

Abstract
We designed a host–guest fusion peptide system, which is completely soluble in water and has a high affinity for biological and lipid model membranes. The guest sequences are those of the fusion peptides of influenza hemagglutinin, which are solubilized by a highly charged unstructured C-terminal host sequence. These peptides partition to the surface of negatively charged liposomes or erythrocytes and elicit membrane fusion or hemolysis. They undergo a conformational change from random coil to an obliquely inserted (≈33° from the surface) α-helix on binding to model membranes. Partition coefficients for membrane insertion were measured for influenza fusion peptides of increasing lengths (n = 8, 13, 16, and 20). The hydrophobic contribution to the free energy of binding of the 20-residue fusion peptide at pH 5.0 is −7.6 kcal/mol (1 cal = 4.18 J). This energy is sufficient to stabilize a “stalk” intermediate if a typical number of fusion peptides assemble at the site of membrane fusion. The fusion activity of the fusion peptides increases with each increment in length, and this increase strictly correlates with the hydrophobic binding energy and the angle of insertion.
Fusion peptides play a central role in facilitating membrane fusion. Fusion peptides are highly conserved and quite hydrophobic sequences of fusion proteins that reside in viral envelope membranes or in the plasma membranes of sperm cells (1). These peptides are thought to be the only segments of the fusion proteins that insert deeply into the lipid bilayer of the target membrane at an early stage of membrane fusion (2). Despite this prominent role and numerous biophysical studies, no precise structure of a fusion peptide in lipid bilayers has yet been determined. Also, the binding of fusion peptides to lipid bilayers could not be studied previously, because these peptides are too hydrophobic to measure their partitioning between aqueous and membrane phases. Obtaining thermodynamic parameters of fusion peptide binding to lipid bilayers would be extremely helpful for the formulation of physically relevant models of energy coupling in membrane fusion. Most previous physical studies of fusion peptide–bilayer interactions were conducted with samples in which peptides and lipids were mixed in organic solvents and hydrated or added from organic solvent to preformed liposomes before spectroscopic analysis. A notable exception was a spin-label EPR spectroscopic study in which an expressed fragment of influenza hemagglutinin (HA) was used to assess the depth of the fusion peptide in lipid bilayers (3). The conformation of hydrophobic peptides in lipid bilayers often depends on the solvent that was used in the reconstitution procedure. Gramicidin A, for example, adopts many different conformations depending on how the samples are prepared (4). Therefore, the current procedures to reconstitute fusion peptide–bilayer complexes may not be adequate for higher-resolution structural studies of fusion peptides in lipid bilayers. To alleviate these problems, we have designed a host–guest peptide system in which the fusion guest peptide is linked via a flexible linker to a host peptide that solubilizes the entire peptide. This design allows us to perform partition experiments and to obtain quantitative data on the binding of fusion peptides to lipid bilayers in physiological buffers. It further opens the possibility to study the structure of such peptides in a presumably more physiological conformation. To prove the validity of this approach, we have applied this concept to study the binding of various peptides that are derived from the fusion peptide sequence of influenza virus HA to lipid bilayers. We also determined the secondary structures of these peptides in solution and in lipid bilayers. The influenza HA fusion peptide resides at the very N terminus of the HA2 subunit of the homotrimeric two-subunit protein. To assess the relative contributions of the different segments of the peptide to the overall binding energy and secondary structure, we have incremented the length of the influenza fusion peptide in four steps. This procedure mimics the progressive growth of the peptide into the lipid bilayer as it may also occur physiologically.
Materials and Methods
Peptides and Liposomes.
All peptides were synthesized by solid phase synthesis by the Biomolecular Research Facility at the University of Virginia by using Fmoc chemistry. Reverse-phase HPLC-purified peptides were >96% pure, and their molecular masses were confirmed by mass spectrometry. Concentrations of stock solutions were determined by quantitative amino acid analysis. Labeling with NBD (90–95% labeled) was performed as described (5).
Small unilamellar vesicles (SUVs) were prepared by sonicating lipid (Avanti Polar Lipids) or lipid–peptide dispersions in buffer [5 mM N-(2-hydroxyethyl)-piperazine-N′-(2-ethanesulfonic acid) (Hepes)/10 mM 2-(4-morpholino)-ethanesulfonic acid (Mes), pH 7.4] with a probe sonicator for 30 min at 50% duty cycle on ice. Phospholipid concentrations were determined by phosphorus assay (6). Large unilamellar vesicles (LUVs; 100 nm in diameter) were prepared by extrusion through polycarbonate membranes as described (7).
Hemolysis and Fusion by Lipid Mixing.
Peptides were incubated with 0.5 ml of washed chicken erythrocytes [2% (vol/vol) hematocrit in PBS]. After the pH had been lowered to 5.0 with an aliquot of 0.15 M citrate buffer (pH 3.5), the mixture was incubated at 37°C for 45 min. Cells were pelleted at 3,000 × g for 1 min, and the OD of the supernatant was determined at 520 nm. Complete hemolysis was obtained after lysing the cells with 0.5% Triton X-100.
Membrane fusion was measured by fluorescence resonance energy transfer (8) on a Spex Industries (Metuchen, NJ) Fluoromax spectrometer. Two populations of 1-palmitoyl-2-oleoyl-3-sn-phosphatidylcholine (POPC):1-palmitoyl-2-oleoyl-3-sn-phosphatidylglycerol (POPG; 4:1 mol/mol) LUVs, one unlabeled and one labeled with 1% each of N-lissamine rhodamine B sulfonyl-egg phosphatidylethanolamine (PE) and N-NBD-egg PE, were mixed at a 9:1 unlabeled:labeled ratio with 100 μM total lipid in 2 ml Hepes/Mes buffer at pH 7.4 or 5.0 at 37°C. Peptide at 5 μM was added after a 5-min preincubation from a 5 mg/ml stock solution in water. The fluorescence before peptide addition and after addition of 20 μl of 10% (vol/vol) Triton X-100 was taken as 0 and 100% fusion, respectively.
CD and Attenuated Total Reflection (ATR)-Fourier Transform Infrared (FTIR) Measurements.
CD spectra of peptides (0.25 mg/ml) in solution and in association with POPC:POPG (4:1, mol/mol) SUVs (prepared by cosonication) were recorded on a Jasco (Easton, MD) 600 spectropolarimeter as described (7). The fraction of residues in α-helical conformation, fH, was estimated from the measured mean residue ellipticity at 222 nm, θ222, by fH = (θ222 − θc)/(θH − θc), where θH = (250T − 44,000)(1 − 3/n) and θc = 2,220 − 53T are the limiting values of θ222 for completely helical and completely random coil peptides, respectively. T is the temperature in centigrade, and n is the number of residues of the peptides (9, 10).
ATR-FTIR spectra of peptides bound to planar phospholipid bilayers supported on germanium ATR plates were recorded on a Nicolet 740 FTIR spectrometer as described (7, 11). The substrate-exposed monolayer was 1,2-myristoyl-3-sn-phosphatidylcholine, and the monolayer facing the liquid cell was composed of POPC and POPG (4:1). Peptides were added to the preformed bilayers in Hepes/Mes pD 7.4 D2O buffer and allowed to bind for 5 min. Unbound peptides were removed by flushing the measuring cell with 3 ml of this buffer. The methods for processing of the infrared spectra and calculations of order parameters have been described (12).
Membrane Binding.
Binding of NBD-labeled peptides to lipid bilayers was measured as described (5). POPC:POPG (4:1, mol/mol) SUVs were added successively to 0.05 μM fluorescent P13H7, P16H7, or P20H7 or to 0.1 μM fluorescent H7 or P8H7 in Hepes/Mes pH 7.4 or 5.0 buffer. Fluorescence intensities were measured with the excitation, emission, and slits set at 490, 530, and 5 nm, respectively. Light scattering was negligible under these conditions. The binding isotherms were analyzed as partition equilibria by using the standard partition equation (13, 14)
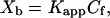 |
1 |
where Xb is defined as the molar ratio of bound peptide per 60% of the total lipid. We assume that the peptides initially partition only into the outer leaflet of the SUVs, which contain about 60% of the total lipid. Kapp is the apparent partition coefficient in units of M−1, and Cf represents the equilibrium concentration of free peptide in solution. To calculate Xb, we first determined the fluorescence intensity F∞, which is the limiting fluorescence when all peptide is bound to lipid. F∞ was obtained by extrapolation of a double-reciprocal plot of the total peptide fluorescence vs the total lipid concentration in the outer leaflet, i.e., 1/F vs. 1/(0.6 CL) (15). Knowing the fluorescence intensities of unbound peptide, F0, the fraction of membrane-bound peptide, fb, was calculated from fb = (F − Fo)/(F∞ − Fo). The free-peptide concentration, Cf, and the concentration of bound peptide, Xb, were then calculated from fb and the total peptide concentration. Plots of Xb vs. Cf are referred to as conventional binding isotherms. Free energies of peptide insertion were determined from standard equations (invoking Gouy–Chapman to account for charge accumulation at the membrane interface) as described in full detail in ref. 11. To calculate the respective incremental free energy changes, ΔΔGins, for each guest sequence, the free energy of insertion of the host peptide was subtracted, i.e.,
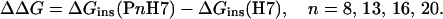 |
2 |
Results
Design of the Host–Guest System.
To study the binding of relatively hydrophobic peptides to lipid bilayers, we designed a host–guest system that should fulfill the following criteria: (i) the peptides should be soluble in aqueous buffers up to concentrations needed for equilibrium binding experiments with lipid bilayers; (ii) the peptides should be monomeric in aqueous buffers to facilitate the analysis of binding equilibria; (iii) the host peptides alone (i.e., without the guest fusion peptides) should bind to membranes with a reasonably high affinity; (iv) the host, but not the guest, segment of the peptides should have a site for attaching a spectroscopic reporter group that is suitable to measure binding to lipid bilayers; and (v) the host and guest segments of the peptides should form independent folding units, i.e., there should be minimal interactions between them. These criteria led to the design of the host peptide H7 and four host–guest peptides PnH7 where n = 8, 13, 16, and 20 stands for the number of residues from the N terminus of the influenza HA (strain X:31) fusion peptide sequence (Table 1). The sequences of the fusion peptides were increased in length to permit us to determine incremental changes in binding and conformation as the peptides were progressively “grown” into the lipid bilayer. The longest peptide consisted of 20 guest residues, because mostly hydrophilic residues follow the first 20 residues of influenza HA2; these subsequent residues do not insert into lipid bilayers as was demonstrated by photoaffinity labeling (2). Good solubility in aqueous buffers was achieved by linking the fusion peptides to a very polar host peptide. The highly charged host peptide further ensured that the peptides remained monomeric in solutions of low ionic strength (see below). The four consecutive lysines facilitated the binding of even the shortest guest peptide to lipid bilayers that contained 20 mol percent acidic phospholipids. This part of our design was guided by detailed studies of the binding of oligolysines to charged lipid bilayers (16, 17). Finally, a linker region consisting of the sequence Gly-Cys-Gly was incorporated into the host peptide. This segment provides a site for the attachment of spectroscopic reporter groups by sulfhydryl alkylation, is expected to be conformationally flexible, and thereby fulfills our criterion for an uncoupling of secondary structure induction across the host and guest peptide boundary. Table 1 also lists for each sequence the predicted hydrophobicities by using two different thermodynamic scales that are based on the transfer of the apolar residues to the membrane interface and the hydrophobic interior, respectively.
Table 1.
Sequences and calculated hydrophobicities of influenza HA host–guest fusion peptides of different lengths
 |
The host segments of each peptide are underlined.
The hydrophobic (negative on the respective scales of ref. 24) residues only are summed by using two different thermodynamic whole-residue hydrophobicity scales that should represent the transfer of unfolded peptides to the bilayer interface and into the hydrophobic interior of the lipid bilayer, respectively (24). Folding into an α-helix adds another −0.14 to −0.41 kcal/mol for each hydrogen-bonded residue in the interface (25, 26) and a likely more negative value in octanol.
Hemolysis and Fusion Activity.
Previous experiments with virus, isolated HA rosettes, and fusion peptides showed that the hemolytic activity of peptide models correlates with viral fusion activity (18–20). The data presented in Fig. 1 demonstrate that the host–guest peptides lyse red blood cells in a pH-, concentration-, and length-dependent manner. The longer peptides P16H7 and P20H7 exhibit strong hemolytic activity at pH 5.0 but not at pH 7.4. H7 and P8H7 are inactive, and P13H7 is marginally active at pH 5.0. The P8 portion of P8H7 consists of only hydrophobic residues (and glycines). It is clearly not sufficient to lyse red blood cells. However, the subsequent sequence of the longer peptides has an amphipathic and increasingly more negatively charged character. One or both of these elements seem to be required for hemolysis. Importantly, the pH-dependent hemolytic activity of our designed fusion peptides mimics very well the pH-dependence of the fusion activity of influenza HA, which fuses viral membranes only below pH ≈5.4 (21).
Figure 1.

Hemolytic activities of host–guest fusion peptides at pH 7.4 (open circles) and 5.0 (closed circles) as a function of peptide concentration.
We also studied the ability of our peptides to fuse liposomes. A common assay for membrane fusion monitors the mixing of lipids between two liposome populations by measuring resonance energy transfer between the fluorescent lipids NBD-eggPE and lissamine rhodamine B sulfonyl-eggPE (8). Fig. 2 shows the fusion of liposomes as measured by lipid mixing and elicited by the five peptides at pH 5.0. The rates and extents of fusion increase with the length of the fusion peptides. Again, H7 and P8H7 were fusion-inactive. Much lower rates and extents of fusion were observed at pH 7.4 for all other peptides (data not shown). These results demonstrate that our designed fusion peptides closely mimic the fusion behavior of influenza virus and, therefore, may be used as a valid model system to study the interaction of fusion peptides with lipid bilayers.
Figure 2.

Induction of fusion of POPC:POPG (4:1, mol/mol) liposomes by host–guest fusion peptides. Lipid mixing induced by 5 μM peptide added to 100 μM lipid was measured at pH 5.0 and 37°C.
Conformation of Peptides in Solution and Bound to Lipid Bilayers.
To assess the secondary structure of the peptides under different conditions, we measured their CD spectra in solution as a function of pH, peptide, and salt concentration. The dashed lines in Fig. 3 show the CD spectra in pH 7.4 and 5.0 buffers without added salt. The CD spectra of all five peptides are characteristic of mostly random coil at both pH values. Only P20H7 may adopt a small amount of α-helix (<10%) at both pH values under these conditions. When designing these peptides, we reasoned that the high charge density at the C termini might disperse the peptides enough by electrostatic repulsion to overcome the tendency of the larger peptides to self-associate by hydrophobic interaction. To test this hypothesis, we measured the CD spectra of all peptides under high salt conditions. Indeed, when electrostatic repulsion between the peptides was screened, they adopted ordered secondary structures, presumably as a result of self-association (11).
Figure 3.

Secondary structures of fusion peptides in solution and bound to lipid bilayers. CD spectra of all peptides in the presence (solid line) or absence (dashed line) of POPC:POPG (4:1, mol/mol) vesicles in 5 mM Hepes/10 mM Mes, pH 7.4 (left column) or pH 5.0 (right column). The peptide concentrations were 0.25 mg/ml, and the lipid/peptide molar ratios were 100. The spectra were recorded at 22 ± 2°C.
The solid lines in Fig. 3 show CD spectra of the five peptides bound to lipid bilayers of POPC/POPG (4:1, mol/mol) at pH 7.4 and 5.0. All spectra were recorded at lipid/peptide molar ratios of 100:1 (mol/mol) and total peptide concentrations of 0.25 mg/ml. We estimate from the partition coefficients (see below) that >99% of the peptides were bound to membranes under these conditions. The spectra indicate that, of the short peptides, H7 and P8H7 still did not adopt any ordered secondary structures after binding to lipid bilayers. However, the longer peptides P13H7, P16H7, and P20H7 adopted mostly α-helical structures on binding to lipid bilayers. Comparison of the spectra shown in Fig. 3 Left and Right shows that a change of the pH from 7.4 to 5.0 further promoted secondary structure formation of the membrane-bound peptides. Table 2 lists the contents of the α-helix of each peptide as calculated from the CD spectra. They range from 26% (P13H7, pH 7.4) to 50% (P16H7, pH 5.0).
Table 2.
Contents of α-helix and orientation of the helical segments of the host–guest fusion peptides bound to lipid bilayers (POPC:POPG, 4:1, mol/mol) at pH 7.4 and 5.0
| P13H7
|
P16H7
|
P20H7
|
||||
|---|---|---|---|---|---|---|
| pH 7.4 | pH 5.0 | pH 7.4 | pH 5.0 | pH 7.4 | pH 5.0 | |
| α-helix, %* | 26 | 33 | 32 | 50 | 41 | 48 |
| R1650ATR† | 1.70 | 1.53 | 1.68 | 1.53 | 1.80 | 1.68 |
| SH‡ | −0.05 | −0.48 | −0.11 | −0.31 | 0.18 | −0.07 |
| θ, degree§ | 57 | 83 | 59 | 69 | 48 | 57 |
α-helix content from CD spectroscopy.
Dicroic ratio of the amide I′ band determined from polarized ATR-FTIR spectroscopy.
Helix order parameter.
Average angle of the α-helix axis of the peptide to the membrane normal.
Binding of Peptides to Lipid Bilayers.
To measure binding of the host–guest fusion peptides to lipid bilayers, we labeled all five peptides at their unique cysteines with the fluorescent probe NBD. NBD is a relatively small amphipathic molecule, which we expect does not significantly perturb the partitioning of the labeled peptides. The quantum yield of NBD strongly depends on the dielectric constant of its environment. This property makes it an excellent spectroscopic probe to measure the binding of NBD-labeled peptides to lipid bilayers (5). All NBD-labeled host–guest peptides exhibited emission maxima at ≈545 nm in solution and at ≈526 nm when bound to lipid bilayers (11). These changes and the severalfold increased fluorescence emission are typical for the translocation of the NBD group to the lipid bilayer interface (5) and were used to measure the binding of the peptides to lipid bilayers. To determine the surface partition coefficients, the fluorescence intensities were converted to moles of bound peptide per moles of lipid and plotted as a function of the free peptide concentration as described in Materials and Methods. At pH 7.4 and without added salt, straight lines were obtained for the binding of all peptides to lipid bilayers composed of POPC:POPG (4:1, mol/mol; Fig. 4, Upper). Only at the highest concentrations, two of the peptides (P8H7 and P20H7) exhibited deviations from straight lines toward increased binding. This behavior is typical for self-association of the peptides at the membrane surface and will be described in more detail in a forthcoming publication (11). The straight lines observed for the other peptides (and for P8H7 and P20H7 at lower concentrations) indicate that the peptides bound to the lipid bilayers as monomers without self-association. Similar data were obtained at pH 5.0 (Fig. 4, Lower). Apparent partition coefficients, Kapp, were determined for each of the NBD-labeled peptides from the slopes of the lines of Fig. 4. They range from 5×104 M−1 for NBD-H7 to 9×105 M−1 for NBD-P20H7 at pH 7.4 and 3×105 to 1.3×106 M−1 at pH 5.0. These data are listed in Table 3. Generally, binding was stronger at the lower pH, and the critical concentrations for self-association were lower at pH 5.0 than at pH 7.4.
Figure 4.

Binding of host–guest fusion peptides to lipid bilayers composed of POPC:POPG (4:1, mol/mol) at pH 7.4 or pH 5.0. The concentrations of the NBD-labeled peptides were 0.05 or 0.1 μM. The binding isotherms were obtained at 25°C by plotting Xb (moles of bound peptide per moles of accessible lipid) vs. the equilibrium concentration of free peptide in solution, Cf (see text).
Table 3.
Partition coefficients and free energies of insertion of the host–guest fusion peptides on interaction with bilayers composed of POPC:POPG (4:1, mol/mol) or POPC in a buffer containing 5 mM HEPES, 10 mM MES, pH 7.4, and pH 5.0
| Peptide | pH | Kapp, M−1 | zp* | K0, M−1† | ΔGins, kcal/mol‡ | ΔΔGins, kcal/mol§ |
|---|---|---|---|---|---|---|
| POPC:POPG (4:1, mol/mol) bilayers | ||||||
| H7 | 7.4 | (5.24 ± 0.79) × 104 | 4 | 0.016 | −0.07 | |
| 5.0 | (3.00 ± 0.73) × 105 | 4 | 0.094 | −0.98 | ||
| H7-ac | 7.4 | (8.75 ± 0.19) × 104 | 4 | 0.027 | −0.24 | 0 |
| 5.0 | (2.57 ± 0.11) × 105 | 4 | 0.081 | −0.89 | 0 | |
| P8H7 | 7.4 | (1.43 ± 0.11) × 105 | 4 | 0.045 | −0.54 | −0.30 |
| 5.0 | (3.81 ± 0.58) × 105 | 4 | 0.12 | −1.12 | −0.23 | |
| P13H7 | 7.4 | (2.87 ± 0.03) × 105 | 3 | 3.81 | −3.17 | −2.93 |
| 5.0 | (5.61 ± 0.15) × 105 | 3 | 7.45 | −3.57 | −2.68 | |
| P16H7 | 7.4 | (6.90 ± 0.66) × 105 | 2 | 3.87 × 102 | −5.91 | −5.67 |
| 5.0 | (7.95 ± 0.21) × 105 | 2 | 4.46 × 102 | −6.00 | −5.11 | |
| P20H7 | 7.4 | (9.15 ± 0.34) × 105 | 1 | 2.16 × 104 | −8.30 | −8.06 |
| 5.0 | (1.29 ± 0.15) × 106 | 1 | 3.05 × 104 | −8.50 | −7.61 | |
| POPC bilayers | ||||||
| P20H7 | 7.4 | — | 1 | 5.50 × 103 | −7.49 | −7.25 |
| 5.0 | — | 1 | 3.78 × 104 | −8.63 | −7.74 |
Net charge of peptides used in Gouy–Chapman calculations.
The intrinsic partition coefficients were calculated from equation K0 = Kappexp(zpFΨ0/RT) by using a surface potential of −96.1 mV which was obtained from the Gouy–Chapman equation sinh[zeΨ0/2kT] = Aσ/(c)1/2 (see ref. 11).
ΔGins = −RT ln(55.5Ko).
The ΔΔGins values were calculated by subtracting ΔGins of H7-ac from the ΔGins values at each pH.
A large component of the “apparent” partition coefficient is caused by electrostatic attraction of the positively charged peptides to the negatively charged bilayers. The local concentration of peptides in the layer of solution very close to the bilayer surface is dramatically enhanced by the negative surface potential of the bilayer. This local concentration can be calculated by means of the Gouy–Chapman equation. It is this local rather than the bulk concentration of peptide that is in direct equilibrium with bound peptide. Therefore, we determined intrinsic partition coefficients, K0, of our peptides with the Gouy–Chapman theory and the surface partition equilibrium. In these calculations, we used the sum of the side chain charges as the formal charge of the peptide. We did not include a charge on the α-amino group of the N terminus because the H7 peptide with an acetylated N terminus yielded very similar partition coefficients as free H7 at pH 7.4 and 5.0 (Table 3). Apparently the α-amino group does not contribute significantly to the electrostatic attraction of these peptides. The binding free energy calculated from the partition coefficient of H7-ac is small and negative (−0.2 kcal/mol at pH 7.4), which indicates a weak chemical attraction of the peptide to the membrane surface. This energy could be the contribution of the NBD moiety to the binding of our peptides to the membrane surface. The binding energy of H7-ac decreases to −0.9 kcal/mol at pH 5.0. The difference of 0.6 to 0.7 kcal/mol may be caused by a local change of the pK values of the peptides in the membrane interface, which is not accounted for by our simplified Gouy–Chapman approach, and/or a true small increase of the intrinsic affinity of these peptides at the lower pH value. Because we are only interested in the contribution of the fusion (guest) peptide moieties to the binding free energies, we subtracted in the last column of Table 3 the respective reference ΔGs of H7-ac from the peptide ΔGs of the longer peptides at each pH value. The ΔΔGins values thus obtained decrease monotonically as the length of the fusion peptide is increased. Although the ΔGs are more negative at low pH than at neutral pH, the ΔΔGs are more negative at neutral pH. To check the validity of the Gouy–Chapman approach to determine intrinsic partition coefficients, we also measured the binding of P20H7 to uncharged POPC bilayers. The directly determined intrinsic partition coefficients of this experiment yielded binding free energies that were, particularly at pH 5.0, very close to those determined when P20H7 was bound to the charged membranes.
Orientation of Peptides in Lipid Bilayers.
We also studied the conformations of the membrane-bound host–guest fusion peptides by polarized ATR-FTIR spectroscopy. When the peptides P13H7, P16H7, and P20H7 were bound to planar supported bilayers composed of 1,2-myristoyl-3-sn-phosphatidylcholine:POPC/POPG (4:1, mol/mol), we observed amide I′ bands with absorbance maxima at 1,650 to 1,652 cm−1 at both pH 7.4 and 5.0 (11). These wavenumbers are consistent with α-helical conformations of the membrane-bound peptides (12). There was no evidence for other amide I′ components in these spectra. Polarized ATR-FTIR spectroscopy was used further to determine the order parameters (reflecting orientational distributions) of the helical segments of the membrane-bound peptides P13H7, P16H7, and P20H7. The measured dichroic ratios of the amide I′ ATR-FTIR bands are listed in Table 2 along with the derived order parameters and average orientation angles of the α-helical segments relative to the bilayer normal (see ref. 12 for definitions). At pH 7.4, the helical order parameters ranged from ≈ −0.1 to 0.18. Under the simplifying assumption that all peptides were aligned at the same angle in a given sample, these order parameters correspond to angles from 59 to 48° to the membrane normal for fusion peptides of increasing lengths. Lowering the pH to 5.0 led to shallower insertion angles. The helical segment of P20H7, for example, was inclined at an angle of 57° from bilayer normal or 33° from the membrane surface. The shorter peptide P13H7 had its helical segment oriented 83° from the normal, i.e., almost parallel to the membrane surface.
Discussion
Much can be learned from thermodynamic studies of the binding of peptides to lipid bilayers (13, 14, 22). Such studies are obviously limited to peptides that are soluble in aqueous buffers. Many membrane-interactive peptides that are of great biological importance are insoluble and thus not amenable to quantitative studies of membrane binding. The fusion peptides of enveloped viruses belong to this class of water-insoluble peptides. These peptides, which often reside at the N terminus of the membrane subunits of the viral fusion proteins, penetrate deeply into the lipid bilayers of cellular target membranes at an early stage of membrane fusion (23). Because fusion peptides are believed to perturb the lipid bilayer structure in preparation for fusion, it is very important to know how much energy is gained on the gradual insertion of these peptides into lipid bilayers. In this work, we have addressed this problem by attaching the fusion peptide of influenza virus HA to a solubilizing host peptide. Detailed binding measurements allowed us to determine the relative contributions of increasingly longer segments of this peptide to the free energy of membrane insertion. We found that at the physiologically relevant pH 5.0, the free energy of insertion of the full-length fusion peptide, P20, is −7.6 kcal/mol. From CD spectroscopy, we further determined that 48%, or 13 residues, are in an α-helical conformation, and from polarized FTIR spectroscopy, we estimated an insertion angle of approximately 33° for the helix axis from the membrane surface.
The experimentally determined energy of insertion of P20 at pH 5.0 or pH 7.4 is intermediate between two theoretical estimates from whole residue scales (24) that consider partitioning of unfolded peptides into the interface or the hydrophobic interior (−6.1 and −10.8 kcal/mol, respectively; Table 1). The ΔG for folding an α-helix in the interface has been estimated to be −0.41 kcal/mol per residue in LUVs (25) and −0.14 kcal/mol in SUVs (26). If 13 residues of P20 fold into an α-helix, the folding contribution to the total free energy in the interface of SUVs would be −1.8 kcal/mol. A total value of −7.9 kcal/mol may thus be expected for the partioning and folding of P20 in the interface. This value is in excellent agreement with the experimental values of −7.6 (pH 5.0) and −8.1 (pH 7.4) kcal/mol that we determined for P20. However, this agreement may be somewhat fortuitous, because we combined values from LUVs (partitioning) and SUVs (folding) in these theoretical calculations. Our experimental values are those of the insertion and folding of a peptide that we know inserts relatively deeply (helical angles of 33° and 42° from the surface at pH 5.0 and 7.4, respectively) into the bilayer of SUVs. An overestimate of partitioning may therefore compensate for an underestimate of folding. The experimental values obtained with P16 (−5.1 and −5.7 kcal/mol; helix 21° and 31° from the membrane surface) are very close to the expectation from the interface scale (−5.5 kcal/mol) but too small if the folding contribution of 13 (−1.8 kcal/mol) or 9 (−1.3 kcal/mol) residues is added. The shorter peptides P13 and P8, which are less helical and strictly located in the interface exhibit free energies of insertion of −2.7 and −0.2 kcal/mol, i.e., much lower values than predicted by the interface scale. The reason for this increasingly larger discrepancy is almost certainly the progressively shallower location of these peptides combined with the use of SUVs. Interfacial scales are limited to specific depths in bilayers of specific average curvatures. Depth and curvature dependencies of the relevant thermodynamic parameters are not known.
It has been proposed by many authors that intermediates with high membrane curvature occur in membrane fusion (27–31). The free energy that is required to form a stalk intermediate has been estimated to be of the order of 100–200 kT depending on the radius of the stalk and the intrinsic curvature of the lipids that are used to form the stalk (32, 33). This energy amounts to an unfavorable 60–120 kcal/mol at room temperature. Therefore, the binding of about 8 to 16 fusion peptides would provide sufficient energy to stabilize such an intermediate. If six trimers of HA participate in a fusion pore (34), the binding of 18 fusion peptides would release −137 kcal/mol, i.e., enough energy to stabilize a stalk-like fusion intermediate.†
The secondary structure of the guest fusion peptide is similar to that reported for influenza fusion peptides by various authors (7, 19, 35, 36). The consensus of the earlier and present studies is that the HA fusion peptide is ≈40–50% α-helical. However, previous measurements of the orientation of the α-helix in the bilayer by polarized ATR-FTIR and EPR spectroscopy have varied widely, ranging from 20 to 60° from the membrane surface (3, 7, 36–38). The large differences in these reports are likely caused by significant differences in sample preparation, including differences in the degree of hydration of the bilayers and whether the peptides were mixed with the lipids in organic solvents to prepare the model membranes. The angle of 33° from the membrane surface we find here from polarized ATR-FTIR spectroscopy for a fusion peptide in fully hydrated bilayers and bound to the membrane from the aqueous phase (without using an organic solvent) is similar to the 25° found for the peptide portion of an expressed HA2 fragment that also was bound to model membranes from solution but measured by EPR spectroscopy. This angle is shallower than the 50 to 60° that we previously reported for peptides that were mixed with the lipids in chloroform/DMSO solutions before vesicles and planar lipid bilayers were formed (7, 36). Because the methods of measurement and data analysis were the same in the present and our previous studies, we believe that the solvent history has a profound effect on the structure of fusion peptides in lipid bilayers. Relatively small contents of β-structure were also reported in some but not all previous studies of fusion peptides in lipid bilayers. As we will show elsewhere (11), the amount of β-structure depends on the peptide concentration in the bilayer. At the relatively small peptide concentrations used in this study, we found no evidence for β-structure.
The host–guest concept presented here should also be useful to study other fusion peptide systems and to assess the effect of mutations on the structure and lipid interactions. We believe that the lipid–peptide complexes are much better defined than those that are generated from solvent mixtures. Therefore, we expect that structural changes caused by specific mutations will be easier to detect and will no longer be masked by solvent-induced structural polymorphisms. Structural studies at higher resolution are also within reach with this system. Host–guest concepts have been proposed previously to study the insertion of transmembrane peptides into lipid bilayers. For example, Moll and Thompson (39) conjugated a hydrophobic peptide to bovine pancreatic trypsin inhibitor and studied its binding to lipid bilayers. Chung and Thompson (40) attached a hexalysine moiety to a proposed transmembrane peptide and, more recently, Wimley and White (41) used the sequence KSKSKS to solubilize another transmembrane model peptide. Even if soluble, these peptides still aggregated in solution where they adopted significant amounts of regular secondary structures. These factors precluded a thorough thermodynamic analysis of their binding to lipid bilayers. In contrast, our host–guest fusion peptides seem to be monomeric random coils in solution. A thermodynamic analysis of their binding as presented here thus was possible. It seems that in many aspects the fusion peptides are intermediate between the purely surface-located peptides such as melittin, magainins, or mitochondrial signal peptides on one hand and true transmembrane peptides on the other hand: The angles of insertion of fusion peptides are oblique; they penetrate deep but only into the cis monolayer; and the hydrophobic components of their binding energies are larger than for the surface peptides and presumably smaller than those of the transmembrane peptides. It is likely that these features are key to their fusogenicity. For example, in Fig. 5, we demonstrate that the free energy of binding and the helix order parameter of the peptides of this study correlate with their fusion activity. The activity increases linearly with the hydrophobic binding energy and the order parameters, reflecting increasingly steeper angles of insertion of these peptides. Although fusion also increases with increasing helicity, this relationship is not linear. It will be interesting to see whether these correlations also hold for fusion peptides with single-site mutations and for lipid bilayers of different lipid compositions.
Figure 5.

Correlation of binding energies, order parameters, and numbers of residues in α-helical conformation of peptides P13H7, P16H7, and P20H7 with their fusion activities measured as the rate of lipid mixing at pH 5.0.
Acknowledgments
This work was supported by National Institutes of Health Grant AI 30557.
Abbreviations
- ATR
attenuated total reflection
- FTIR
Fourier transform infrared
- HA
hemagglutinin
- LUVs
large unilamellar vesicles
- POPC
1-palmitoyl-2-oleoyl-3-sn-phosphatidylcholine
- POPG
1-palmitoyl-2-oleoyl-3-sn-phosphatidylglycerol
- SUVs
small unilamellar vesicles
- NBD
7-nitrobenz-2-oxa-1,3-diazol
- PE
phosphatidylethanolamine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.230212097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.230212097
Bound fusion peptides may alter the intrinsic curvature of the bilayer significantly. The magnitude of this effect is not known and, therefore, has not been taken into account in this over-simplified estimate.
References
- 1.Hernandez L D, Hoffman L R, Wolfsberg T G, White J M. Annu Rev Cell Dev Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 2.Durrer P, Galli C, Hoenke S, Corti C, Gluck R, Vorherr T, Brunner J. J Biol Chem. 1996;271:13417–13421. doi: 10.1074/jbc.271.23.13417. [DOI] [PubMed] [Google Scholar]
- 3.Macosko J C, Kim C-H, Shin Y-K. J Mol Biol. 1997;267:1139–1148. doi: 10.1006/jmbi.1997.0931. [DOI] [PubMed] [Google Scholar]
- 4.Killian J A. Biochim Biophys Acta. 1992;1113:391–425. doi: 10.1016/0304-4157(92)90008-x. [DOI] [PubMed] [Google Scholar]
- 5.Frey S, Tamm L K. Biochem J. 1990;272:713–719. doi: 10.1042/bj2720713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ames B N. Methods Enzymol. 1966;8:115–118. [Google Scholar]
- 7.Han X, Steinhauer D A, Wharton S A, Tamm L K. Biochemistry. 1999;38:15052–15059. doi: 10.1021/bi991232h. [DOI] [PubMed] [Google Scholar]
- 8.Struck D K, Hoekstra D, Pagano R E. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- 9.Rohl C A, Baldwin R L. Biochemistry. 1997;36:8435–8442. doi: 10.1021/bi9706677. [DOI] [PubMed] [Google Scholar]
- 10.Luo P, Baldwin R L. Biochemistry. 1997;36:8413–8421. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 11.Han, X. & Tamm, L. K., J. Mol. Biol., in press.
- 12.Tamm L K, Tatulian S. Q Rev Biophys. 1997;30:365–429. doi: 10.1017/s0033583597003375. [DOI] [PubMed] [Google Scholar]
- 13.Tamm L K. In: Membrane Protein Structure: Experimental Approaches. White S H, editor. New York: Oxford Univ. Press; 1994. pp. 284–286. [Google Scholar]
- 14.White S H, Wimley W C, Ladokhin A S, Hristova K. Methods Enzymol. 1998;295:62–87. doi: 10.1016/s0076-6879(98)95035-2. [DOI] [PubMed] [Google Scholar]
- 15.Schwarz G, Stankowski S, Rizzo V. Biochim Biophys Acta. 1986;861:141–151. doi: 10.1016/0005-2736(86)90573-0. [DOI] [PubMed] [Google Scholar]
- 16.Kim J, Mosior M, Chung L A, Wu H, McLaughlin S. Biophys J. 1991;60:135–148. doi: 10.1016/S0006-3495(91)82037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-Tal N, Honig B, Peitzsch R M, Denisov G, McLaughlin S. Biophys J. 1996;71:561–575. doi: 10.1016/S0006-3495(96)79280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato S B, Kawasaki K, Ohnishi S-I. Proc Natl Acad Sci USA. 1983;80:3153–3157. doi: 10.1073/pnas.80.11.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wharton S A, Martin S R, Ruigrok R W H, Skehel J J, Wiley D C. J Gen Virol. 1988;69:1847–1857. doi: 10.1099/0022-1317-69-8-1847. [DOI] [PubMed] [Google Scholar]
- 20.Wharton S A, Skehel J J, Wiley D C. Virology. 1986;149:27–35. doi: 10.1016/0042-6822(86)90083-8. [DOI] [PubMed] [Google Scholar]
- 21.Stegmann T, Hoekstra D, Scherphof G, Wilschut J. J Biol Chem. 1986;261:10966–10969. [PubMed] [Google Scholar]
- 22.Seelig J. Biochim Biophys Acta. 1997;1331:103–116. doi: 10.1016/s0304-4157(97)00002-6. [DOI] [PubMed] [Google Scholar]
- 23.Durell S R, Martin I, Ruysschaert J-M, Shai Y, Blumenthal R. Mol Membr Biol. 1997;14:97–112. doi: 10.3109/09687689709048170. [DOI] [PubMed] [Google Scholar]
- 24.White S H, Wimley W C. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 25.Ladokhin A S, White S H. J Mol Biol. 1999;285:1363–1369. doi: 10.1006/jmbi.1998.2346. [DOI] [PubMed] [Google Scholar]
- 26.Wieprecht T, Apostolov O, Beyermann M, Seelig J. J Mol Biol. 1999;294:785–794. doi: 10.1006/jmbi.1999.3268. [DOI] [PubMed] [Google Scholar]
- 27.Markin V S, Kozlov M M, Borovjagin V L. Gen Physiol Biophys. 1984;5:361–377. [PubMed] [Google Scholar]
- 28.Chernomordik L V, Chanturiya A, Green J, Zimmerberg J. Biophys J. 1995;69:922–929. doi: 10.1016/S0006-3495(95)79966-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melikyan G B, White J M, Cohen F S. J Cell Biol. 1995;131:679–691. doi: 10.1083/jcb.131.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J, Lentz B R. Biochemistry. 1997;36:6251–6259. doi: 10.1021/bi970404c. [DOI] [PubMed] [Google Scholar]
- 31.Kozlov M M, Chernomordik L V. Biophys J. 1998;75:1384–1396. doi: 10.1016/S0006-3495(98)74056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siegel D P. Biophys J. 1993;65:2124–2140. doi: 10.1016/S0006-3495(93)81256-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siegel D P. Biophys J. 1999;76:291–313. doi: 10.1016/S0006-3495(99)77197-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blumenthal R, Sarkar D P, Durell S, Howard D E, Morris S J. J Cell Biol. 1996;135:63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lear J D, DeGrado W F. J Biol Chem. 1987;262:6500–6505. [PubMed] [Google Scholar]
- 36.Gray C, Tatulian S A, Wharton S A, Tamm L K. Biophys J. 1996;70:2275–2286. doi: 10.1016/S0006-3495(96)79793-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishiguro R, Kimura N, Takahashi S. Biochemistry. 1993;32:9792–9797. doi: 10.1021/bi00088a034. [DOI] [PubMed] [Google Scholar]
- 38.Lüneberg J, Martin I, Nüssler F, Ruysschaert J-M, Herrmann A. J Biol Chem. 1995;270:27606–27614. doi: 10.1074/jbc.270.46.27606. [DOI] [PubMed] [Google Scholar]
- 39.Moll T S, Thompson T E. Biochemistry. 1994;33:15469–15482. doi: 10.1021/bi00255a029. [DOI] [PubMed] [Google Scholar]
- 40.Chung L A, Thompson T E. Biochemistry. 1996;35:11343–11354. doi: 10.1021/bi960080c. [DOI] [PubMed] [Google Scholar]
- 41.Wimley W C, White S H. Biochemistry. 2000;39:4432–4442. doi: 10.1021/bi992746j. [DOI] [PubMed] [Google Scholar]