

Abstract
Rationale: In chronic heart failure, the lung endothelial permeability response to angiotensin II or thapsigargin-induced store depletion is ablated, although the mechanisms are not understood. Objectives: To determine whether the ablated permeability response to store depletion during heart failure was due to impaired expression of store operated Ca2+ channels in lung endothelium. Methods: Heart failure was induced by aortocaval fistula in rats. Permeability was measured in isolated lungs using the filtration coefficient and a low Ca2+/Ca2+ add-back strategy to identify the component of the permeability response dependent on Ca2+ entry. Main Results: In fistulas, right ventricular mass and left ventricular end diastolic pressure were increased and left ventricular shortening fraction decreased compared with shams. Thapsigargin-induced store depletion increased lung endothelial permeability in shams, but not in fistulas. Permeability increased in both groups after the Ca2+ ionophore A23187 or 14,15-epoxyeicosatrienoic acid, independent of store depletion. A diacylglycerol analog had no impact on permeability. Increased distance between the endoplasmic reticulum and the plasmalemmal membrane was ruled out as a mechanism for the loss of the permeability response to store depletion. Endothelial expression of the endoplasmic reticulum Ca2+ ATPase was not altered in fistulas compared with shams, whereas the store-operated canonical transient receptor potential channels 1, 3, and 4 were downregulated in extraalveolar vessel endothelium. Conclusions: We conclude that the adaptive mechanism limiting store depletion–induced endothelial lung injury in the aortocaval model of heart failure involves downregulation of store-operated Ca2+ channels.
Keywords: calcium channels, permeability, secondary pulmonary hypertension
Although sustained pulmonary venous hypertension and high levels of edemogenic factors such as angiotensin II are common findings in chronic heart failure (CHF), alveolar edema and the resultant respiratory distress do not always occur (1–3). Structural remodeling of the pulmonary endothelial barrier appears to play a role in this process, because thickening of both the endothelial and the alveolar epithelial layers of the septal network has been reported in several animal models of heart failure and in humans with chronic pulmonary venous hypertension (4–8). However, endothelial adaptations to CHF and chronic pulmonary venous hypertension appear to contribute more than simple mechanical advantage due to structural remodeling. Ca2+ entry in lung endothelium activates signaling cascades, which promote the formation of endothelial gaps and increase the filtration coefficient (Kf) (8–10), a sensitive index of water conductance through the endothelial barrier in the intact lung. An important pathway for Ca2+ entry in lung endothelium is that linking depletion of the endoplasmic reticulum Ca2+ stores to activation of store-operated Ca2+ channels (SOCs) in the plasmalemmal membrane (9, 10). In the canine lung, store depletion–induced increases in endothelial permeability require epoxyeicosatrienoic acids (EETs) (11–13), P450 derivatives of arachidonic acid that have been implicated as soluble mediators of store depletion–induced Ca2+ entry in systemic endothelium (14). Moreover, in the rapid-ventricular-pacing canine model of CHF, store-dependent increases in permeability were ablated, a finding that correlated with downregulation of P450 epoxygenases and decreased synthesis of EETs (15).
We hypothesized that CHF could confer similar protection against thapsigargin-induced endothelial injury in rat lung. Because, in normal rat lung, EETs do not mediate thapsigargin-induced increase in permeability (16), we reasoned that any adaptation in regulation of endothelial permeability in this species after CHF must involve other mechanisms. Our study was designed to address three potential mechanisms: (1) displacement of endothelial endoplasmic reticulum away from the plasma membrane, which could impair physical coupling to SOCs (17, 18); (2) downregulation of canonical transient receptor potential proteins (TRPs), which are putative components of SOCs (19–23) and have been shown to be expressed in cultured rat lung endothelium (20, 24); and (3) global attenuation of Ca2+ entry and/or responsiveness to Ca2+. We used the aortocaval fistula model of CHF and the measure of Kf in lungs isolated 3 to 5 wk after sham surgery or creation of the fistula to evaluate altered regulation of endothelial permeability. Some of the results of these studies have been previously reported in the form of abstracts (25, 26).
METHODS
Aortocaval Fistula Model of CHF
Infrarenal aortocaval fistulas were created in CD40 rats (250–275 g) anesthetized with pentobarbital sodium (50 mg/kg, intraperitoneally), as described previously (27). Sham rats were subjected to surgery but no fistula was created. Use of animals adhered to the NIH guidelines for the care and use of laboratory animals; protocols were approved by the Institutional Animal Care and Use Committee. At 3 to 5 wk after surgery, animals were anesthetized for terminal studies. Terminal bodyweight and the wet weights of the right ventricle and left ventricle plus septum were measured. Hemodynamics (Millar catheter), left ventricular dimensions, and left ventricular shortening fraction (both via M-mode echocardiography) were evaluated. Lungs were isolated for ex vivo perfusion (16).
Assessment of Endothelial Permeability
Isolated lungs were perfused at constant flow with buffer containing 1.8 mmol/L CaCl2 and 4% albumin unless noted (16, 28). Endothelial permeability was assessed by paired measurements of Kf at baseline and after treatment with the following: (1) 150 nmol/L thapsigargin to promote Ca2+ entry via SOCs (29, 30), (2) the Ca2+ ionophore A23187 (10 μmol/L) to test whether molecular targets critical for regulation of endothelial permeability retained their Ca2+ sensitivity after CHF, or (3) 14,15-EET (3 μmol/L) to assess a Ca2+ entry pathway regulated independent of store depletion (16). To confirm that 14,15-EET increased endothelial permeability in a Ca2+ entry–dependent manner after CHF, lungs from fistulas were perfused with a low Ca2+ buffer containing 0.02 mmol/L CaCl2 and 4% bovine serum albumin (16). Kf was measured at baseline and after addition of 14,15-EET in low Ca2+, and then again after Ca2+ add-back to restore physiologic Ca2+ concentration (2.2 mmol/L). To evaluate the involvement of receptor-operated Ca2+ entry channels (31, 32), Kf was measured in normal rat lungs challenged with 100 or 300 μmol/L 1-oleoyl-2-acetyl-sn-glycerol (OAG), a diacylglycerol analog (23, 32); buffers contained 3% dextran and 1% albumin to limit OAG–albumin binding (16). To determine whether lung injury induced by 300 μmol/L OAG was due to Ca2+ entry, we used the low Ca2+/Ca2+ add-back strategy in a separate group of lungs: Kf was measured before and after addition of OAG at low Ca2+, and then again after Ca2+ add-back.
Endoplasmic Reticulum–Plasmalemmal Membrane Distance
Lung tissue was fixed by immersion in 3% glutaraldehyde, postfixed in osmium tetroxide, and embedded in PolyBed 812 resin (Polysciences, Inc., Warrington, PA). Sections (80 nm) were stained with uranyl acetate and Reynolds lead citrate and examined using transmission electron microscopy (Phillips CM 100; FEI Company, Hillsboro, OR). The minimal distance between the endoplasmic reticulum and the apical or basolateral plasmalemmal membrane was measured from random micrographs of endothelial cells in pulmonary arteries and microvessels (33).
Immunohistochemistry
Lungs from shams (n = 4) and fistulas (n = 5) were perfusion-fixed with 4% paraformaldehyde, paraffin-embedded, and sectioned (5 μm). Primary antibodies were used to detect expression of TRPC1 and TRPC4 (1:20; Santa Cruz Biotechnology, Santa Cruz, CA), TRPC6/TRPC7 (1:10; Santa Cruz Biotechnology), and TRPC3 (1:50; Alomone Laboratories, Jerusalem, Israel). To rule out loss of thapsigargin's pharmacologic target, the sarcoplasmic endoplasmic reticulum Ca2+ ATPase (SERCA), we also evaluated endothelial expression of SERCA2 and SERCA3 (1:10; Affinity BioReagents, Golden, CO).
Statistics
Data are means ± SEM. Means were compared using an unpaired t test or one-way analysis of variance with Tukey's post hoc t test. p values less than 0.05 were considered significant.
RESULTS
Documentation of CHF
Fistula patency was confirmed using transabdominal pulsed-Doppler ultrasound (Figures 1A and 1C) or direct visualization of oxygenated blood permeating into the inferior vena cava at terminal experiments. Left ventricular dimensions and left ventricular shortening fraction in sham animals (Table 1) were similar to those previously reported for normal rats (34–36). In contrast, fistulas exhibited left ventricular dilation (Figures 1B and 1D) and mild ventricular dysfunction, similar to that previously reported in this model (37, 38). Left ventricular end diastolic pressure increased 2.3-fold and left ventricular shortening fraction was reduced by 21% compared with shams. Secondary pulmonary hypertension was evidenced by increased right ventricular mass and the right ventricular/(left ventricular + septum) mass ratio in fistulas (Table 1). The increase in right ventricular mass positively correlated with left ventricular end diastolic pressure (r2 = 0.64, p < 0.05).
Figure 1.

Echocardiographic assessment of fistula patency and left ventricular function in the chronic heart failure (CHF) model. (A, C) Angio-mode Doppler images from representative sham and fistula animals, respectively. High-flow velocity limited to the abdominal aorta (a) was observed in shams but could also be seen in the inferior vena cava (vc) in fistulas. (B, D) M-mode echos in representative sham and fistula animals, respectively. Left ventricular dimensions were increased in the fistula group compared with shams (see Table 1 for detail).
TABLE 1.
Chronic heart failure assessment at 3 to 5 wk after fistula creation
| Shams | Fistulas | |
|---|---|---|
| Body weight, g | 438 ± 9 (n = 30) | 454 ± 6 (n = 39) |
| RV mass, g | 0.20 ± 0.01 (n = 22) | 0.30 ± 0.01 (n = 31)* |
| RV/(LV + S) mass ratio | 0.26 ± 0.01 (n = 22) | 0.32 ± 0.01 (n = 31)* |
| Heart rate, beats/min | 379 ± 17 (n = 6) | 335 ± 16 (n = 11) |
| Mean arterial pressure, mm Hg | 111 ± 5 (n = 6) | 107 ± 4 (n = 11) |
| LVEDP, mm Hg | 3.4 ± 0.6 (n = 5) | 7.8 ± 1.5 (n = 8)* |
| LV shortening fraction, % | 44.1 ± 2.6 (n = 6) | 34.7 ± 1.3 (n = 12)* |
| LVESD, cm | 0.37 ± 0.03 (n = 6) | 0.53 ± 0.02 (n = 12)* |
| LVEDD, cm | 0.67 ± 0.03 (n = 6) | 0.81 ± 0.02 (n = 12)* |
Definition of abbreviations: LV = left ventricular; LVEDD = left ventricular end diastolic diameter; LVEDP = left ventricular end diastolic pressure; LVESD = left ventricular end systolic diameter; RV = right ventricular mass; RV/(LV + S) = ratio of right ventricle to left ventricle plus septum mass.
Data are means ± SE.
p < 0.05 versus shams.
Baseline Hemodynamics in the Isolated Lung
At baseline, pulmonary arterial pressure (Pa), capillary pressure, total vascular resistance (Rt), and Kf were not significantly different in shams and fistulas, when compared under the same perfusate conditions (Table 2). A tendency for increased Pa in the fistula group can be attributed to the higher isogravimetric flow rate when compared with shams (16). No differences were found in the hemodynamics or Kf between the fistula groups perfused with low or normal Ca2+ buffer.
TABLE 2.
Isolated lung hemodynamics at baseline
| Perfusate
|
|||
|---|---|---|---|
| Normal Ca2+
|
Low Ca2+
|
||
| Shams | Fistulas | Fistulas | |
| n | 16 | 15 | 5 |
| Body weight, g | 418 ± 9 | 433 ± 9 | 455 ± 18 |
| Q, ml/min | 12.3 ± 0.5 | 14.1 ± 0.6* | 12.7 ± 0.4 |
| Pa, cm H2O | 13.8 ± 0.5 | 16.9 ± 1.4 | 13.9 ± 1.0 |
| Pc, cm H2O | 8.7 ± 0.2 | 9.4 ± 0.6 | 9.0 ± 0.6 |
| Pv, cm H2O | 4.2 ± 0.1 | 4.3 ± 0.1 | 4.2 ± 0.3 |
| Rt, cm H2O/ml/min | 0.79 ± 0.04 | 0.92 ± 0.13 | 0.78 ± 0.08 |
| Kf, ml/min/cm H2O/g dry wt | 0.009 ± 0.001 | 0.008 ± 0.001 | 0.009 ± 0.001 |
Definition of abbreviations: Kf = filtration coefficient; Pa = pulmonary artery pressure; Pc = pulmonary capillary pressure; Pv = pulmonary venous pressure; Q = perfusate flow; Rt = total pulmonary vascular resistance.
Data are means ± SE.
p < 0.05 versus shams.
Impact of CHF on Store Depletion–induced Endothelial Injury
Thapsigargin increased Kf approximately fivefold in lungs from shams, but had no impact on permeability in the fistula group (Figure 2). Thapsigargin modestly increased Pa in shams from 13.8 ± 0.7 cm H2O at baseline to 18.5 ± 2.2 cm H2O after treatment (p < 0.05), whereas in fistulas, Pa did not change (18.0 ± 2.2 vs. 19.3 ± 3.1 cm H2O, respectively). Nonetheless, no significant differences were observed in Rt in shams or fistulas as a result of treatment with thapsigargin (from 0.91 ± 0.06 to 1.41 ± 0.04 and from 1.22 ± 0.34 to 1.32 ± 0.27 cm H2O/ml/min, comparing baseline and final, respectively), suggesting that the permeability effect of thapsigargin was unrelated to a hemodynamic insult.
Figure 2.

Effect of store depletion via thapsigargin on endothelial permeability. Baseline (BL) and final Kf (F) were compared after treatment with thapsigargin. Thapsigargin increased endothelial permeability in shams (open bars, n = 7) but not in fistulas (hatched bars, n = 5). *p < 0.05 versus BL. Kf = filtration coefficient.
Effect of Store-independent Ca2+ Entry on Endothelial Injury during CHF
The Ca2+ ionophore A23187 increased Kf in both shams and fistulas (Figure 3A), suggesting that the molecular targets for Ca2+-dependent regulation of endothelial permeability remained intact. The 14,15-EET increased permeability in shams, but the final Kf was not statistically different from baseline in the fistula group (Figure 3A) due to an exaggerated response in one experiment. Nonetheless, 14,15-EET increased Kf by a minimum of 2.2-fold in both groups. We had previously found that the endothelial permeability response to 14,15-EET involved both Ca2+ entry–dependent and Ca2+-independent mechanisms (16). To ensure that the maintenance of a permeability response to 14,15-EET after CHF was not due to the loss of a Ca2+ entry pathway coupled with upregulation of the Ca2+-insensitive component, we reevaluated the effect of 14,15-EET in the fistula group using a low Ca2+ buffer, with Ca2+ add-back to restore a physiologic Ca2+ concentration. The pattern of the permeability response to 14,15-EET in this experiment (Figure 3B) mirrored that previously reported in normal rat lung (16).
Figure 3.

Effect of the Ca2+ entry agonists A23187 and 14,15–epoxyeicosatrienoic acid (14,15-EET) on endothelial permeability. (A) Kf was measured at baseline (BL) and after treatment (F) in shams (open bars) and fistulas (hatched bars); n = 5 in each group. A23187 significantly increased endothelial permeability in both groups. The 14,15-EET increased endothelial permeability in shams (n = 4) but not significantly in fistulas (n = 5) due to an exaggerated response in one of the fistula experiments. *p < 0.05 versus BL. (B) To show dependence of the 14,15-EET permeability response on Ca2+ entry, in fistulas (n = 5) we measured Kf using a low Ca2+/Ca2+ add-back strategy. The 14,15-EET increased endothelial permeability at low Ca2+, with further injury after Ca2+ add-back. *p < 0.05 versus BL.
We concluded that CHF in the aortocaval fistula model specifically ablated regulation of endothelial permeability via the store-operated Ca2+ entry pathway. Because we had previously shown that thapsigargin's effect in normal rat lung was completely dependent on Ca2+ entry (16), and our current results showed that the endothelial barrier remained sensitive to increased intracellular Ca2+, we turned our investigation to the study of mechanisms linking store depletion and Ca2+ entry.
Physical Coupling Mechanism
Physical coupling of the endoplasmic reticulum and the putative SOCs appears to be required for Ca2+ entry after depletion of the endothelial Ca2+ store (18, 21). Relative displacement of the endoplasmic reticulum from the plasmalemmal membrane in cultured lung microvascular endothelium has been believed to explain the resistance of this cell type to store-operated Ca2+ entry (33), compared with that in pulmonary artery endothelium. However, the distances between these two structures were no different in shams and fistulas in either the pulmonary artery or the septal capillary endothelium (Figure 4). This suggests that, despite the documented endothelial thickening after CHF (4–7), physical uncoupling by a means of increased distance between the store and SOCs is not likely to explain the adaptive protection observed after CHF in rat lung.
Figure 4.

Distances between the endoplasmic reticulum and the endothelial plasmalemmal membrane in intact lungs from shams (n = 4) and fistulas (n = 3). Data points and horizontal lines represent individual and average measurements, respectively, in pulmonary artery (left) and septal microvessel (right) endothelial cells (EC). Although the endoplasmic reticulum and the plasma membrane tended to be more distant from each other at the basolateral aspect compared with that in the apical compartment of pulmonary artery endothelium, this trend was not significant. We found no significant differences between shams and fistulas at either site in either cell population.
Expression of SOCs
Thapsigargin, used as a pharmacologic tool to evoke store depletion, acts by inhibiting the SERCA, leading to activation of SOCs. Because TRPC1 and TRPC4 have been implicated as components of SOCs, we first asked whether CHF downregulated the expression of SOCs. We used immunohistochemistry to probe for TRPC1 and TRPC4 in lungs from shams and fistulas, and compared these data to expression of the receptor-operated channels TRPC3 and TRPC6/7. In shams, endothelial expression of all four isoforms was confirmed in extraalveolar arteries and veins larger than 25 μm in diameter (Figure 5), as well as in septal capillaries (not shown). TRPC1 and TRPC3 were diffusely expressed in vascular smooth muscle, whereas TRPC4 showed a punctuate expression pattern in this cell type (Figure 5). TRPC1 was expressed at the apical aspect of airway epithelium, whereas TRPC4 was present at the basal compartment in the same cell type (not shown). TRPC3 was expressed in interstitial cells and alveolar macrophages. TRPC6/7 was expressed almost exclusively in endothelium. In contrast, after fistula-induced CHF, expression of TRPC1 or TRPC3 was not observed in endothelium of any sized vessel. TRPC4 was downregulated in endothelium of small extraalveolar vessels and septal capillaries, whereas variable expression was observed in conduit vessels. In vascular smooth muscle, expression of TRPC4 appeared to be more robust in the media and adventitia in fistulas compared with that of shams. TRPC6/7 expression did not appear to differ between the two groups. We ruled out loss of SERCA expression as well. Endothelial expression of neither SERCA2 nor SERCA3 changed after CHF (Figure 6). Collectively, these data support the notion that the specific loss of Ca2+ store depletion–induced endothelial injury after CHF may be explained by the endothelial downregulation of TRPC1, TRPC3, and/or TRPC4.
Figure 5.
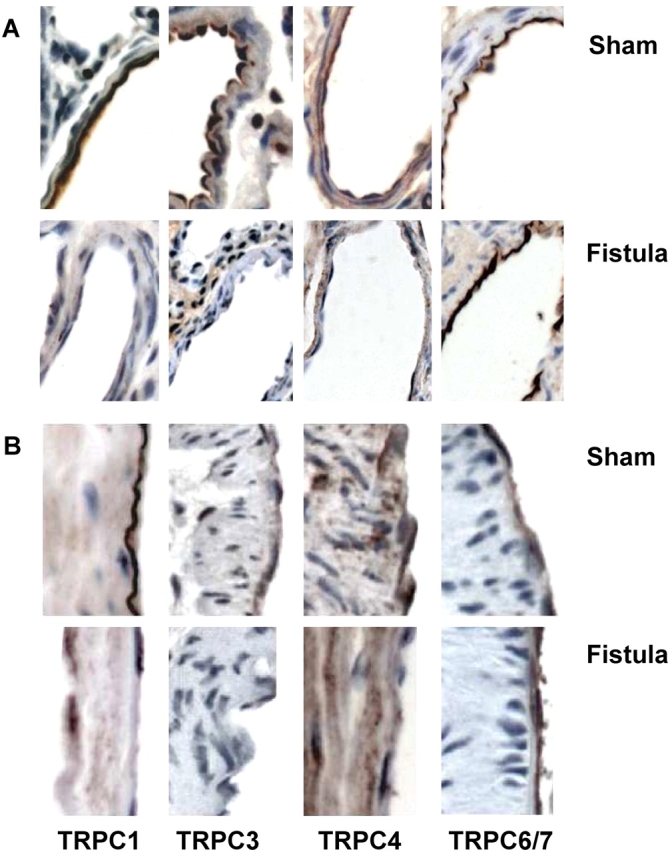
Immunostaining for transient receptor potential proteins TRPC1, TRPC3, TRPC4, and TRPC6/7 in rat lungs. Expression of each TRPC isoform in endothelium was confirmed in small muscular or partially muscularized extraalveolar vessels (A) and conduit artery (B) from shams and fistulas. Expression of TRPC1, TRPC3, and TRPC4 in endothelial cells was downregulated in small pulmonary arterioles after CHF, whereas TRPC6/7 expression was unchanged. TRPC4 expression in the medial layer of large conduit pulmonary vessels was increased in fistulas. Original magnification = 40×. See text for more detail.
Figure 6.

Expression of sarcoplasmic endoplasmic reticulum Ca2+ ATPase (SERCA) isoforms in rat lung. Expression of SERCA2 (left) and SERCA3 (right) in endothelium was confirmed in small muscular or partially muscularized extraalveolar vessels and conduit artery from shams (top row) and fistulas (bottom row). There were no apparent differences in endothelial SERCA expression after CHF. Original magnification = 40×.
Evaluation of Receptor-operated Ca2+ Entry on Endothelial Permeability
As a first approach to determining whether downregulation of TRPC3 could explain loss of the permeability response to store depletion, we asked whether activation of TRPCs via diacylglycerol could increase endothelial permeability in normal rat lung. In this group, baseline hemodynamics and Kf (0.011 ± 0.001 ml/min/cm H2O/g dry wt) were not significantly different from that in shams (Table 2). OAG (100 μmol/L), a concentration that promotes Ca2+ entry via receptor-operated TRPCs (32, 39, 40), did not alter permeability in the normal rat lung (Figure 7). At 300 μmol/L, OAG increased permeability, although this was associated with a significant pressor response: 10 min after treatment with OAG, Pa increased from 17.2 ± 0.4 to 28.4 ± 0.8 cm H2O (p < 0.05). Importantly, lungs treated with 300 μmol/L OAG using the low Ca2+/Ca2+ add-back strategy exhibited neither a pressor response nor an increase in endothelial permeability (Figure 7), suggesting that the permeability response was related to pressure-induced barotrauma.
Figure 7.

Effect of receptor-operated Ca2+ entry on endothelial permeability. Baseline (BL) and final (F) Kf were compared after treatment with 1-oleoyl-2-acetyl-sn-glycerol (OAG). With physiologic extracellular Ca2+, OAG increased endothelial permeability at 300 but not at 100 μmol/L (n = 3 in each group). Using the low Ca2+/Ca2+ add-back strategy, 300 μmol/L OAG (n = 3) was found to have no impact on endothelial permeability. See text for more detail. *p < 0.05 versus BL.
DISCUSSION
This study provides the first evidence of a protective adaptation limiting store depletion–induced acute lung injury in an aortocaval fistula model of CHF, associated with downregulated expression of key store-operated TRPC channels in pulmonary endothelial cells. Although alteration of Ca2+ homeostasis has been reported to occur in the lung endothelium and in the myocardium during CHF (12, 41, 42), little is known about the mechanisms responsible for the protection against pulmonary edema (12, 13). Adaptive remodeling in idiopathic pulmonary arterial hypertension or chronic hypoxia upregulate TRPC expression and receptor- or store-operated Ca2+ entry in pulmonary vascular smooth muscle (TRPC6) or pulmonary artery endothelium (TRPC4), respectively (24, 43). In contrast, our data suggest that the adaptive mechanism limiting store depletion–induced endothelial injury in rat lung after CHF relates to decreased Ca2+ entry. Expression of putative SOCs, including TRPC1, TRPC3, and TRPC4, was downregulated in fistulas compared with shams, whereas expression of SERCA isoforms was not altered. Because OAG, an agonist which evokes Ca2+ entry via receptor-operated TRPC channels, failed to increase endothelial permeability in the normal rat lung, we suggest that downregulated expression of TRPC1 and TRPC4 in lung endothelium after CHF provides a plausible explanation for the ablation of store depletion–induced endothelial injury.
In the rapid-ventricular-pacing canine model of CHF, the store depletion–dependent permeability response to both angiotensin II and thapsigargin was ablated (12, 13), a mechanism linked to decreased EET synthesis (15). Because in the aortocaval fistula model, elevation of pulmonary vascular pressures and plasma angiotensin II concentrations are common findings (2, 44), we asked whether the resistance to store depletion–induced lung injury is a protective adaptation that occurs exclusively in the canine model or if it is a more generic mechanism inherent to CHF. In the present study, we observed an approximately fivefold increase in Kf after treatment with thapsigargin in shams (Figure 2). However, thapsigargin did not alter endothelial permeability in fistulas, suggesting that the protection against store depletion–mediated lung injury may occur irrespective of the species or the model. Nonetheless, the mechanism appears to be species-dependent because EETs provide a link between store depletion and increased endothelial permeability in canine lung, but not in rat lung (11, 16).
Our data support the notion that CHF in the rat specifically leads to downregulation of SOCs in pulmonary endothelium, conferring protection against store depletion–induced lung injury. TRPCs have been postulated as putative SOCs (45, 46), and all but TRPC2 have been shown to be expressed in endothelium (24, 46). Functional SOCs require assembly of four TRPC subunits to form a tetrameric channel (31). Although the contribution of each isoform to Ca2+ entry initiated by store depletion is controversial (21, 45), heteromultimers formed by the association of TRPC1 and TRPC4 can be regulated by store depletion (20, 22). In contrast, heteromultimers of TRPC3 and TRPC6 or TRPC7 typically form a channel regulated by diacylglycerol (31, 47). However, functional channels composed of TRPC1 and TRPC3 subunits have been also described (32, 48, 49), and when expressed at low levels, TRPC3 may act as a SOC (23). In cultured pulmonary endothelium and vascular smooth muscle, mRNA encoding for TRPC1, TRPC3, TRPC4, TRPC6, and TRPC7 has been identified (24, 50, 51), and in the present study, protein expression in vivo was confirmed by immunohistochemistry. Overexpression of TRPC1 increased Ca2+ influx after store depletion in human umbilical vein endothelium (52), whereas antibodies against TRPC1 blocked a store-operated Ca2+ current in pulmonary artery endothelium (20). Furthermore, thrombin-induced store-operated Ca2+ entry and the resultant pulmonary endothelial permeability response are impaired in TRPC4−/− mice (22). In fistulas, expression of TRPC1 and TRPC3 was not observed in the lung endothelium. TRPC4 expression was modestly maintained in endothelium from conduit vessels but it was not expressed in endothelium of small muscular or partially muscularized extraalveolar vessels. This is significant because thapsigargin-induced endothelial injury is localized to small extraalveolar vessels in the rat lung (9, 53, 54). Thus, downregulation of endothelial TRPC1, TRPC3, and/or TRPC4 in the fistula group could potentially explain the observed resistance to store depletion–induced lung injury.
Although our data show loss of endothelial TRPC3 as well as TRPC1 and TRPC4, in conjunction with loss of the store depletion–induced permeability response, we conclude that the TRPC3/TRPC6/TRPC7 subfamily does not play a direct role in regulation of endothelial permeability in rat lung. We base this conclusion on the observation that OAG had no impact on endothelial permeability in the isolated rat lung (Figure 7). OAG is often used as a tool to activate Ca2+ entry via TRPCs in heterologous expression systems, although the specific TRPC channels targeted are the subject of debate (23, 32, 39, 40). Complicating this issue is the potential for concomitant activation of protein kinase C (PKC) by OAG and phosphorylation-dependent downregulation of TRPC channels (39, 55). Whether these paradigms pertain to regulation of Ca2+ entry via TRPCs in endothelial cells may well depend on the isoforms of PKC and TRPCs endogenously expressed. OAG promoted endothelial Ca2+ entry via TRPC6 and increased hydraulic permeability (a component of the Kf) in frog mesenteric microvessels (56). In contrast, thapsigargin and thrombin induced TRPC1 phosphorylation via PKCα in endothelium, Ca2+ entry, and barrier disruption (57). TRPC1, TRPC3 and TRPC6/7 are clearly expressed in endothelium of rat lung, yet OAG did not alter the filtration coefficient. Although 300 μmol/L OAG increased endothelial permeability in the rat lung, this was associated with a significant pressor response and neither effect was observed in the low Ca2+/Ca2+ add-back experiment, suggesting that the injury evoked by high-dose OAG at normal Ca2+ was secondary to mechanical endothelial injury (58). Collectively, our data, taken in light of other published work, provide strong inferential support for the conclusion that loss of endothelial TRPC1 and TRPC4 expression, but not TRPC3, in the aortocaval fistula model explains the resistance against store depletion–induced lung injury.
Store-operated Ca2+ entry appears to require conformational coupling between the endoplasmic reticulum and adjacent plasmalemmal membrane Ca2+ channels in some models (17, 21, 45). Furthermore, Cioffi and colleagues (21) and King and colleagues (33) found that increased endoplasmic reticulum–plasmalemmal membrane distance in cultured lung microvascular endothelium, compared with that in pulmonary artery endothelium, correlated with the reduced permeability response of microvascular endothelium to store depletion. In contrast, we found no difference in the distance at the apical aspect and a trend toward a smaller distance at the basolateral aspect of pulmonary microvascular endothelium in shams. This may relate to tensional forces on the microvasculature in intact lung, because peripheral vessels are subject to radial traction. In fact, endothelial thickness is less in small, nonmuscular, extraalveolar vessels from rat lung compared with that in pulmonary artery (59). Nonetheless, because endothelial thickness is increased in humans with CHF, and in the rapid ventricular pacing and aortic banding models of CHF (4–8), it was important to evaluate altered mechanical coupling between these two endothelial structures. However, we found no significant difference in distance in either the apical or the basolateral compartment of pulmonary or microvascular endothelium in the intact lung from shams and fistulas (Figure 4).
We also addressed whether the ablation of acute lung injury after store depletion in the aortocaval fistula model of CHF was due to decreased sensitivity of Ca2+ targets in endothelium critical for regulation of permeability or to a generic impairment of Ca2+ entry. Treatment with the Ca2+ ionophore A23187, a drug that produces a global rise in intracellular Ca2+, increased endothelial permeability in shams and fistulas. This suggests that once Ca2+ entry is promoted, the Ca2+ response of the endothelial contractile apparatus and junctional complexes is maintained after CHF, a conclusion also supported by the preserved response to 14,15-EET after CHF. A further implication of our results is that in rat lung endothelium, 14,15-EET activates a Ca2+ entry pathway distinct from that mediated by TRPC1, TRPC3, or TRPC4.
In conclusion, our study shows that ablation of store depletion–induced lung injury occurs in a high-output model of CHF in the rat, similar to previous reports in the canine CHF model (12, 13). Resistance to store depletion–induced pulmonary endothelial injury, but not to that evoked by 14,15-EET, indicates that the adaptation in CHF selectively targets Ca2+ entry regulated by store depletion in rat lung. Our data support the notion that loss of the store depletion permeability response is likely due to downregulated expression of TRPC1 and TRPC4 in pulmonary endothelial cells after CHF.
Acknowledgments
The authors thank Dr. Greg Brower (Auburn University) and Sue Barnes (University of South Alabama) for their assistance with the fistula model of heart failure. They also thank Virginia Parks and Freda McDonald (University of South Alabama) for their technical assistance with ultrasound and electron microscopy, respectively.
Supported by National Heart, Lung, and Blood Institute grant HL-61955 and American Heart Association Southeast Affiliate predoctoral fellowship 0315049B.
Conflict of Interest Statement: None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Rame JE, Dries DL, Drazner MH. The prognostic value of the physical examination in patients with chronic heart failure. Congest Heart Fail 2003;9:170–178. [DOI] [PubMed] [Google Scholar]
- 2.Riegger GA, Liebau G, Bauer E, Kochsiek K. Vasopressin and renin in high output heart failure of rats: hemodynamic effects of elevated plasma hormone levels. J Cardiovasc Pharmacol 1985;7:1–5. [DOI] [PubMed] [Google Scholar]
- 3.Stevenson LW, Perloff JK. The limited reliability of physical signs for estimating hemodynamics in chronic heart failure. JAMA 1989;261:884–888. [PubMed] [Google Scholar]
- 4.Huang W, Kingsbury MP, Turner MA, Donnelly JL, Flores NA, Sheridan DJ. Capillary filtration is reduced in lungs adapted to chronic heart failure: morphological and haemodynamic correlates. Cardiovasc Res 2001;49:207–217. [DOI] [PubMed] [Google Scholar]
- 5.Kay JM, Edwards FR. Ultrastructure of the alveolar-capillary wall in mitral stenosis. J Pathol 1973;111:239–245. [DOI] [PubMed] [Google Scholar]
- 6.Townsley MI, Fu Z, Mathieu-Costello O, West JB. Pulmonary microvascular permeability: responses to high vascular pressure after induction of pacing-induced heart failure in dogs. Circ Res 1995;77:317–325. [DOI] [PubMed] [Google Scholar]
- 7.Kingsbury MP, Huang W, Donnelly JL, Jackson E, Needham E, Turner MA, Sheridan DJ. Structural remodelling of lungs in chronic heart failure. Basic Res Cardiol 2003;98:295–303. [DOI] [PubMed] [Google Scholar]
- 8.Chetham PM, Babal P, Bridges JP, Moore TM, Stevens T. Segmental regulation of pulmonary vascular permeability by store-operated Ca2+ entry. Am J Physiol 1999;276:L41–L50. [DOI] [PubMed] [Google Scholar]
- 9.Norwood N, Moore TM, Dean DA, Bhattacharjee R, Li M, Stevens T. Store-operated calcium entry and increased endothelial cell permeability. Am J Physiol 2000;279:L815–L824. [DOI] [PubMed] [Google Scholar]
- 10.Stevens T, Garcia JG, Shasby DM, Bhattacharya J, Malik AB. Mechanisms regulating endothelial cell barrier function. Am J Physiol 2000;279:L419–L422. [DOI] [PubMed] [Google Scholar]
- 11.Ivey CL, Stephenson AH, Townsley MI. Involvement of cytochrome P-450 enzyme activity in the control of microvascular permeability in canine lung. Am J Physiol 1998;275:L756–L763. [DOI] [PubMed] [Google Scholar]
- 12.Ivey CL, Roy BJ, Townsley MI. Ablation of lung endothelial injury after pacing-induced heart failure is related to alterations in Ca++ signaling. Am J Physiol 1998;275:H844–H851. [DOI] [PubMed] [Google Scholar]
- 13.Roy BJ, Pitts VH, Townsley MI. Pulmonary vascular response to angiotensin II in canine pacing-induced heart failure. Am J Physiol 1996;271:H222–H227. [DOI] [PubMed] [Google Scholar]
- 14.Graier WF, Simecek S, Sturek M. Cytochrome P450 mono-oxygenase-regulated signalling of Ca++ entry in human and bovine endothelial cells. J Physiol 1995;482:259–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Townsley MI, Leuwerke S, Jacobs ER. Depressed EET synthesis does not explain enhanced pressor response to norepinephrine (NE) after heart failure (HF) [abstract]. FASEB J 1999;13:A500. [Google Scholar]
- 16.Alvarez DF, Gjerde EA, Townsley MI. Role of EETs in regulation of endothelial permeability in rat lung. Am J Physiol 2004;286:L445–L451. [DOI] [PubMed] [Google Scholar]
- 17.Berridge MJ. Capacitative calcium entry. Biochem J 1995;312:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu S, Sangerman J, Li M, Brough GH, Goodman SR, Stevens T. Essential control of an endothelial cell ISOC by the spectrin membrane skeleton. J Cell Biol 2001;154:1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nilius B. From TRPs to SOCs, CCEs, and CRACs: consensus and controversies. Cell Calcium 2003;33:293–298. [DOI] [PubMed] [Google Scholar]
- 20.Brough GH, Wu S, Cioffi D, Moore TM, Li M, Dean N, Stevens T. Contribution of endogenously expressed Trp1 to a Ca++-selective, store-operated Ca++ entry pathway. FASEB J 2001;15:1727–1738. [PubMed] [Google Scholar]
- 21.Cioffi DL, Wu S, Stevens T. On the endothelial cell ISOC. Cell Calcium 2003;33:323–336. [DOI] [PubMed] [Google Scholar]
- 22.Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4−/− mice interferes with increase in lung microvascular permeability. Circ Res 2002;91:70–76. [DOI] [PubMed] [Google Scholar]
- 23.Vazquez G, Wedel BJ, Trebak M, St John BG, Putney JW Jr. Expression level of the canonical transient receptor potential 3 (TRPC3) channel determines its mechanism of activation. J Biol Chem 2003;278:21649–21654. [DOI] [PubMed] [Google Scholar]
- 24.Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, Yuan JX. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol 2003;285:L1233–L1245. [DOI] [PubMed] [Google Scholar]
- 25.Alvarez DF, Townsley MI. Are P450 epoxygenase arachidonic acid derivatives (EETs) involved in lung endothelial permeability after heart failure? [abstract] FASEB J 2002;16:A402–A403. [Google Scholar]
- 26.Alvarez DF, King JA, Townsley MI. Adaptive mechanism underlying resistance to acute lung injury in heart failure [abstract]. Am J Respir Crit Care Med 2004;169:A157. [Google Scholar]
- 27.Garcia R, Diebold S. Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc Res 1990;24:430–432. [DOI] [PubMed] [Google Scholar]
- 28.Perry M, Taylor AE. Phorbol myristate acetate-induced injury of isolated perfused rat lungs: neutrophil dependence. J Appl Physiol 1988;65:2164–2169. [DOI] [PubMed] [Google Scholar]
- 29.Mikkelsen EO, Thastrup O, Christensen SB. Effects of thapsigargin in isolated rat thoracic aorta. Pharmacol Toxicol 1988;62:7–11. [DOI] [PubMed] [Google Scholar]
- 30.Takemura H, Hughes AR, Thastrup O, Putney JW Jr. Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells: evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J Biol Chem 1989;264:12266–12271. [PubMed] [Google Scholar]
- 31.Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA 2002;99:7461–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, Groschner K. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-senstitive cation channels. J Biol Chem 2000;275:27799–27805. [DOI] [PubMed] [Google Scholar]
- 33.King J, Hamil T, Creighton J, Wu S, Bhat P, McDonald F, Stevens T. Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc Res 2004;67:139–151. [DOI] [PubMed] [Google Scholar]
- 34.Derumeaux G, Mulder P, Richard V, Chagraoui A, Nafeh C, Bauer F, Henry JP, Thuillez C. Tissue Doppler imaging differentiates physiological from pathological pressure-overload left ventricular hypertrophy in rats. Circulation 2002;105:1602–1608. [DOI] [PubMed] [Google Scholar]
- 35.Slama M, Susic D, Varagic J, Ahn J, Frohlich ED. Echocardiographic measurement of cardiac output in rats. Am J Physiol 2003;284:H691–H697. [DOI] [PubMed] [Google Scholar]
- 36.Reffelmann T, Kloner RA. Transthoracic echocardiography in rats: evalution of commonly used indices of left ventricular dimensions, contractile performance, and hypertrophy in a genetic model of hypertrophic heart failure (SHHF-Mcc-facp-Rats) in comparison with Wistar rats during aging. Basic Res Cardiol 2003;98:275–284. [DOI] [PubMed] [Google Scholar]
- 37.Brower GL, Henegar JR, Janicki JS. Temporal evaluation of left ventricular remodeling and function in rats with chronic volume overload. Am J Physiol 1996;271:H2071–H2078. [DOI] [PubMed] [Google Scholar]
- 38.Flaim SF, Minteer WJ, Nellis SH, Clark DP. Chronic arteriovenous shunt: evaluation of a model for heart failure in rat. Am J Physiol 1979;236:H698–H704. [DOI] [PubMed] [Google Scholar]
- 39.Venkatachalam K, Ma HT, Ford DL, Gill DL. Expression of functional receptor-coupled TRPC3 channels in DT40 triple receptor InsP3 knockout cells. J Biol Chem 2001;276:33980–33985. [DOI] [PubMed] [Google Scholar]
- 40.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999;397:259–263. [DOI] [PubMed] [Google Scholar]
- 41.Esposito G, Santana LF, Dilly K, Cruz JD, Mao L, Lederer WJ, Rockman HA. Cellular and functional defects in a mouse model of heart failure. Am J Physiol 2000;279:H3101–H3112. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt AG, Zhai J, Carr AN, Gerst MJ, Lorenz JN, Pollesello P, Annila A, Hoit BD, Kranias EG. Structural and functional implications of the phospholamban hinge domain: impaired SR Ca2+ uptake as a primary cause of heart failure. Cardiovasc Res 2002;56:248–259. [DOI] [PubMed] [Google Scholar]
- 43.Kunichika N, Landsberg JW, Yu Y, Kunichika H, Thistlethwaite PA, Rubin LJ, Yuan JX. Bosentan inhibits transient receptor potential channel expression in pulmonary vascular myocytes. Am J Respir Crit Care Med 2004;170:1101–1107. [DOI] [PubMed] [Google Scholar]
- 44.Scheuermann-Freestone M, Freestone NS, Langenickel T, Hohnel K, Dietz R, Willenbrock R. A new model of congestive heart failure in the mouse due to chronic volume overload. Eur J Heart Fail 2001;3:535–543. [DOI] [PubMed] [Google Scholar]
- 45.Zitt C, Halaszovich CR, Luckhoff A. The TRP family of cation channels: probing and advancing the concepts on receptor-activated calcium entry. Prog Neurobiol 2002;66:243–264. [DOI] [PubMed] [Google Scholar]
- 46.Nilius B, Droogmans G, Wondergem R. Transient receptor potential channels in endothelium: solving the calcium entry puzzle? Endothelium 2003;10:5–15. [DOI] [PubMed] [Google Scholar]
- 47.Schilling WP, Goel M. Mammalian TRPC channel subunit assembly. Novartis Found Symp 2004;258:18–30. [PubMed] [Google Scholar]
- 48.Wu X, Babnigg G, Villereal ML. Functional significance of human trp1 and trp3 in store-operated Ca2+ entry in HEK-293 cells. Am J Physiol 2000;278:C526–C536. [DOI] [PubMed] [Google Scholar]
- 49.Wu X, Zagranichnaya TK, Gurda GT, Eves EM, Villereal ML. A TRPC1/TRPC3 mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J Biol Chem 2004;279:43392–43402. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Shimoda LA, Sylvester JT. Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am J Physiol 2004;286:L848–L858. [DOI] [PubMed] [Google Scholar]
- 51.Ng LC, Gurney AM. Store-operated channels mediate Ca2+ influx and contraction in rat pulmonary artery. Circ Res 2001;89:923–929. [DOI] [PubMed] [Google Scholar]
- 52.Paria BC, Malik AB, Kwiatek AM, Rahman A, May MJ, Ghosh S, Tiruppathi C. Tumor necrosis factor-α induces nuclear factor-kB-dependent TRPC1 expression in endothelial cells. J Biol Chem 2003;278:37195–37203. [DOI] [PubMed] [Google Scholar]
- 53.Alvarez DF, King JA, Townsley MI. Evaluation of endothelial permeability by a corrosion casting technique. Microsc Microanal 2004;10:200–201. [Google Scholar]
- 54.Gebb S, Stevens T. On lung endothelial cell heterogeneity. Microvasc Res 2004;68:1–12. [DOI] [PubMed] [Google Scholar]
- 55.Trebak M, Hempel N, Wedel BJ, Smyth JT, Bird GJ, Putney JW Jr. Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol Pharmacol 2005;67:558–563. [DOI] [PubMed] [Google Scholar]
- 56.Pocock TM, Foster RR, Bates DO. Evidence of a role for TRPC channels in VEGF-mediated increased vascular permeability in vivo. Am J Physiol Heart Circ Physiol 2004;286:H1015–H1026. [DOI] [PubMed] [Google Scholar]
- 57.Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, Malik AB. Protein kinase Cα phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem 2004;279:20941–20949. [DOI] [PubMed] [Google Scholar]
- 58.Parker JC, Ivey CL, Tucker JA. Gadolinium prevents high airway pressure-induced permeability increases in isolated rat lungs. J Appl Physiol 1998;84:1113–1118. [DOI] [PubMed] [Google Scholar]
- 59.Meyrick B, Reid L. Ultrastructural features of the distended pulmonary arteries of the normal rat. Anat Rec 1979;193:71–98. [DOI] [PubMed] [Google Scholar]