

Abstract
A major question in chromatin involves the exact organization of nucleosomes within the 30-nm chromatin fiber and its structural determinants of assembly. Here we investigate the structure of histone octamer helical tubes via the method of iterative helical real-space reconstruction. Accurate placement of the x-ray structure of the histone octamer within the reconstructed density yields a pseudoatomic model for the entire helix, and allows precise identification of molecular interactions between neighboring octamers. One such interaction that would not be obscured by DNA in the nucleosome consists of a twofold symmetric four-helix bundle formed between pairs of H2B-α3 and H2B-αC helices of neighboring octamers. We believe that this interface can act as an internucleosomal four-helix bundle within the context of the chromatin fiber. The potential relevance of this interface in the folding of the 30-nm chromatin fiber is discussed.
Introduction
Access to the genetic code in eukaryotes is granted through a higher epigenetic code, enforced by chromatin structure and modulated via the pattern of posttranslational modifications of the underlying histone proteins (1). Such modifications can alter various chromatin-folding hierarchies, either directly through charge alteration, or indirectly through the recruitment of various chromatin-associated proteins, including chromatin-remodeling factors. The fundamental repeating unit of chromatin, the nucleosome, is well-characterized, and the various x-ray structures of the core particle provide considerable insights regarding the functioning of the nucleosome as a regulatory unit of transcription, particularly through the mechanisms of regulated nucleosome mobility (2) and histone-variant substitution (3). However, extending a similar functional understanding to higher folding levels first requires an elucidation of how arrays of nucleosomes are further folded into the helical chromatin fiber.
The histone octamer forms the central protein scaffold of chromatin structure, and is responsible for its organization into both primary and secondary levels. These different roles are mediated through distinct portions of the octamer structure: DNA binding is predominately mediated via the histone fold domains, whereas chromatin-fiber folding is mediated via the N-terminal tails and octamer face of nucleosomes. Within the globular core, the heterodimer units H2A-H2B and H3-H4 (formed by handshake pairing of the individual core histones) serve as the basic structural elements of DNA binding, and are repetitively placed on a spiral path by three four-helix bundles (4,5). The first bundle forms over the twofold axis, to create the central (H3-H4)2 tetramer, whereas the remaining two bundles add the H2A-H2B dimers to either side of this tetramer. This final assembly is what then serves as a binding platform for 147 bp of DNA as 1.67 left-handed supercoils, to complete the nucleosome core particle (6).
Elucidation of the higher-order arrangement of the chromatin fiber is proving to be a considerably more difficult task. Several classes of competing models have been built to explain a wide range of differing experimental evidence (7). In the original solenoid model (8,9), bent linker DNA continues the superhelical trajectory set up within the nucleosome core particle, connecting adjacent nucleosomes in a simple one-start arrangement. Subsequent models based on the one-start helical model also proposed the interdigitation of nucleosomes, where nucleosomes from consecutive gyres of the fiber superhelix partially slot in between each other (10). Alternatively, in two-start models, linker DNA is essentially straight, and joins two separate nucleosome stacks. The two subforms of the two-start model are simply twisted or supercoiled topological variants, i.e., the crossed-linker (11) and helical-ribbon (12) models, respectively. The major discriminating features of the solenoid and two-start models are therefore the start number, linker DNA conformation, and internucleosomal contacts. In these respects, most modern evidence using defined nucleosomal arrays favors a two-start, crossed-linker model (13). The start number was convincingly demonstrated via disulfide cross-linking to be two-start, and was shown to be preserved for a variety of nucleosome repeat lengths (NRLs) and array lengths, independent of linker histone content (14). However, that experiment did not distinguish between twisted and supercoiled two-start models, and this issue was resolved by the crystal structure of a tetranucleosome (15). Here the conformation of linker DNA is visualized as essentially straight, and as joining two opposite stacks of nucleosome cores in a truncated two-start arrangement. This demonstrates that for at least a 167-bp NRL, the fiber is in the twisted form.
There is intense interest in possible nucleosome interactions observed as crystal contacts in the structures of both core particles (5,16) and histone octamers (17,18), because such interactions could provide clues as to how nucleosomes associate in the chromatin fiber. So far only one interaction, wherein the base of the H4 tail contacted the H2A-H2B acidic patch of a neighboring nucleosome (5), was demonstrated to be directly relevant to chromatin fibers. This contact proved crucial for both the crystallization of core particles (5) and the compaction of nucleosome arrays (19), and the site was subsequently used in the disulfide cross-linking experiment mentioned above. Unfortunately, the nucleosome interface evident within the tetranucleosome crystal is incompatible with this interaction. Instead, nucleosomes stack via a pseudo-twofold interaction involving H2B-α1, H2B-αC, and H2A-α2 helices, thereby obscuring the acidic patch. Simply building a fiber model using this interface was not possible because of steric clashes, and an idealized model had to be constructed using the proven H4 tail-acidic patch interaction (15). However, because this contact is small and flexible, it can only act as a relatively weak modeling constraint in this regard (after all, in the crystal structure of the nucleosome core (5), the H4 tail stabilizes adjacent nucleosomes, with their dyad axes reversed). Apart from the tails, other short-range interactions are expected to come into play as nucleosomes closely approach and octamer faces dock upon full fiber compaction. These would provide important constraints for 30-nm fiber modeling, and would offer important predictions about its mechanistic behavior.
Here, we reinvestigate the structure of histone octamer helical tubes, first studied by Klug et al. more than 25 years ago (20), using electron microscopy and Fourier-Bessel methods (21). Their reconstruction provided valuable insights into the overall dimensions of the histone octamer, a rough assignment of core histone positions, and the potential superhelical wrapping surface for DNA. Here we reexamine the structure with the benefit of a modern method of iterative helical real-space reconstruction (IHRSR) (22) and knowledge of the x-ray structure of the histone octamer (4). Our aim was to build on earlier work, and identify the quaternary interactions within the helical tubes. Because octamers in the absence of DNA are known to form these helical tubes readily at high salt concentrations, there is a question of whether the associating surfaces present in these tubes bear any direct relevance to the structure of the chromatin fiber.
Although these helical tubes are well-ordered and therefore suitable for Fourier-Bessel reconstruction, the IHRSR approach still offers numerous advantages over its counterpart (23), i.e., the procedure is largely automated, and requires less user intervention. It can use more images to produce better averaging and resolution (especially when the signal/noise ratio is low). The segmentation of helices into short boxes compensates for long-range disorder and bending. Finally, helical symmetry is better determined iteratively, versus deciding a priori upon a symmetry based on an integer fraction.
Methods
Protein purification
The sample was prepared by methods similar to those described previously (24). Briefly, histone proteins were isolated from soluble chromatin prepared from chicken red blood cell nuclei. Free core histones were separated from DNA and linker histones via hydroxyapatite chromatography, and assembled into complete octamers by incubation with 2 M NaCl. Histone octamers were then isolated from excess dimers by Sephadex G-100 gel exclusion chromatography, and encouraged to form helices by double dialysis against a slowly exchanging gradient of 20–40% (NH4)2SO4 during 2 weeks.
Negative stain microscopy
Precipitate was stabilized by the addition of 0.4% uranyl acetate and applied to glow-discharged carbon-coated grids, and washed twice with 0.2% uranyl acetate. Fifty micrographs were taken at 50,000 × magnification, using the minimum-dose technique (≤100 e−/Å2) on a Leo 912 transmission electron microscope (Zeiss, Oberkochen, Germany) operating at 120 kV with an in-column energy filter set for zero-loss imaging. We digitized SO-163 negatives at 4 Å/pixel with a Leafscan 45 scanner (Ilford Ltd., Cheshire, UK).
Iterative real-space reconstruction
Helices were semiautomatically segmented into 3500 overlapping boxes of 128 × 128 pixels, using the program Boxer from the EMAN package (25), with an overlap difference corresponding to the axial rise (65 Å). Segments were then translationally prealigned in the x-dimension, using a reference-free approach (26) to minimize the search range needed. Three-dimensional reconstruction was then performed using SPIDER (27) and the iterative helical real-space reconstruction method (22). Because of the 11-fold symmetry, and an azimuthal increment of 4.1°, this resulted in eight reference projections used as templates in a projection-matching approach (26). This procedure was iterated until convergence was indicated by a stable structure and helical symmetry values.
Docking
The original x-ray structure of the chicken histone octamer (Protein Data Bank (PDB)-2HIO) was used for all docking procedures (4). Initial six-dimensional rigid body docking was performed using Situs (28). Two-dimensional correlation-based docking was implemented in Fortran 95, and used SPIDER (27) to calculate the cross-correlation between densities (with the atomic electron density filtered to 16 Å). Final van der Waals optimization of the helical diameter was implemented in Fortran 95, using a 6–12 Lennard-Jones potential.
Visualization
Electrostatic potentials were calculated using the program APBS (29), implemented within the molecular graphics program PMV (30). Potentials were calculated using a protein dielectric of 3, solvent dielectric of 78.5, ion-exclusion radius of 1.4 Å, and monovalent salt added to a concentration of 150 mM. Potentials were mapped to the solvent-accessible surface (31), colored by a range of −7.0 (red) to +7.0 (blue) kcal · mol−1 · e−1. Conjugate gradient energy minimization was performed using Hyperchem version 7.5 (32) and AMBER force fields (33). All figures were prepared with Chimera (34), with volumes contoured to enclose 78.6 kDa per octamer at 813 Å3/kDa, which excluded 28% of the molecular mass contained in the disordered tail regions.
The reconstructed volume and docked atomic model were deposited into the electron microscopy data bank and the PDB under the accession codes EMD-1469 and PDB-3C9K, respectively.
Results
Three-dimensional reconstruction
Images of negatively stained histone octamer tubes are shown in Fig. 1 A. These tubes are 300 Å in diameter and hollow, and possess strong horizontal striations. These striations arise because the helices are composed of stacked, closed rings of histone octamer subunits, each of which has a consistent angular offset relative to the preceding ring in the stack. Subdivision of the images of individual helices into overlapping boxes resulted in a data set of 3500 helical segments, from which an unbiased averaged power spectrum was calculated by adding together the rotationally aligned power spectra from each segment. The result (Fig. 1 B) was a first meridional reflection at 1/65 Å−1, and a first off-meridional at 1/280 Å−1, enabling the rise and pitch of the helix to be determined unambiguously. Further meridional reflections are evident at 1/33 Å−1 and 1/22 Å−1, and weakly at 1/16 Å−1. Visually these helices and the positions of reflections in the power spectrum are indistinguishable from those of Klug et al. (20). Similarly, the number of octamers per ring could not be judged directly from the images. Klug et al. (20) described heterogeneity, with fibers having either 10 or 11 octamers per ring. The axial rotation value (ΔΦ) is different in each of these two cases. The power spectrum alone is unable to distinguish this type of point-group heterogeneity, which was instead resolved by inspection of the phases of the complex Fourier transform for eight extended filaments that all displayed an odd Bessel order for layer line 4 (at 1/280 Å−1) (Supplementary Material, Fig. S1). This demonstrates that for these fibers, the rotational symmetry is 11-fold, as opposed to the 10-fold symmetry of the fibers originally reconstructed (20). No evidence for heterogeneity was found using a multimodel IHRSR refinement approach with both 10-fold and 11-fold symmetrized reference volumes (data not shown).
Figure 1.
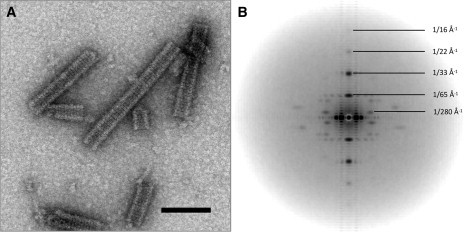
(A) Electron micrograph of negatively stained histone octamer tubes. Scale bar, 100 nm. (B) Averaged power spectrum created from 3500 helical segments. Potential resolution is gauged as 16 and 23 Å in meridional and axial directions, respectively.
Three-dimensional reconstruction was performed via the IHRSR method (22), which is essentially a single-particle technique involving an iterative projection-matching approach (26) coupled to a search and imposition of helical symmetry on the asymmetric, back-projected volume at the end of each iteration. Because the cyclic point group is fixed as 11-fold, an asymmetric wedge of only (360/11)° needs to be filled, requiring just eight reference projections for a maximally achievable resolution of 11 Å. This high symmetry aided the rapid convergence of the helical parameters, and a stable reconstruction was achieved after just seven iterations. The resulting structure is composed of closed rings, with 11 octamer subunits further stacked into a helix by a left-handed axial rotation (ΔΦ) of −7.6°, a rise (Δz) of 65.1 Å, and a pitch of 281 Å/turn or 4.36 rings/turn (Fig. 2 A). Thus the rise and pitch are consistent with the values directly measured from the power spectrum (Fig. 1 B). These helical parameters were further validated by performing two separate reconstructions, both using a featureless cylinder of 300-Å diameter as a template, but with different starting estimates of ΔΦ (1° on either side of the final value). Both converged upon the same solution (Fig. S2 A). The Fourier shell correlation of these two independently generated structures was 0.5 at 20 Å (Fig. S2 B).
Figure 2.
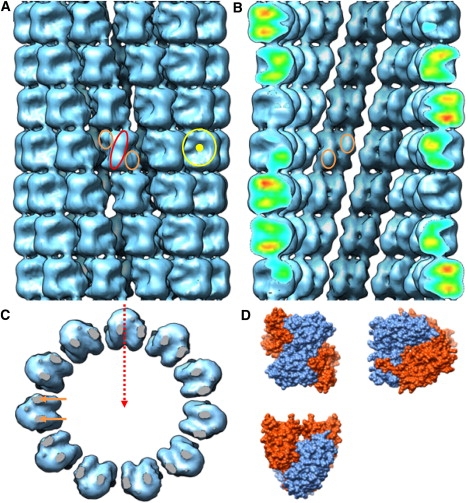
Histone octamer helical tube reconstruction. (A) Exterior view shows characteristic H3-H4 propeller blades (ellipsoid) with flanking H2A-H2B dimer hips (small circles), as well as disc-shaped octamer side views (large circle). (B) Cross section reveals the inner lattice surface, with clear H2A-H2B dimer protuberances (small circles) across the dyad axis. (C) Top view of a single octamer ring displays D11 point group symmetry and wedge-shaped octamer views. Two regions of density (short arrows) form stacking connections between the octamers of successive rings. Twofold dyad axis is indicated by the dashed arrow, and intersects the helical axis. (D) For comparison, the same three orthogonal views (propeller, disc, and wedge) of histone octamer molecular surface were generated from the x-ray structure (4).
The reconstructed electron density is well-defined, and the subunits are easily identifiable as histone octamers by their three characteristic orthogonal views (Fig. 2) (4). When viewed directly toward the helical axis, octamers display the central H3-H4 “propeller”, flanked on either side by H2A-H2B dimer “hips”. In side profile toward the edges of helices, the 65-Å disc shape is apparent, and an indent reflects the spiral axis. Finally, the wedge view is seen when rings are viewed from above. The dyad axis of the octamer, which was not enforced during reconstruction, is also clearly preserved, serving as an independent validity check: this dyad must lie perpendicular to the helical axis, as required by the constraints laid out for permissible helical line groups (35,36). Accordingly, entire rings have dihedral symmetry, and can be described by the helical line group D11S−4.36. Fig. 3 shows the helical nets constructed for both the D10 and D11 helices. The shared helical lattice explains why both helices generate the same diffraction patterns, despite their differences in point group symmetry.
Figure 3.
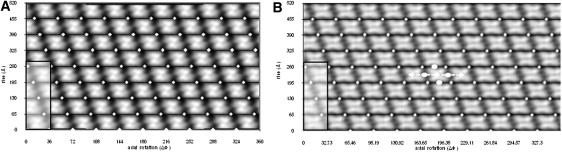
Helical nets overlaid onto cylindrical projections for (A) an atomic model created from the helical parameters of Klug et al. (20) with D10 point group symmetry, and (B) the reconstruction according to this study with D11 point group symmetry. The 280-Å pitch of both helices is identical, as indicated by shaded boxes. The dyad axis (central dot), spiral axis (arrows), and degenerate twofold axes (elliptical dots) are shown for a single unit cell.
Docking
Interpretation of the quaternary interactions occurring within the helix required docking the atomic structure of the histone octamer (4). This particular structure was chosen because it lacked the disordered tail regions, and contained a crystallographic twofold axis coinciding with the molecular twofold axis of the histone octamer. An initial full six-dimensional cross-correlation search was performed to identify the binary orientation of this dyad axis, and this search was possible because the octamer density is well-segregated in the map, avoiding partial occupancies. Nonetheless, accuracy could be increased by docking an atomic model of the entire helix, generated by applying the helical symmetry determined by IHRSR and the constraints of the helical line group. Forcing the dyad axis of the octamer to intersect and lie perpendicular to the helical axis reduces the search to just two-dimensional degrees of freedom, i.e., translations and rotations about this axis (Fig. 4 A). Fig. 4 B shows the resulting correlation surface, with a best fit achieved at a diameter of 315 Å and a 0° rotation, indicating that the spiral axis of the octamer (describing the path of histone heterodimers, analogous to the superhelical axis of the nucleosome) lies in-plane with the octamer rings, and could act as an additional docking constraint (although this was not known a priori). Furthermore, this intersection of spiral axes from neighboring octamers implies that a pseudocontinuous spiraling of histone heterodimer units occurs within individual octamer rings.
Figure 4.
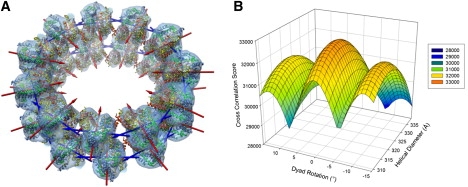
(A) Optimal two-dimensional correlation docked result of multiple copies of the histone octamer crystal structure into the reconstruction, using both helical and dyad axis constraints. The result is truncated to two rings for display purposes. Dyad axes (red arrows) point toward the helical axis, whereas adjacent spiral axes (blue arrows) intersect each other to form a pseudocontinuous left-handed heterodimer spiral. (B) Two-dimensional cross-correlation surface, produced by the constrained docking shown in A. The surface displays a peak at a helical diameter of 320 Å, and a dyad rotation of 0°.
Flattening is one of the major artifacts to affect hollow, negatively stained helices, and is evidenced by asymmetries in reflections across the meridian in the power spectrum (Fig. 1 B). Because flattened helices present a wider diameter in projection, the entire reconstruction becomes dilated, leading to a reduction of resolution in the radial dimension. Hence correlation-based docking could never achieve an optimal result. As an alternative, with the fitting now reduced to just the dimension of the tube diameter, the search was reevaluated using the van der Waals energy alone, and was found to be a minimum at a reduced diameter of 295 Å.
Interoctamer contacts
Connectivity between octamer subunits can be divided into two major components: inter-ring, representing axial interactions between rings; and intra-ring, reflecting azimuthal interactions within rings. Inter-ring contacts are well-resolved in the electron density map, and form over two small surface areas on either side of a twofold axis. Fitting reveals that octamers are joined vertically in these regions via helix dipole-dipole coupling between the carboxyl-terminus of H2A-α2 and the amino terminus of H3′-α1 (Fig. 5 A). This is a successful way of satisfying the electrostatics in part of the octamer surface usually engaged in DNA-binding, and is ultimately what sets the relative rotation between rings, providing a molecular basis for the pitch shared by both 10-start and 11-start helices. This interaction is not directly relevant to chromatin structure, however, because it occurs on the octamer surface normally obscured by DNA within the context of the nucleosome.
Figure 5.
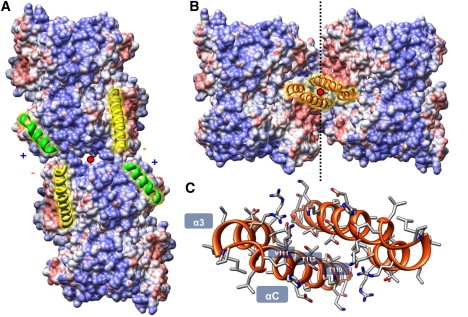
(A) Vertical inter-ring contacts are stabilized by helix dipoles formed between H2A-α2 (long helices) and H3-α1 (short helices). (B) Horizontal intra-ring contacts are formed by a four-helix bundle composed of H2B-α3 and H2B-αC helices. Twofold interaction axes are labeled as dots. Surface-mapped electrostatic potentials indicate an overall twofold charge complementarily for this interface. (C) H2B-α3/αC four-helix bundle viewed down the twofold axis, with key residues labeled.
Part of the motivation for optimizing the helical diameter arose from the obvious shape complementary exhibited by intra-ring interactions, through the potential overlap of adjacent octamer H2A-H2B dimer “hips”. Through van der Waals optimization, the αC accessory helices of H2B from neighboring octamers come together in a scissor-like action over a twofold axis, and associate as a four-helix bundle formed out of twofold related pairs of H2B-α3 and H2B-αC helices (Fig. 5 B). These two V-shaped α-helices are approximately aligned, allowing the basic α-helical grooves (i ± 1n) to extend across both these helices and form a corrugated triangular surface. The ∼70° angle set between the αC helices leads to a “ridges into grooves” mode of interaction (37), whereby the i ± 1n and i ± 4n helical grooves from both octamers intercalate with each other across the twofold axis (Fig. 5 C). This helix-packing mode is a way of satisfying the intercalation of both these helical grooves simultaneously, and sets the angle between adjacent octamers and thus the 11-fold rotational symmetry of the helix. The flexibility of this interface is indicated by the possibility of forming both 10-start and 11-start helices. Further energy minimization was performed to optimize the side-chain conformations at this rigid body location, to gain information regarding theoretical residue-specific interactions. Thr-115 and Thr-115′ pack together over the twofold axis, whereas Thr-119 and Thr-122 insert their methyl groups between the pockets formed in the V-shaped cleft of opposing α3′ and αC′ helices. Val-118 cements both α3 and αC helices and the opposite αC′ via its branched methyl groups.
Discussion
We reconstructed the structure of negatively stained histone octamer helical tubes via IHRSR to a resolution of 20 Å. Our reconstruction is of a different helical class at an improved resolution, compared with a previous reconstruction that used Fourier-Bessel methods (20). The improvement in resolution can be attributed not only to the larger image set used for the reconstruction, but also to the slight differences in helical symmetry, more accurately determined here via the iterative method. It was also demonstrated that care should be taken when dealing with point-group heterogeneities, because ambiguities present in the power spectrum can sometimes only be resolved through an examination of the phases of the Fourier transform. Finally, energy minimization, using the constraints determined in the reconstruction, led to an accurate molecular model of the histone octamer helix, enabling a rationalization of the quaternary interactions.
Of real interest is whether the intra-ring four-helix bundle interaction between octamers may be relevant to chromatin structure. Numerous lines of evidence suggest that this may be likely. Unlike a simple crystal contact, this interaction forms over a large surface area with extended shape complementarity, and engages a well-known tertiary protein motif with a distinct ridges-into-grooves mode of helical packing. Furthermore, the pseudocontinuous histone heterodimer spiraling observed within each ring, implied by the intersection of spiral axes and propagated through this four-helix bundle, mimics the internal tetramer and octamer assemblies formed by the H3-H3′ and H2B-H4 four-helix bundles, respectively. In fact, it was previously suggested that a remaining four-helix bundle formed by the C-terminal half of H2A-α2 and H2A-α3 helices might mediate an internucleosomal contact (5). We think that the H2B-α3 and H2B-αC helices are better candidates, because they are more peripheral and form the predominant shape of the octamer surface. In addition, they are not involved in either octamer assembly or DNA binding, yet they exhibit strong sequence conservation and have no assigned function. Unlike the inter-ring contact found within octamer tubes, this interaction would not be occluded by DNA, because it occurs on the open lateral face of the nucleosome. To substantiate our idea further, we suggest that a similar interface occurs in the presence of DNA in the nucleosome bilayers studied by Leforestier et al. (38). In their observed columnar-lamellar phase, nucleosome core particles stack into columns, such that their superhelical axes intersect and their dyads axes are approximately parallel. We believe by implication that the same four-helix bundle may stabilize these quasicrystalline arrays as it does within our octamer crystal tubes, further lending support to the idea that this bundle is not exclusive to naked octamers, and can occur between bona fide nucleosome core particles. The interface in our model may also be likened to that found in the tetranucleosome, because it also involves a pseudo-twofold contact involving H2B-αC helices packing together in a parallel fashion. The difference involves the use of a different side of the αC helices in the interface that we suggest, and importantly, this mode of αC engagement now reveals the acidic patch.
We therefore attempted to use this H2B-α3/αC four-helix bundle as a new static constraint to model the 30-nm chromatin fiber. We encountered one obstacle, i.e., within octamer tubes, the dyad axes are wrongly orientated, and the chromatin fiber would require more regular wedge-shaped packing between nucleosomes, with the thin edge of wedges (H3-H3′) facing the lumen. Hinging the four-helix bundle to this new angle results in axial displacement because of the scissor-action and ∼10° angle of the αC helices on the surface of the octamer. However, by using more of the length of these helices, this packing in fact achieves a better overall shape complementarily than within the octamer tubes. The idealized model of Schalch et al. (15), as derived from their tetranucleosome structure, was used a starting point for modeling the fiber. This model was empirically adjusted via decreases in fiber twist and rise, to produce a more compact fiber containing the new H2B-α3/αC internucleosomal four-helix bundle (Fig. 6 A). This leads to a model with a nucleosome compaction ratio more consistent with electron microscope measurements (10), and a gyre spacing of 110 Å, matching the small angle x-ray scattering data of native chromatin (9). Beyond the H2B-α3/αC bundle, this fiber model also brings together the previous constraint of the H4-charge patch interaction (14). Combined, these two constraints make a strong case for a two-start crossed-linker model. Our attempts to model this four-helix bundle within a one-start solenoid model failed because of irreconcilable clashes between the DNA strands of neighboring nucleosomes.
Figure 6.
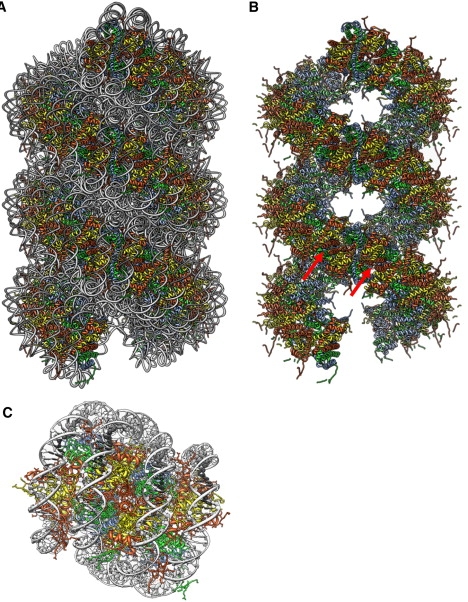
(A) A 30-nm fiber model, built using the H2B-α3/αC four-helix bundle as a constraint. (B) Underlying histone octamer scaffold in same view as A, with DNA removed and with octamer interfaces indicated by arrows. (C) Zoom view of internucleosomal interface contained in A, with two αC helices seen at center of the interface. PDB structure 1ZBB (15) was used as a template for creating these models.
Apart from achieving a convenient internucleosomal contact to maintain two-start connectivity, this model also makes some unique mechanistic predictions. Stripping the DNA away reveals the underlying histone scaffold (Fig. 6 B), and shows that these interfacial contacts are what largely determine the bulk architecture of the fiber. The extrapolation of internal histone octamer construction rules to the higher-order structure means that the fiber is not really composed of separate histone octamers, but rather a continuous histone oligomer formed by the repetitive spiraling of histone heterodimers joined by three types of four-helix bundles (H3-H3′, H2B-H4, and H2B-H2B′). This suggests a molecular mechanism wherein the formation of a 30-nm fiber is in part driven by the symmetry operation of successively placing histone heterodimers along a spiral path, where these twinned histone spirals represent the primary determinants of the final compact chromatin fiber. Flexibility at each of these four-helix bundle sites may also allow the fiber to tolerate NRL variability and aid in the compaction process, as similarly suggested for the H3-H3′ bundle by the nucleosome gaping model (39).
Our model may also help explain the transition observed between loose and compact fiber states (40), insofar as these states occur via distinct mechanisms of tail and face associations, respectively. Because the final determinant of fiber-folding is the octamer interface, our model clarifies why tails are partially redundant in vitro and may be compensated for by high salt (19) (although the base of the H4 tail is different, because it forms part of this interface). Bertin et al. demonstrated that the H4 N-terminal tail was required for attractive interactions between monodisperse, reconstituted nucleosome core particles (41). Because the tails do not determine the relative geometry of the fiber, they may be seen as mere facilitators of folding and not as outright determinants, acting more through charge neutralization to allow the close approach of DNA strands. Although the compaction process may still occur via a reduction in entry/exit angles, we distinguish the final compact structure from that of the two-angle model (42), wherein the linker length and entry/exit trajectories are the primary determinants of architecture of the compact structure. In our model, trajectories are a consequence of joining adjacent nucleosome stacks.
Linker-length, fiber-diameter interdependence is often cited as a requirement of the crossed-linker model (10), but this may be a simplistic notion, because the structural details of this model are not fully elucidated. The emerging picture is of a more complex helix displaying different connectivities between protein and DNA components, flexible tails, variable supercoiling, and uneven linker DNA lengths. Coupled to these variabilities, however, the histone scaffold may provide some needed regularity to the structure. By constraining helical parameters through invariant internucleosomal contacts, increases in linker DNA length need not be accommodated by simple changes in diameter, but rather through topological changes in fiber geometry between a twisted and a supercoiled form (as suggested elsewhere) (13,15).
Although the significance of the interface we have identified remains to be demonstrated, in analogous fashion to the H4 charge patch interaction, it is tempting to speculate that histone modifications to the H2B-αC region play a role in regulating the structure of the 30-nm chromatin fiber, and may also serve as a platform for the recognition of nucleosomes by various chromatin-associated factors.
Supporting Material
Two figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00487-1.
Supporting Material
Acknowledgments
The authors thank Professor Edward Egelman for his considerable input, and Dr. Anthony Crowther for constructive comments on an early version of the manuscript.
We are grateful for financial support received from the National Research Foundation and the Carnegie Corporation of New York.
Footnotes
Timothy D. Frouws' present address is Institut für Molekularbiologie und Biophysik ETHZ, ETH-Hönggerberg, Zürich, Switzerland.
References
- 1.Strahl B.D., Allis C.D. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 2.Cosgrove M.S., Boeke J.D., Wolberger C. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 2004;11:1037–1043. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 3.Luger K. Dynamic nucleosomes. Chromosome Res. 2006;14:5–16. doi: 10.1007/s10577-005-1026-1. [DOI] [PubMed] [Google Scholar]
- 4.Arents G., Burlingame R.W., Wang B.C., Love W.E., Moudrianakis E.N. The nucleosomal core histone octamer at 3.1 Å resolution: a tripartite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci. USA. 1991;88:10148–10152. doi: 10.1073/pnas.88.22.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 6.Richmond T.J., Davey C.A. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 7.Robinson P.J., Rhodes D. Structure of the “30 nm” chromatin fibre: a key role for the linker histone. Curr. Opin. Struct. Biol. 2006;16:336–343. doi: 10.1016/j.sbi.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Finch J.T., Klug A. Solenoidal model for superstructure in chromatin. Proc. Natl. Acad. Sci. USA. 1976;73:1897–1901. doi: 10.1073/pnas.73.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Widom J., Klug A. Structure of the 300 Å chromatin filament: x-ray diffraction from oriented samples. Cell. 1985;43:207–213. doi: 10.1016/0092-8674(85)90025-x. [DOI] [PubMed] [Google Scholar]
- 10.Robinson P.J., Fairall L., Huynh V.A., Rhodes D. EM measurements define the dimensions of the “30-nm” chromatin fiber: evidence for a compact, interdigitated structure. Proc. Natl. Acad. Sci. USA. 2006;103:6506–6511. doi: 10.1073/pnas.0601212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams S.P., Athey B.D., Muglia L.J., Schappe R.S., Gough A.H. Chromatin fibers are left-handed double helices with diameter and mass per unit length that depend on linker length. Biophys. J. 1986;49:233–248. doi: 10.1016/S0006-3495(86)83637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodcock C.L., Frado L.L., Rattner J.B. The higher-order structure of chromatin: evidence for a helical ribbon arrangement. J. Cell Biol. 1984;99:42–52. doi: 10.1083/jcb.99.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu C., Bassett A., Travers A. A variable topology for the 30-nm chromatin fibre. EMBO Rep. 2007;8:1129–1134. doi: 10.1038/sj.embor.7401115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorigo B., Schalch T., Kulangara A., Duda S., Schroeder R.R. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science. 2004;306:1571–1573. doi: 10.1126/science.1103124. [DOI] [PubMed] [Google Scholar]
- 15.Schalch T., Duda S., Sargent D.F., Richmond T.J. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 16.White C.L., Suto R.K., Luger K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001;20:5207–5218. doi: 10.1093/emboj/20.18.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chantalat L., Nicholson J.M., Lambert S.J., Reid A.J., Donovan M.J. Structure of the histone-core octamer in KCl/phosphate crystals at 2.15 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 2003;59:1395–1407. doi: 10.1107/s0907444903011880. [DOI] [PubMed] [Google Scholar]
- 18.Wood C.M., Nicholson J.M., Lambert S.J., Chantalat L., Reynolds C.D. High-resolution structure of the native histone octamer. Acta Crystallogr. F. 2005;61:541–545. doi: 10.1107/S1744309105013813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorigo B., Schalch T., Bystricky K., Richmond T.J. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 2003;327:85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 20.Klug A., Rhodes D., Smith J., Finch J.T., Thomas J.O. A low resolution structure for the histone core of the nucleosome. Nature. 1980;287:509–516. doi: 10.1038/287509a0. [DOI] [PubMed] [Google Scholar]
- 21.DeRosier D.J., Moore P.B. Reconstruction of three-dimensional images from electron micrographs of structures with helical symmetry. J. Mol. Biol. 1970;52:355–369. doi: 10.1016/0022-2836(70)90036-7. [DOI] [PubMed] [Google Scholar]
- 22.Egelman E.H. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- 23.Egelman E.H. The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J. Struct. Biol. 2007;157:83–94. doi: 10.1016/j.jsb.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 24.von Holt C., Brandt W.F., Greyling H.J., Lindsey G.G., Retief J.D. Isolation and characterization of histones. Methods Enzymol. 1989;170:431–523. doi: 10.1016/0076-6879(89)70061-6. [DOI] [PubMed] [Google Scholar]
- 25.Ludtke S.J., Baldwin P.R., Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 26.Penczek P., Radermacher M., Frank J. Three-dimensional reconstruction of single particles embedded in ice. Ultramicroscopy. 1992;40:33–53. [PubMed] [Google Scholar]
- 27.Frank J., Radermacher M., Penczek P., Zhu J., Li Y. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 28.Wriggers W., Milligan R.A., McCammon J.A. Situs: a package for docking crystal structures into low-resolution maps from electron microscopy. J. Struct. Biol. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- 29.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanner M.F. Python: a programming language for software integration and development. J. Mol. Graph. Model. 1999;17:57–61. [PubMed] [Google Scholar]
- 31.Sanner M.F., Olson A.J., Spehner J.C. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 32.Froimowitz M. HyperChem: a software package for computational chemistry and molecular modeling. Biotechniques. 1993;14:1010–1013. [PubMed] [Google Scholar]
- 33.Ponder J.W., Case D.A. Force fields for protein simulations. Adv. Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 34.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 35.Klug A., Crick F.H.C., Wyckoff H.W. Diffraction by helical structures. Acta Crystallogr. 1958;11:199–213. [Google Scholar]
- 36.Caspar D.L., Makowski L. The symmetries of filamentous phage particles. J. Mol. Biol. 1981;145:611–617. doi: 10.1016/0022-2836(81)90549-0. [DOI] [PubMed] [Google Scholar]
- 37.Chothia C., Levitt M., Richardson D. Structure of proteins: packing of alpha-helices and pleated sheets. Proc. Natl. Acad. Sci. USA. 1977;74:4130–4134. doi: 10.1073/pnas.74.10.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leforestier A., Dubochet J., Livolant F. Bilayers of nucleosome core particles. Biophys. J. 2001;81:2414–2421. doi: 10.1016/S0006-3495(01)75888-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mozziconacci J., Victor J.M. Nucleosome gaping supports a functional structure for the 30nm chromatin fiber. J. Struct. Biol. 2003;143:72–76. doi: 10.1016/s1047-8477(03)00102-3. [DOI] [PubMed] [Google Scholar]
- 40.Hansen J.C. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu. Rev. Biophys. Biomol. Struct. 2002;31:361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- 41.Bertin A., Renouard M., Pedersen J.S., Livolant F., Durand D. H3 and H4 histone tails play a central role in the interactions of recombinant NCPs. Biophys. J. 2007;92:2633–2645. doi: 10.1529/biophysj.106.093815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woodcock C.L., Grigoryev S.A., Horowitz R.A., Whitaker N. A chromatin folding model that incorporates linker variability generates fibers resembling the native structures. Proc. Natl. Acad. Sci. USA. 1993;90:9021–9025. doi: 10.1073/pnas.90.19.9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.