ angles at Leu-8 and Gly-12, 73° and 80°, respectively, in the solution state. Conversely, two independent CD spectra (17,41) recorded on statherin's N-terminal binding domain indicate that helical conformers are populated in solution. Because ssNMR measurements predict a helical structure for statherin's N-terminal binding domain in the adsorbed state (24,25), disagreements with respect to the solution state have led some to report large-scale HAp induced folding, whereas others suggest little or no difference between the states. Based on our findings, we suggest an intermediate model where statherin solution states are partially prestructured for adsorption. We find that Asp-1 and Sep-2 are not part of any regular secondary structure and that a folding event occurs near Gly-12. These models agree with CD data and suggest that the macroscopic structure of the N-terminal binding domain is partially helical in solution.
Structural differences at the three HAp surfaces
The mean score at the (001), (010), and (100) faces is −77 ± 7, −77 ± 6, and −77 ± 6 REU, respectively (see Materials and Methods in the Supporting Material). These small differences in score suggest promiscuous binding at three HAp surfaces. Similarly, predicted structures at the three HAp faces agree approximately equally well with ssNMR experimental measurements (data not shown for HAp (010) and (100) surfaces). The chemistry and geometry at the three HAp surfaces is similar, and small changes in statherin's backbone and side-chain torsion angles can complement the three HAp surfaces in similar ways with small energetic barriers. But, because the simulation of all physiological conditions during biomineralization is not feasible (protein concentration, protein-protein interactions, salt concentration, simultaneous crystallization of all relevant faces, etc.), the relevant free energy of adsorption cannot be calculated. It is therefore difficult to assess the effects such small structural perturbations might impart on specificity.
The vast knowledge obtained from studying crystallized protein-protein complexes from the Protein Data Bank (58) makes it tempting to assume proteins generally interact in a highly specific manner. But there may be considerable differences in the way proteins behave at phase boundaries compared to globular protein-protein association. For instance, biomineral inhibition could be accomplished by adsorbing protein to all or several faces of a growing biomineral, the relative rates depending on the intrinsic energy of each exposed face. Also, the presence of an adsorbate protein may be sufficient to inhibit biomineralization, regardless of whether a single structure dominates the free energy minimum. With an increased number of ssNMR measurements, it may be possible to constrain biased simulations to determine the relevant binding face(s). For instance, the arrangement of ionic side chains in constrained structures may electrostatically repel the relative calcium and phosphate geometries at some HAp surfaces, potentially identifying such faces as incompatible with statherin binding.
Ability to predict adsorbed-state structures de novo
Results from unbiased simulations agree well with experiment considering the conformational space associated with folding a protein at an interface. Also, adsorption of a control peptide resulted in an anticipated unfolding event between solution and adsorbed states. The largest departure from experiment was the absence of predicted structures in agreement with ssNMR measured long-range Pro-23–Pro-33 and Pro-23–Tyr-34 distances and the intermolecular HAp–Phe-14 distance.
Proline rich segments, such as the statherin segment comprising residues 23–34, often give rise to structural dispersion due to torsional constraints imposed by proline's imide bond and an absence of backbone hydrogen-bond donors (59). Given that the magnitude of the long-range distance measurements span the HAp unit cell (long-range distance measurements are 8.0–11.5 Å; HAp unit cell a = 9.4 Å), one possible explanation for this discrepancy is that the ssNMR experiments detected protein-protein intermolecular interactions from adjacently adsorbed statherin molecules. Also, the isotopic labeling scheme at Pro-23 included fluorine (20), and fluorine labeled proline residues can change and/or stabilize protein folds via electronegative inductive effects (60). Rosetta sampled proline cis- and trans-isomers and alternate ring puckers and still did not avoid these steric clashes.
Another possible explanation for discrepancies between ssNMR measurements and our predictions is the RosettaSurface energy function and representation of the system. RosettaSurface accounts for solvent implicitly rather than explicitly representing individual water molecules and salt ions, and water molecules and salt ions can influence protein adsorption (11). Furthermore, the EEF-1 implicit solvent model (43) employed by RosettaSurface was not originally developed to capture solvent effects at interfaces. Finally, the charge density at the statherin-HAp interface is significant, and it has not been demonstrated that the distant-dependent-dielectric method applied here can correctly account for electrostatic effects at such an interface.
Unbiased RosettaSurface simulation did not predict the C-terminal folding event that biased simulation predicted. Structures predicted from biased simulation place Pro-36 in the middle of an α-helix. Proline residues are rare in the middle of α-helices and always produce a kink of ∼20–30° or greater (61). Unbiased simulation predicts that the C-terminal α-helix initiates at Pro-36 (Fig. 1 a), in agreement with solution-state NMR measurements for statherin in TFE (40).
It is difficult to infer detailed protein structure from minimal experimental constraints. RosettaSurface-predicted structures present a concern for interpreting ssNMR long-range distance measurements as intramolecular, suggest alternative interpretations consistent with the data, and propose experiments that could determine the correct interpretation(s). Similarly, RosettaSurface predicted Phe-14 adsorption only after ssNMR bias was added to the simulation, highlighting the benefit experimental measurements can impart in computational structure prediction. The synergy between ssNMR and RosettaSurface may be particularly useful because adsorbed-state structures cannot be determined via alternative methods.
Prospects for a combined ssNMR RosettaSurface method
The concluding remarks of Goobes et al. (18) in their 2007 review on ssNMR spectroscopy and protein-surface interactions highlight the importance of the methods developed for this study: “Current protein structure prediction programs cannot predict the folded state of a protein in the presence of another macromolecule or surface starting from an unfolded or random coil conformation. The ultimate goal of computational technique development would be to assert the experimental observation of a transition from an unfolded state to an active state upon exposure of the potential energy of the surface.” For the work presented in this study, we developed a program capable of accomplishing such a goal.
Future RosettaSurface NMR collaborations on new systems may take the following form: educate initial placement of isotopically labeled amino-acid pairs by analysis of unbiased RosettaSurface output, considering areas of high and low confidence. The initial NMR measurements would in turn confirm or refute high-confidence predictions and inform low-confidence predictions for subsequent rounds of biased simulations. Given the results of this work, it may be possible to converge on plausible structures with only a few iterations of a combined method. We wish not to understate the difficulty of such a collaboration, but to emphasize the unique capability it possess to address the difficult and important problem of macromolecular structure prediction at interfaces.
Supporting Material
Materials and methods, four figures, and references are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00508-6.
Supporting Material
Acknowledgments
We thank Gary Drobny, Gil Goobes, Moise Ndao, Sid Chaudhury, Rahul Bhowmik, Monica Berrondo, Aroop Sircar, Elizabeth Specht, and Sarah Schrier for reviewing this manuscript.
This work was funded by the Arnold and Mabel Beckman Foundation through a young investigator grant and the National Institutes of Health, grant No. T32 GM008403.
Contributor Information
David L. Masica, Email: david.masica@gmail.com.
Jeffrey J. Gray, Email: jgray@jhu.edu.
References
- 1.Gray J.J. The interaction of proteins with solid surfaces. Curr. Opin. Struct. Biol. 2004;14:110–115. doi: 10.1016/j.sbi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Latour R.A. Molecular simulation of protein-surface interactions: benefits, problems, solutions, and future directions. Biointerphases. 2008;3:FC2–FC12. doi: 10.1116/1.2965132. (Review) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raut V.P., Agashe M.A., Stuart S.J., Latour R.A. Molecular dynamics simulations of peptide-surface interactions. Langmuir. 2005;21:1629–1639. doi: 10.1021/la047807f. [DOI] [PubMed] [Google Scholar]
- 4.Wierzbicki A., Dalal P., Cheatham T.E., Knickelbein J.E., Haymet A.D.J. Antifreeze proteins at the ice/water interface: three calculated discriminating properties for orientation of type I proteins. Biophys. J. 2007;93:1442. doi: 10.1529/biophysj.107.105189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X., Wang J.C., Lacki K.M., Liapis A.I. Molecular dynamics simulation studies of the transport and adsorption of a charged macromolecule onto a charged adsorbent solid surface immersed in an electrolytic solution. J. Colloid Interface Sci. 2004;277:483–498. doi: 10.1016/j.jcis.2004.04.048. [DOI] [PubMed] [Google Scholar]
- 6.Gerbaud V., Pignol D., Loret E., Bertrand J.A., Berland Y. Mechanism of calcite crystal growth inhibition by the N-terminal undecapeptide of lithostathine. J. Biol. Chem. 2000;275:1057–1064. doi: 10.1074/jbc.275.2.1057. [DOI] [PubMed] [Google Scholar]
- 7.Huq N.L., Cross K.J., Reynolds E.C. Molecular modelling of a multiphosphorylated sequence motif bound to hydroxyapatite surfaces. J. Mol. Model. 2000;6:35–47. [Google Scholar]
- 8.Dong X., Wang Q., Wu T., Pan H. Understanding adsorption-desorption dynamics of BMP-2 on hydroxyapatite (001) surface. Biophys. J. 2007;93:750. doi: 10.1529/biophysj.106.103168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen J.W., Wu T., Wang Q., Pan H.H. Molecular simulation of protein adsorption and desorption on hydroxyapatite surfaces. Biomaterials. 2008;29:513–532. doi: 10.1016/j.biomaterials.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 10.Sun Y., Dominy B.N., Latour R.A. Comparison of solvation-effect methods for the simulation of peptide interactions with a hydrophobic surface. J. Comput. Chem. 2007;28:1883. doi: 10.1002/jcc.20716. [DOI] [PubMed] [Google Scholar]
- 11.Castells V., Yang S., Van Tassel P.R. Surface-induced conformational changes in lattice model proteins by Monte Carlo simulation. Phys. Rev. E. 2002;65:31912. doi: 10.1103/PhysRevE.65.031912. [DOI] [PubMed] [Google Scholar]
- 12.Liu S.M., Haynes C.A. Mesoscopic analysis of conformational and entropic contributions to nonspecific adsorption of HP copolymer chains using dynamic Monte Carlo simulations. J. Colloid Interface Sci. 2004;275:458–469. doi: 10.1016/j.jcis.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 13.Zhdanov V.P., Kasemo B. Folding of bundles of-helices in solution, membranes, and adsorbed overlayers. Proteins. 2001;42:481–494. doi: 10.1002/1097-0134(20010301)42:4<481::aid-prot70>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 14.Phillips R., Dittrich M., Schulten K. Quasi-continuum representations of atomic-scale mechanics: from proteins to dislocations. Annu. Rev. Mater. Res. 2002;32:219–233. [Google Scholar]
- 15.Villa E., Balaeff A., Mahadevan L., Schulten K. Multiscale method for simulating protein-DNA complexes. Multiscale Model. Simul. 2004;2:527–553. [Google Scholar]
- 16.Shaw W.J., Ferris K., Tarasevich B., Larson J.L. The structure and orientation of the C-terminus of LRAP. Biophys. J. 2008;94:3247. doi: 10.1529/biophysj.107.119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen P.H., Tseng Y.H., Mou Y., Tsai Y.L., Guo S.M. Adsorption of a statherin peptide fragment on the surface of nanocrystallites of hydroxyapatite. J. Am. Chem. Soc. 2008;130:2862–2868. doi: 10.1021/ja076607y. [DOI] [PubMed] [Google Scholar]
- 18.Goobes G., Stayton P.S., Drobny G.P. Solid state NMR studies of molecular recognition at protein–mineral interfaces. Prog. Nucl. Magn. Reson. Spectrosc. 2007;50:71–85. doi: 10.1016/j.pnmrs.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raghunathan V., Gibson J.M., Goobes G., Popham J.M., Louie E.A. Homonuclear and heteronuclear NMR studies of a statherin fragment bound to hydroxyapatite crystals. J. Phys. Chem. B. 2006;110:9324–9332. doi: 10.1021/jp056644g. [DOI] [PubMed] [Google Scholar]
- 20.Goobes G., Goobes R., Schueler-Furman O., Baker D., Stayton P.S. Folding of the C-terminal bacterial binding domain in statherin upon adsorption onto hydroxyapatite crystals. Proc. Natl. Acad. Sci. USA. 2006;103:16083–16088. doi: 10.1073/pnas.0607193103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibson J.M., Popham J.M., Raghunathan V., Stayton P.S., Drobny G.P. A solid-state NMR study of the dynamics and interactions of phenylalanine rings in a statherin fragment bound to hydroxyapatite crystals. J. Am. Chem. Soc. 2006;128:5364–5370. doi: 10.1021/ja056731m. [DOI] [PubMed] [Google Scholar]
- 22.Gibson J.M., Raghunathan V., Popham J.M., Stayton P.S., Drobny G.P. A REDOR NMR study of a phosphorylated statherin fragment bound to hydroxyapatite crystals. J. Am. Chem. Soc. 2005;127:9350–9351. doi: 10.1021/ja050910m. [DOI] [PubMed] [Google Scholar]
- 23.Shaw W.J., Campbell A.A., Paine M.L., Snead M.L. The COOH terminus of the amelogenin, LRAP, is oriented next to the hydroxyapatite surface. J. Biol. Chem. 2004;279:40263. doi: 10.1074/jbc.C400322200. [DOI] [PubMed] [Google Scholar]
- 24.Long J.R., Shaw W.J., Stayton P.S., Drobny G.P. Structure and dynamics of hydrated statherin on hydroxyapatite as determined by solid-state NMR. Biochemistry. 2001;40:15451–15455. doi: 10.1021/bi010864c. [DOI] [PubMed] [Google Scholar]
- 25.Shaw W.J., Long J.R., Dindot J.L., Campbell A.A., Stayton P.S. Determination of statherin N-terminal peptide conformation on hydroxyapatite crystals. J. Am. Chem. Soc. 2000;122:1709–1716. [Google Scholar]
- 26.Long J.R., Dindot J.L., Zebroski H., Kiihne S., Clark R.H. A peptide that inhibits hydroxyapatite growth is in an extended conformation on the crystal surface. Proc. Natl. Acad. Sci. USA. 1998;95:12083. doi: 10.1073/pnas.95.21.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlesinger D.H., Hay D.I. Complete covalent structure of statherin, a tyrosine-rich acidic peptide which inhibits calcium phosphate precipitation from human parotid saliva. J. Biol. Chem. 1977;252:1689–1695. [PubMed] [Google Scholar]
- 28.Moreno E.C., Varughese K., Hay D.I. Effect of human salivary proteins on the precipitation kinetics of calcium phosphate. Calcif. Tissue Int. 1979;28:7–16. doi: 10.1007/BF02441212. [DOI] [PubMed] [Google Scholar]
- 29.Johnsson M., Richardson C.F., Bergey E.J., Levine M.J., Nancollas G.H. The effects of human salivary cystatins and statherin on hydroxyapatite crystallization. Arch. Oral Biol. 1991;36:631–636. doi: 10.1016/0003-9969(91)90014-l. [DOI] [PubMed] [Google Scholar]
- 30.Robinson C., Connell S., Kirkham J., Shore R., Smith A. Dental enamel—a biological ceramic: regular substructures in enamel hydroxyapatite crystals revealed by atomic force microscopy. J. Mater. Chem. 2004;14:2242–2248. [Google Scholar]
- 31.Fischetti V.A., Amano A., Nakamura T., Kimura S., Morisaki I. Molecular interactions of porphyromonas gingivalis fimbriae with host proteins: kinetic analyses based on surface plasmon resonance. Infect. Immun. 1999;67:2399–2405. doi: 10.1128/iai.67.5.2399-2405.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berrondo M., Ostermeier M., Gray J.J. Structure prediction of domain insertion proteins from structures of the individual domains. Structure. 2008;16:513–527. doi: 10.1016/j.str.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradley P., Misura K.M.S., Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 34.Daily M.D., Masica D., Sivasubramanian A., Somarouthu S., Gray J.J. CAPRI rounds 3–5 reveal promising successes and future challenges for RosettaDock. Proteins. 2005;60:181–186. doi: 10.1002/prot.20555. [DOI] [PubMed] [Google Scholar]
- 35.Gray J.J., Moughon S., Wang C., Schueler-Furman O., Kuhlman B. Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 36.Kuhlman B., Dantas G., Ireton G.C., Varani G., Stoddard B.L. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 37.Sivasubramanian A., Maynard J.A., Gray J.J. Modeling the structure of mAb 14B7 bound to the anthrax protective antigen. Proteins. 2008;70:218–230. doi: 10.1002/prot.21595. [DOI] [PubMed] [Google Scholar]
- 38.Meiler J., Baker D. Rosettaligand: protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 39.Makrodimitris K., Masica D.L., Kim E.T., Gray J.J. Structure prediction of protein-solid surface interactions reveals a molecular recognition motif of statherin for hydroxyapatite. J. Am. Chem. Soc. 2007;129:13713–13722. doi: 10.1021/ja074602v. [DOI] [PubMed] [Google Scholar]
- 40.Naganagowda G.A., Gururaja T.L., Levine M.J. Delineation of conformational preferences in human salivary statherin by 1H, 31P NMR and CD studies: sequential assignment and structure-function correlations. J. Biomol. Struct. Dyn. 1998;16:91–107. doi: 10.1080/07391102.1998.10508230. [DOI] [PubMed] [Google Scholar]
- 41.Raj P.A., Johnsson M., Levine M.J., Nancollas G.H. Salivary statherin. Dependence on sequence, charge, hydrogen bonding potency, and helical conformation for adsorption to hydroxyapatite and inhibition of mineralization. J. Biol. Chem. 1992;267:5968–5976. [PubMed] [Google Scholar]
- 42.Aurora R., Rose G.D. Helix capping. Protein Sci. 1998;7:21. doi: 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goobes R., Goobes G., Shaw W.J., Drobny G.P., Campbell C.T. Thermodynamic roles of basic amino acids in statherin recognition of hydroxyapatite. Biochemistry. 2007;46:4725–4733. doi: 10.1021/bi602345a. [DOI] [PubMed] [Google Scholar]
- 44.Marqusee S., Robbins V.H., Baldwin R.L. Unusually stable helix formation in short alanine-based peptides. Proc. Natl. Acad. Sci. USA. 1989;86:5286. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haynes C.A., Norde W. Globular proteins at solid/liquid interfaces. Colloids Surf. B Biointerfaces. 1994;2:517–566. [Google Scholar]
- 46.Ball A., Jones R.A.L. Conformational changes in adsorbed proteins. Langmuir. 1995;11:3542–3548. [Google Scholar]
- 47.Buijs J., Speidel M., Oscarsson S. The stability of lysozyme adsorbed on silica and gallium arsenide surfaces: preferential destabilization of part of the lysozyme structure by gallium arsenide. J. Colloid Interface Sci. 2000;226:237–245. [Google Scholar]
- 48.Kim D.T., Blanch H.W., Radke C.J. Direct imaging of lysozyme adsorption onto mica by atomic force microscopy. Langmuir. 2002;18:5841–5850. [Google Scholar]
- 49.Steadman B.L., Thompson K.C., Russell Middaugh C., Matsuno K., Vrona S. The effects of surface adsorption on the thermal stability of proteins. Biotechnol. Bioeng. 1992;40:8–15. doi: 10.1002/bit.260400103. [DOI] [PubMed] [Google Scholar]
- 50.Xia N., May C.J., McArthur S.L., Castner D.G. Time-of-flight secondary ion mass spectrometry analysis of conformational changes in adsorbed protein films. Langmuir. 2002;18:4090–4097. [Google Scholar]
- 51.Sicheri F., Yang D.S.C. Ice-binding structure and mechanism of an antifreeze protein from winter flounder. Nature. 1995;375:427–431. doi: 10.1038/375427a0. [DOI] [PubMed] [Google Scholar]
- 52.Leinala E.K., Davies P.L., Jia Z. Crystal structure of β-helical antifreeze protein points to a general ice binding model. Structure. 2002;10:619–627. doi: 10.1016/s0969-2126(02)00745-1. [DOI] [PubMed] [Google Scholar]
- 53.Ko T.P., Robinson H., Gao Y.G., Cheng C.H.C., DeVries A.L. The refined crystal structure of an eel pout type III antifreeze protein RD1 at 0.62-A resolution reveals structural microheterogeneity of protein and solvation. Biophys. J. 2003;84:1228–1237. doi: 10.1016/S0006-3495(03)74938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reyes-Grajeda J.P., Moreno A., Romero A. Crystal structure of ovocleidin-17, a major protein of the calcified gallus gallus eggshell: implications in the calcite mineral growth pattern. J. Biol. Chem. 2004;279:40876. doi: 10.1074/jbc.M406033200. [DOI] [PubMed] [Google Scholar]
- 55.Dowd T.L., Rosen J.F., Li L., Gundberg C.M. The three-dimensional structure of bovine calcium ion-bound osteocalcin using 1H NMR spectroscopy. Biochemistry. 2003;42:7769–7779. doi: 10.1021/bi034470s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frazao C., Simes D.C., Coelho R., Alves D., Williamson M.K. Structural evidence of a fourth Gla residue in fish osteocalcin: biological implications. Biochemistry. 2005;44:1234–1242. doi: 10.1021/bi048336z. [DOI] [PubMed] [Google Scholar]
- 57.Hoang Q.Q., Sicheri F., Howard A.J., Yang D.S. Bone recognition mechanism of porcine osteocalcin from crystal structure. Nature. 2003;425:977–980. doi: 10.1038/nature02079. [DOI] [PubMed] [Google Scholar]
- 58.Berman, H.M., J. Westbrook, Z. Feng, G. Gilliland, T. Bhat, H. Weissig, I.N. Shindyalov, and P.E. Bourne. The Protein Data Bank. logo 58:899–907. [DOI] [PMC free article] [PubMed]
- 59.Williamson M.P. The structure and function of proline-rich regions in proteins. Biochem. J. 1994;297:249–260. doi: 10.1042/bj2970249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Renner C., Alefelder S., Bae J.H., Budisa N., Huber R. Fluoroprolines as tools for protein design and engineering. Angew. Chem. Int. Ed. Engl. 2001;40:923–925. doi: 10.1002/1521-3773(20010302)40:5<923::AID-ANIE923>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 61.Barlow D.J., Thornton J.M. Helix geometry in proteins. J. Mol. Biol. 1988;201:601–619. doi: 10.1016/0022-2836(88)90641-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.