

Abstract
STAT6-mediated chemokine production in the lung is required for Th2 lymphocyte and eosinophil homing into the airways in allergic pulmonary inflammation, and thus is a potential therapeutic target in asthma. However, the critical cellular source of STAT6-mediated chemokine production has not been defined. Here we demonstrate that STAT6 in bone marrow-derived myeloid cells was sufficient for the production of CCL17, CCL22, CCL11, and CCL24 and for Th2 lymphocyte and eosinophil recruitment into the allergic airway. In contrast, STAT6 in airway lining cells did not mediate chemokine production or support cellular recruitment. Selective depletion of CD11b+ myeloid cells in the lung identified these cells as the critical cellular source for the chemokines CCL17 and CCL22. These data reveal that CD11b+ myeloid cells in the lung help orchestrate the adaptive immune response in asthma, in part, through the production of STAT6-inducible chemokines and the recruitment of Th2 lymphocytes into the airway.
Keywords: Th2 cells, allergy, chemokines, eosinophils
Introduction
Asthma is a complex syndrome broadly defined by inflammation of the airways associated with airways hyper-responsiveness (AHR) and mucus hypersecretion (1). In most cases, the airway inflammation characteristic of asthma results from an allergic-type reaction to an inhaled substance from the environment (allergic asthma). In response to this antigenic challenge, the airways develop eosinophilic inflammation with edema and mucus production.
The homing of activated Th2 lymphocytes into the airways is necessary to establish and orchestrate the asthmatic inflammatory response (2, 3). In support of this central role of Th2 cells, levels of Th2 lymphocytes and their cytokines are elevated in the airways of humans with asthma (4-6). In addition, murine models of asthma have demonstrated that Th2 cells and the cytokines they secrete are essential for the development of airway eosinophilia and AHR (7-10). Consistent with this, adoptive transfer of effector Th2 cells into naive mice followed by exposure to inhaled antigen induces features of asthma, including eosinophilic inflammation, mucus hypersecretion and AHR (11-14), demonstrating that these cells are fully capable of producing the asthma phenotype.
The accumulation of Th2 cells in the airways in this model has been shown to be due to recruitment of Th2 cells that have proliferated in the thoracic lymph nodes, rather than due to proliferation of Th2 cells already in the lung (14, 15). The subsequent homing of eosinophils is then dependent on Th2 cell accumulation in the airways (16). Taken together, these data reveal that homing of Th2 cells to the airways is central to the pathogenesis of asthma. It follows that a therapeutic strategy that interrupts the recruitment of Th2 cells into the lung should effectively interrupt airway inflammation in asthma.
In prior studies, we determined that Th2 cell trafficking into the airway is controlled by G protein-coupled chemoattractant receptors (16). Multiple chemokines and lipid chemoattractants and their receptors have been identified as having the potential to direct Th2 cell recruitment into the asthmatic airway. However, studies inhibiting or deleting single chemoattractant receptors expressed on Th2 cells (e.g. CCR4, or CCR8) or chemoattractants active on them have revealed conflicting results (17-22). The preservation of Th2 cell trafficking in the absence of individual chemoattractants or their receptors has led us to examine potential master regulators of Th2 cell recruitment, such as transcription factors that regulate chemokine secretion in allergic inflammation. Previous research by our group and others has shown that deletion of STAT6 in the lung eliminates >80% of Th2 cell trafficking into the airways and eliminates airway eosinophilia following allergen challenge (23-25). Our prior study also demonstrated that STAT6 expression in a resident lung cell is necessary for effective Th2 cell and eosinophil recruitment into the airways. Consistent with this, STAT6-/- mice had significantly lower production of the Th2 cell-active chemokines CCL17 and CCL22, and the eosinophil-active chemokines CCL11 and CCL24. In the present study, we sought to determine the critical mediators of chemokine production and Th2 cell recruitment in this model of asthma.
Here we demonstrate that transplantation of wild-type or RAG1-/- bone marrow into STAT6-/- mice restored the production of the chemokines CCL17, CCL22, and CCL24 and reestablished Th2 lymphocyte and eosinophil recruitment into the lung following Th2 cell transfer and antigen challenge. In contrast, STAT6 in airway lining cells in the lung did not restore T cell or eosinophil trafficking, despite the restoration of mucus hypersecretion following Th2 cell transfer and antigen challenge. We then demonstrate that a distinct population of CD11c+/CD11b+ myeloid cells in the lung is the primary source of CCL17 and CCL22 and that these cells contribute to the recruitment of Th2 cells into the airways during allergic inflammation. Our data suggests that STAT6-dependent chemokine production from CD11b+ myeloid cells in the lung is important for the recruitment of Th2 lymphocytes in this model and may represent an effective target for asthma therapy.
Materials and Methods
Mice
Wild-type mice, DO11.10 TCR transgenic mice, RAG1-/- mice, and STAT6-/- mice (all in a BALB/c background) were purchased from Jackson Laboratories (Bar Harbor, ME). The EpiSTAT6 strain was provided by Drs. Kuperman and Dr. Erle in a BALB/c background. The CD11b-DTR strain was provided by Dr. Duffield. This strain was provided in an FVB background and was crossed one generation to BALB/c wild-type mice. Mice were used at 6-8 weeks of age and were age and sex matched for all experiments. For the bone marrow transplant protocol, mice were transplanted at 6 weeks of age and then allowed to reconstitute for 8 weeks prior to use in the asthma model. All protocols were approved by the Massachusetts General Hospital Subcommittee on Research and Animal Care.
Mouse models
Allergic airway inflammation was induced in mice as previously described (16, 23). Briefly, naive mice received intravenous or intratracheal transfer of 5 × 106 in vitro-differentiated OVA-specific Th2 cells obtained from the DO11.10 transgenic mouse strain. Th2 polarization was performed as described (16, 23) using irradiated splenocytes as antigen presenting cells. In some experiments, single cell suspensions of lung cells were obtained as described (26, 27), irradiated and then used as antigen presenting cells to stimulate naïve OVA-specific Th2 cells or in vitro Th2-polarized OVA specific T cells as described (16, 23). Th2 differentiation was confirmed by intracytoplasmic cytokine staining for both IL-4 and IFNγ (BD Pharmingen, San Diego, CA), which demonstrated 40-60% of cells positive for IL-4 and 0% positive for IFNγ. In some experiments the Th2 cells were labeled with CFSE as previously described (28). 24 hours after injection, mice underwent aerosol challenge with OVA (50 mg/ml in PBS) or PBS alone daily for 4 days. Mice were sacrificed 20-24 h after the last aerosol challenge.
Bone marrow transplant
Bone marrow was isolated from STAT6-/- mice, RAG1-/- mice, and wild-type BALB/c mice. Donor mice were killed in 100% CO2. Bone marrow was obtained by flushing of the femurs and tibiae of donor mice with RPMI using a 24-gauge needle. Recovered cells were filtered, counted, and resuspended in sterile RPMI. Recipient wild-type BALB/c or STAT6-/- mice were lethally irradiated with 7 Gy whole-body γ-irradiation using a 137Cs source. Following irradiation, the recipient mice were then injected intravenously with 5 × 106 bone marrow cells from STAT6-/- mice, RAG1-/- mice, or wild-type BALB/c mice in a volume of 0.5 ml via the lateral tail vein. The mice were then housed for 8 weeks to allow full engraftment of the bone marrow. At this point the mice received intravenous adoptive transfer of 5 × 106 in vitro-differentiated OVA-specific Th2 cells obtained from the DO11.10 transgenic mouse strain followed by 4 daily OVA aerosol challenges as outlined previously.
Diphtheria injection
CD11b-DTR mice were crossed for 1 generation to BALB/c mice. Mice were injected intraperitoneally with 25 ng/g diphtheria toxin (Sigma-Aldrich, St. Louis, MO) resuspended in 250 μl of PBS or PBS alone. Some mice then received intravenous adoptive transfer of 5 × 106 in vitro differentiated OVA-specific Th2 cells obtained from the DO11.10 transgenic mouse strain the next day. The following day the mice received a second injection of DT or PBS. The same day the mice also received the first of 4 daily OVA or PBS aerosol challenges as outlined previously. Mice were harvested 24 hours after the last OVA dose.
Bone marrow derived dendritic cell culture and transfer
Bone marrow was isolated from wild-type BALB/c and STAT6-/- mice and cultured in media with 20 ng/ml GM-CSF (R&D Systems, Minneapolis, MN) as previously described (29). After culturing the cells for 10 days, the non-adherent cells were collected, washed and resuspended in PBS. CD11b-DTR mice which had been given DT and OVA-specific Th2 cells as described above, received 2 million wild-type or STAT6-/- dendritic cells in 50 μl of PBS intratracheally 3 hours after the first OVA challenge. Mice then received the second injection of DT and 3 more daily OVA challenges. Control mice received no dendritic cells and just DT or PBS injections. Mice were harvested 24 hours after the last challenge.
IL-13 administration
CD11b-DTR/BALB/c hybrid mice were injected with DT or PBS as indicated above. Three days after the last injection, the mice received 5 μg of recombinant murine IL-13 (Peprotech, Rocky Hill, NJ) via intranasal injection and were harvested 24 hours later.
Mouse harvest and analysis
Mice were anesthetized with ketamine and a BAL and single cell suspensions of the thoracic lymph node and lung were obtained as previously described (26, 27). Cells recovered from the BAL, lymph node and lung were blocked and then stained with fluorescently labeled antibodies to the OVA-specific TCR (KJ-126 from Caltag, Carlsbad, CA), CD4, and CD8 (BD Biosciences, San Jose, CA). Flow cytometry was performed after gating on the lymphocyte population utilizing a FACS Caliber analytical flow cytometer (BD Biosciences) and analyzed using Flowjo software (Treestar, Ashland, OR). The differential cell count on cells isolated from the BAL was determined by enumerating macrophages, neutrophils, eosinophils, and lymphocytes on cytocentrifuge preparations of the cells stained with Diff-Quick (Dade Behring, Newark, DE). For histopathologic examination lungs were flushed free of blood, inflated with 10% buffered formalin and were prepared and evaluated as previously described (26). For immunohistochemistry sections were stained with anti-GFP (Abcam, Cambridge, MA) according to a published protocol (30). RNA was purified from the lung and analyzed by quantitative PCR as previously described (26). In some mice, blood was obtained by RV puncture and suspended in PBS with EDTA to prevent clotting. Red blood cells were lysed with resuspension in RBC lysis buffer (Sigma-Aldrich, St. Louis, MO) and the remaining leukocytes were blocked and stained with an antibody to murine CCR3 (R&D Systems). Flow cytometry was performed after gating on the granulocyte population utilizing a FACS Caliber analytical flow cytometer (BD Biosciences) and analyzed using Flowjo software (Treestar). BAL chemokine protein levels were assessed by commercial ELISA kits (R&D Systems). For lung cell isolation, a single cell suspension of total lung cells were prepared as described (26, 27) and then stained with antibodies to CD11b and CC11c. Cells were sorted using a FACSAria Cell-Sorting System (BD Biosciences).
Data analysis
Data are expressed as mean ± SEM. Differences in results were considered to be statistically significant when p < 0.05 using a student’s t-test or ANOVA.
Results
STAT6 in airway lining cells is insufficient to induce Th2 cell and eosinophil recruitment
We used adoptive transfer of OVA-specific in vitro polarized Th2 cells followed by OVA challenge to model allergic airway inflammation. This model has been shown to lead to prominent eosinophil and Th2 cell recruitment into the airways without the need for immunization (23), and thus is an ideal system to study mechanisms of Th2 cell trafficking independent of other processes such as in vivo T cell activation. We used this model in wild-type BALB/c mice, STAT6-/- mice (in a BALB/c background), and the previously described EpiSTAT6 mice (in a BALB/c background), which are STAT6-/- mice that express STAT6 driven by the Clara cell specific promoter CC10 (31). These mice express STAT6 only in the airway lining and not in other cells of the lung. After transfer of Th2 cells and antigen challenge, there was prominent recruitment of OVA-specific Th2 cells (identified by an antibody specific to the transgenic T cell receptor, referred to as KJ) into the airways of wild-type mice. However, transfer of Th2 cells into STAT6-/- or EpiSTAT6 mice followed by antigen challenge resulted in nearly 10-fold less OVA-specific Th2 cell recruitment into the airways (Figure 1A). There was also a significant defect in total CD4+ T cells and eosinophils recruited into the airways (Figure 1B, C, D). As shown previously (23), PBS challenge did not lead to significant recruitment of OVA-specific Th2 cells, total CD4+ T cells or eosinophils into the airways of wild-type or STAT6-/- mice (Figures 1A, 1B and data not shown). Prominent eosinophilic airway inflammation was seen in wild-type mice but not in the STAT6-/- mice, EpiSTAT6 mice, or in PBS challenged mice following Th2 cell transfer and antigen challenge (Figure 1E and data not shown).
Figure 1. STAT6 expression in airway lining cells does not support Th2 cell and eosinophil trafficking into the airways.

A) Number of CD4+ and KJ+ OVA-specific Th2 cells, and B) CD4+ and CD8+ T cells in the BAL of wild-type, STAT6-/- and EpiSTAT6 mice following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA or PBS challenges (n = 4-6 mice per group, from 2 experiments). C) Percentage and D) number of eosinophils in the BAL of wild-type, STAT6-/- and EpiSTAT6 mice following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 6 mice per group, from 2 experiments). E) Representative lung histology (stained with hematoxylin and eosin) from wild-type (i, iv - 100x and 400x respectively), STAT6-/- mice (ii, v), and EpiSTAT6 (iii, vi) mice following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 6 mice per group, from 2 experiments). Black bars are 100 μm, arrows demonstrate clusters of eosinophils only in the section from wild-type lungs. F) Lung chemokine RNA copies normalized to copies of GAPDH RNA and G) BAL chemokine protein levels in wild-type, STAT6-/- and EpiSTAT6 mice following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA or PBS challenges (n = 4-6 mice per group, from 2 experiments).
Decreased levels of chemokines in STAT6-/- and EpiSTAT6 mice
We quantified the levels of RNA transcripts for the major eosinophil- and Th2 lymphocyte-active chemokines CCL11, CCL24, CCL17, CCL22, and CCL1 in RNA isolated from lungs of wild-type, STAT6-/-, and EpiSTAT6 mice following Th2 cell transfer and antigen or PBS challenge (Figure 1F). As shown previously (23), Th2 cell transfer and OVA challenge led to increased levels of CCL11, CCL24, and CCL17 RNA in wild-type mice relative to PBS challenged mice. However, there were reduced RNA levels of these chemokines in the lungs of STAT6-/- mice and EpiSTAT6 mice following Th2 cell transfer and OVA challenge, with transcript levels similar to levels in PBS challenged wild-type and STAT6-/- mice. CCL1 levels were low and unaffected. Levels of CCL22 were relatively low in wild-type mice and were reduced further in the STAT6-/- mice but not in the EpiSTAT6 mice, suggesting that the airway lining cells can produce CCL22 RNA in the EpiSTAT6 mice. However, this did not lead to detectable levels of CCL22 protein in the bronchoalveolar lavage fluid (BAL) (Figure 1G) or recruitment of Th2 cells comparable to levels in wild-type mice. Protein levels of CCL24, CCL22, and CCL17 in the BAL were reduced in the STAT6-/- and EpiSTAT6 mice compared to wild-type mice (Figure 1G). Levels of these proteins were undetectable in PBS challenged mice (data not shown). We did not detect significant protein levels of CCL11 or CCL1 in the BAL in any of these mice. These data demonstrate that the critical source of STAT6-dependent chemokines is not the airway lining, but rather, another resident lung cell.
STAT6 in airway lining cells is sufficient for mucus cell hypertrophy
Despite the lack of airway inflammation we did see increased Periodic-Acid-Schiff (PAS) staining of the airway epithelium in the EpiSTAT6 (Figure 2C) comparable to wild-type mice (Figure 2A), but this staining was absent in STAT6-/- mice and PBS challenged wild-type and STAT6-/- mice (Figure 2B and data not shown). These data indicate that the small number of transferred OVA-specific Th2 cells that were recruited into the lung were likely producing Th2-type cytokines in response to antigen, which resulted in mucus hypersecretion in the EpiSTAT6 mice, but did not lead to amplification of Th2 cell recruitment and effective airway recruitment of Th2 cells. These data suggests that STAT6 functions are anatomically compartmentalized, and that STAT6 in a non-airway lining cell controls the recruitment of Th2 cells and eosinophils, while STAT6 in epithelial cells is sufficient for mucus production.
Figure 2. Mucus hyper-secretion in wild-type mice, STAT6-/- mice, EpiSTAT6 mice, and STAT6-/- mice following bone marrow transplant.
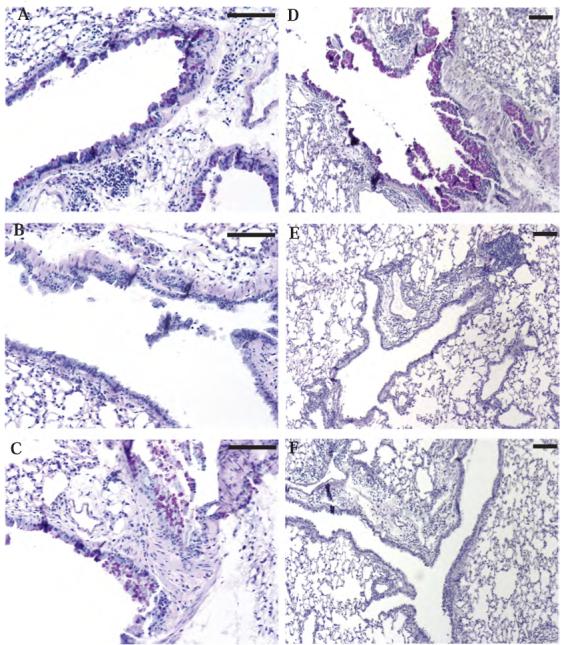
Representative histologic sections stained with PAS to evaluate mucus hypersecretion following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges. Sections are from wild-type BALB/c mice (A), STAT6-/- mice (B), EpiSTAT6 mice (C), wild-type BALB/c mice after bone marrow transplant with wild-type bone marrow (D), STAT6-/- mice after bone marrow transplant with wild-type bone marrow (E), or STAT6-/- mice after bone marrow transplant with RAG1-/- bone marrow (F). Black bars are 100 μm.
Intratracheal transfer of OVA-specific Th2 lymphocytes into STAT6-/- mice did not restore eosinophil trafficking
In a previous study, we demonstrated that intratracheal transfer of OVA-specific Th2 cells into naïve wild-type mice followed by antigen challenge led to eosinophil recruitment into the airways (16). PBS challenge in these mice did not lead to significant eosinophil recruitment (16). To determine if the defect in eosinophil recruitment in STAT6-/- mice is a result solely from the defect in Th2 cell trafficking or if there is a direct effect of STAT6 deletion on eosinophil recruitment, we transferred 5 × 106 OVA-specific in vitro polarized Th2 cells intratracheally into wild-type and STAT6-/- mice followed by three OVA or PBS aerosol challenges. Following adoptive transfer and challenge, STAT6-/- mice had significantly more OVA-specific Th2 cells retained in the BAL compared to wild-type recipients (Figure 3A and B). The reasons for this are unclear but may relate to a defect in Th2 cell exit from the airways of STAT6-/- mice. As expected, PBS challenge did not result in eosinophils in the BAL in either the wild-type or STAT6-/- mice. Despite the increased numbers of Th2 cells in the BAL, STAT6-/- mice did not recruit eosinophils into the airways following OVA challenge (Figure 3C). Both STAT6-/- mice and wild-type mice did however develop increased eosinophils in the blood with OVA challenge (Figure 3D), indicating that the reduced number of eosinophils found in the airways was due to a recruitment defect, rather than a defect in eosinophil production and release from the bone marrow. Consistent with this, there were reduced RNA levels of CCL11, CCL24, and CCL17 in the lungs isolated from STAT6-/- mice compared to wild-type mice in these experiments (Figure 3E). The results indicate that STAT6 expression in the lung is also necessary for eosinophil recruitment into the airways in this model.
Figure 3. Intratracheal transfer of Th2 cells does not restore eosinophil trafficking into the airways in STAT6-/- mice.
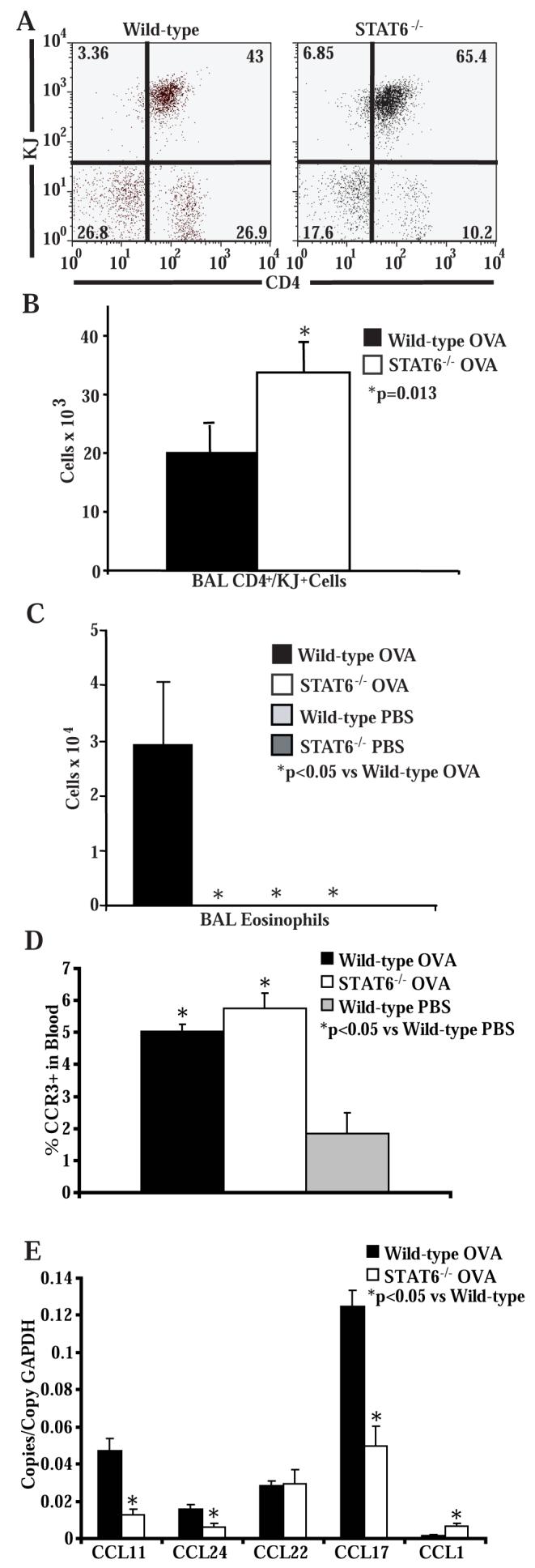
A) Representative flow cytometry of BAL cells stained for CD4 and KJ following intratracheal transfer of OVA-specific in vitro polarized Th2 lymphocytes and 3 OVA challenges (n = 6 mice per group, from 2 experiments). B) Number of CD4+ and KJ+ OVA-specific Th2 cells in the BAL of wild-type and STAT6-/- mice following intratracheal transfer of OVA-specific in vitro polarized Th2 lymphocytes and 3 OVA challenges (n = 6 mice per group, from 2 experiments). C) Number of eosinophils in the BAL of wild-type and STAT6-/-mice following intratracheal transfer of OVA-specific in vitro polarized Th2 lymphocytes and 3 OVA challenges (n = 6 mice per group, from 2 experiments). D) Percentage of CCR3+ cells in the granulocyte gate of cells isolated from blood of wild-type and STAT6-/- mice following intratracheal transfer of OVA-specific in vitro polarized Th2 lymphocytes and 3 OVA or PBS challenges (n = 3 mice per group, from 1 experiment). E) Lung chemokine RNA copies normalized to copies of GAPDH RNA in wild-type and STAT6-/- mice following intratracheal transfer of OVA-specific in vitro polarized Th2 lymphocytes and 3 OVA challenges (n = 6 mice per group, from 2 experiments).
Th2 lymphocyte and eosinophil trafficking is restored in STAT6-/- mice reconstituted with wild-type or RAG1-/- bone marrow
To investigate if the critical source of STAT6-dependent chemokines was a bone marrow derived cell in the lung, we reconstituted STAT6-/- mice with wild-type, RAG1-/-, or STAT6-/- bone marrow. Eight weeks after bone marrow transplant, we adoptively transferred OVA-specific Th2 cells into these mice and then challenged them with OVA. STAT6-/- mice reconstituted with STAT6-/- bone marrow had significantly less OVA-specific Th2 cells, total CD4+ T cells, and eosinophils recruited into the BAL, compared to STAT6-/- mice reconstituted with wild-type or RAG1-/- bone marrow (Figure 4A, B, C, D). In addition, there was prominent eosinophilic airway inflammation in the STAT6-/- mice reconstituted with wild-type or RAG1-/- bone marrow followed by Th2 cell transfer and antigen challenge compared to STAT6-/- mice reconstituted with STAT6-/- bone marrow (Figure 4E). Despite the eosinophilic airway inflammation in the STAT6-/- mice reconstituted with wild-type or RAG1-/- bone marrow, we did not see increased PAS staining of the airway epithelium in these mice (Figure 2E, F), compared to wild-type mice reconstituted with wild-type bone marrow (Figure 2D). This is consistent with our findings demonstrating that STAT6 expression in epithelial cells controls mucous production. Of note, the numbers of lymphocytes recruited into the BAL in these mice were 3-fold greater compared to mice with allergic airway inflammation that had not received a bone marrow transplant. However, the relative difference in Th2 cell recruitment between STAT6-/- mice that received wild-type or RAG1-/- bone marrow and those that received STAT6-/- bone marrow was similar to the differences seen between wild-type mice and STAT6-/- mice that did not receive bone marrow transplants. In addition, a similar increase in BAL cell numbers was obtained when we reconstituted wild-type BALB/c mice with wild-type bone marrow followed by adoptive transfer of OVA-specific Th2 cells and OVA challenges (Figure 4A). This suggests that the general increase in Th2 cell recruitment following bone marrow transplant may relate to the use of older mice (on average 8 weeks older) and/or the effects of radiation treatment.
Figure 4. STAT6 expression in myeloid derived cells in the lung does support Th2 cell and eosinophil trafficking into the airways.

A) Number of OVA-specific Th2 cells in the BAL of wild-type BALB/c mice after bone marrow transplant with wild-type bone marrow or STAT6-/- mice after bone marrow transplant with wild-type, STAT6-/- or RAG1-/- bone marrow, and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 4-8 mice per group, from 3 experiments). B) Number of CD4+ T cells, and CD8+ T cells in the BAL of STAT6-/- mice after bone marrow transplant with wild-type, STAT6-/-, or RAG1-/- bone marrow, and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 8 mice per group, from 2 experiments). C) Percentage and D) number of eosinophils in the BAL of STAT6-/- mice after bone marrow transplant with wild-type, STAT6-/- or RAG1-/- bone marrow, and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 8 mice per group, from 2 experiments). E) Representative lung histology from STAT6-/- mice after bone marrow transplant with wild-type (i, iv - 100x and 400x respectively), STAT6-/- (ii, v), or RAG1-/- (iii, vi) bone marrow, and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 8 mice per group, from 2 experiments). Black bars are 100 μm. F) Lung chemokine RNA copies normalized to copies of GAPDH RNA and G) BAL chemokine protein levels in STAT6-/- mice after bone marrow transplant with wild-type, STAT6-/- or RAG1-/- bone marrow, and following transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 8 mice per group, from 2 experiments).
RNA transcript and protein levels of CCL17, CCL22, and CCL24 in the lungs of STAT6-/- mice reconstituted with STAT6-/- bone marrow followed by Th2 cell transfer and antigen challenge were significantly lower than levels in the lungs of STAT6-/- mice reconstituted with wild-type or RAG1-/- bone marrow (Figure 4F, G). However, we did not see any difference in the expression levels of CCL11 in the lungs of STAT6-/- mice reconstituted with wild-type or RAG1-/- bone marrow compared to STAT6-/- mice reconstituted with STAT6-/- bone marrow, suggesting that CCL11 may be primarily produced in the lung by a non-bone marrow derived cell, and is not critical for eosinophil accumulation in the airway. Thus, restoration of STAT6 expression in a myeloid-derived cell in the lung is sufficient to restore Th2 cell and eosinophil trafficking in STAT6-/- mice but not mucus hyper-secretion.
CD11c+/CD11b+ myeloid cells are the major producers of Th2-cell active chemokines
Myeloid cells in the lung can be broadly categorized by the expression of CD11c and CD11b. In general, CD11c+/CD11b- cells are considered macrophages and CD11c+/CD11b+ cells are considered dendritic cells (32). A recent study has demonstrated that the CD11c+/CD11bhigh population of myeloid cells are potent producers of chemokines during allergic inflammation (33), thus we were interested in determining if these myeloid populations produce the relevant chemokines in our model of asthma. We sorted the CD11c+/CD11b+ and CD11c+/CD11b- populations from wild-type naïve mice and wild-type mice following Th2 cell transfer and OVA challenge using flow cytometry. We then analyzed levels of CCL17, CCL22, CCL11, and CCL24 RNA in these cells (Figure 5A). The CD11c+/CD11b+ population from mice following Th2 cell transfer and OVA challenge had very high RNA levels of CCL17 and CCL22 compared to CD11c+/CD11b+ from naïve mice. In addition, CD11c+/CD11b+ cells from mice following Th2 cell transfer and OVA challenge produced more CCL17 and CCL22 compared to CD11c+/CD11b- cells from the same mice. These data clearly demonstrate that the CD11c+/CD11b+ population of myeloid cells in the lung make high levels of Th2-active chemokines and are much more potent source of these chemokines compared to CD11c+/CD11b- myeloid cells. Interestingly, CD11c+/CD11b+ cells and CD11c+/CD11b- cells from these mice both made CCL24. CCL11 expression followed a similar pattern of expression as the CCL17 and CCL22 with the highest levels of expression in CD11c+/CD11b+ cells from the OVA challenged mice, however levels were 100-fold less than CCL17 and CCL22 and thus do not appear on the graph (values are 0.0015 copies/copy GAPDH for naïve CD11c+/CD11b- cells, 0.0026 copies/copy GAPDH for naïve CD11c+/CD11b+ cells, 0.0006 copies/copy GAPDH for OVA CD11c+/CD11b- cells, 0.004 copies/copy GAPDH for OVA CD11c+/CD11b+ cells). These data are consistent with our bone marrow reconstitution experiments, which did not demonstrate high-level CCL11 expression from myeloid derived cells.
Figure 5. DT administration selectively depletes myeloid dendritic cells in CD11b-DTR mice.
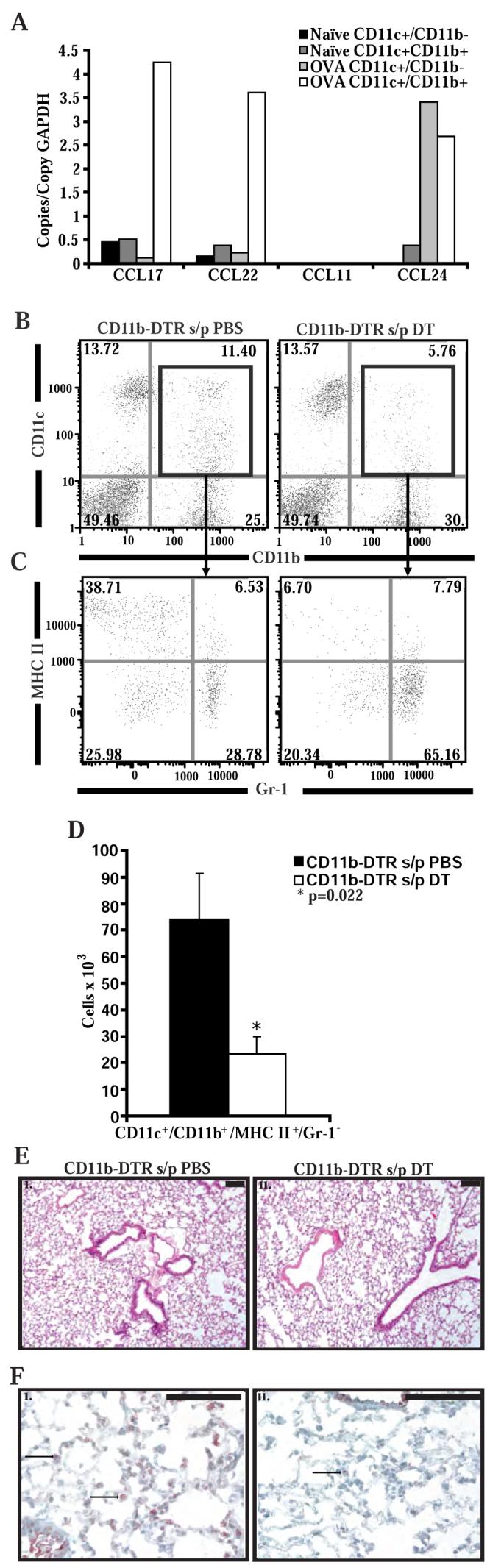
A) Chemokine RNA copies normalized to copies of GAPDH from CD11c+/CD11b+ and CD11c+/CD11b- cells sorted from the lungs of wild-type naïve mice and wild-type mice following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 4 pooled mice per group, from 1 experiment). B) Representative flow cytometry of lung cells isolated from CD11b-DTR mice 4 days after 2 treatments with PBS or DT. Cells are stained with labeled antibodies to CD11b and CD11c and gated on the monocytes/macrophages. C) Representative flow cytometry of lung cells isolated from CD11b-DTR mice 4 days after 2 treatments with PBS or DT. Cells were stained with labeled antibodies to Gr-1 (RB6-8C5 from BD Pharmingen), MHC II, CD11b and CD11c and gated on monocytes/macrophages and on CD11b+/CD11c+ cells. D) Number of CD11c+/CD11b+/MHC-II+/Gr-1- cells in the lungs of CD11b-DTR mice as measured by flow cytometry after treatment with PBS or DT (n = 6 mice per group, from 2 experiments). E) Representative lung histology from CD11b-DTR mice 4 days after 2 treatments with PBS (i) or DT (ii). Lung sections are stained with hematoxylin and eosin. Black bar is 100 μm. F) Representative immunohistochemistry for GFP in lungs taken from CD11b-DTR mice 4 days after 2 treatments with PBS (i) or DT (ii).
Selective depletion of CD11b+ cells in the lung reduces Th2 lymphocyte and eosinophil recruitment
To better characterize the myeloid cell necessary for STAT6-dependent Th2 cell recruitment, we utilized transgenic mice that allow selective depletion of CD11b+ cells (34). These mice have been engineered to express the human diphtheria toxin receptor (DTR) and green fluorescent protein (GFP) driven by the CD11b promoter (CD11b-DTR mice). Since diphtheria toxin (DT) is normally not toxic to murine cells, injection of DT into mice with this transgene will cause selective death of CD11b+ cells. Previous experiments with these mice have shown selective depletion of macrophages in the liver, peritoneum, and blood vessels after treatment of the mice with two doses of diphtheria toxin (DT) without adverse effects (30, 34). Interestingly, granulocytes do not seem to be deleted by DT in these mice (34). Myeloid derived cells in the lung include dendritic cells, macrophages, and granulocytes, all of which may express CD11b (32, 35). To define the effects in the lung of DT injection in these mice, we dosed CD11b-DTR mice with 2 doses of DT or vehicle (PBS) alone 1 day apart, and isolated the lungs from these mice 1, 2, 3, and 4 days following the second injection. The mice appeared normal during this study period. Single cells suspensions from the lungs were made after collagenase digestion and stained for both CD11c and CD11b expression. Analysis 4 days after the second DT injection revealed a 2-fold decrease in the percentage of CD11c+/CD11b+ cells compared to CD11b-DTR mice injected with PBS alone (Figure 5B). This difference was evident 1 day following the second DT injection and persisted on days two and three (data not shown). To better define the depleted cell population, we stained single cell suspensions of lungs from these mice 4 days after two DT injections with a panel of leukocyte markers (CD11b, CD11c, Gr-1, MHC II, CD8, CD68, CD3, CD103 and B220) and performed multi-color flow cytometry. After gating on the CD11c+/CD11b+ population, we found a selective 3-fold decrease in a subpopulation of lung cells that were CD11b+/CD11c+/MHC II+/Gr-1- (Figures 5C and 5D). These cells have been classified as myeloid dendritic cells in other studies (15, 36, 37). Cells that were CD11b+/CD11c+/MHC II+/Gr-1+ (plasmacytoid dendritic cells), CD11b-/CD11c+/F4/80+/CD68+ (macrophages), CD11b-/CD11c+/CD103+ (a subclass of airway dendritic cells (33)) and CD11b+/CD11c-/Gr-1+ (granulocytes) were not significantly depleted by DT injections (Figure 5B, 5C and data not shown).
Lung histology from CD11b-DTR mice 4 days after 2 DT injections did not disclose any abnormalities (Figure 5Eii) or differences compared to mice treated with injections of PBS alone (Figure 5Ei). Using immunohistochemistry on frozen sections of lungs from these mice stained with an antibody to GFP we found that PBS treated mice had GFP+ cells in the lung and around airways as previously reported (Figure 5Fi) (30). Consistent with the flow data, immunohistochemistry also revealed that there were reduced numbers of GFP+ cells in the lungs of mice treated with DT (Figure 5Fii).
To investigate the role of this subpopulation of CD11b+ cells in the lung during allergic airway inflammation, we transferred OVA-specific Th2 cells into CD11b-DTR mice. Mice were injected with DT or PBS on the day before and the day after Th2 cell transfer. OVA or PBS challenges were given starting on the day after Th2 cell transfer. The DT injections were timed to eliminate the CD11b+ cells during the effector phase of the response to allergen. As expected CD11b-DTR mice challenged with PBS did not recruit OVA-specific Th2 cells or eosinophils into the airways (data not shown). Mice challenged with OVA and injected with DT had significantly less OVA-specific Th2 cell, total CD4+ and CD8+ T cell, and eosinophil recruitment into the BAL when compared to mice challenged with OVA and injected with PBS (Figure 6A, B, C, D). The percentage of eosinophils in the blood was not affected by DT injection (data not shown), suggesting that the reduced eosinophil levels in the BAL were due to a recruitment defect rather than deletion of cells through the action of DT. Consistent with this interpretation, analysis of chemokine production demonstrated reduced transcript levels of CCL17, CCL22, CCL11, and CCL24 in RNA isolated from lungs of OVA challenged CD11b-DTR mice treated with DT compared to OVA challenged CD11b-DTR mice treated with PBS (Figure 6E). RNA levels of CCL17, CCL22 and CCL24 in the OVA challenged and DT treated mice were similar to levels in the lungs of PBS challenged CD11b-DTR mice treated with PBS. The magnitude of airway inflammation was markedly attenuated in CD11b-DTR mice treated with DTR, compared to CD11b-DTR mice treated with PBS (Figure 6F). In addition, immunohistochemistry for GFP in lungs from CD11b-DTR mice treated with DT or PBS after Th2 cell transfer and OVA challenge showed increased numbers of GFP+ cells around the airways of PBS-treated mice compared to DT-treated mice (Figure 6G). Consistent with this we found a nearly 2-fold reduction in the number of CD11b+/CD11c+ cells in the lungs of these mice treated with DT when compared to mice treated with PBS (Figure 6H).
Figure 6. DT administration to CD11b-DTR mice impairs Th2 cell and eosinophil recruitment into the airways in an adoptive transfer model of allergic airway inflammation.

A) Number of OVA-specific Th2 cells and B) CD4+ and CD8+ T cells in the BAL of CD11b-DTR mice after treatment with PBS or DT and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 8 mice per group, from 3 experiments). C) Percentage and D) number of eosinophils in the BAL of CD11b-DTR mice after treatment with PBS or DT and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 8 mice per group, from 3 experiments). E) Lung chemokine RNA copies normalized to copies of GAPDH RNA in CD11b-DTR mice after treatment with PBS or DT and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA or PBS challenges (n = 3-6 mice per group, from 2 experiments). F) Representative lung histology from CD11b-DTR mice treated with PBS (i) or DT (ii), and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 8 mice per group, from 3 experiments). Black bars are 100 μm. G) Representative immunohistochemistry for GFP in lungs taken from CD11b-DTR mice after treatment with PBS or DT and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 4 mice per group, from 1 experiment). Black bars are 10 μm. H) Number of CD11c+/CD11b+ cells in the lungs of CD11b-DTR mice as measured by flow cytometry after treatment with PBS or DT and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 8 mice per group, from 3 experiments).
To demonstrate that the depleted CD11c+/CD11b+ cell population is sufficient for Th2 cell recruitment, we transferred wild-type or STAT6-/- bone marrow-derived dendritic cells intratracheally into DT treated CD11b-DTR mice several hours after the first OVA challenge. This technique has been used by others to restore dendritic cells in the lung after depletion by DT (35). Flow cytometry revealed that the majority of transferred cells were CD11c+/CD11b+/MHC II+/Gr-1- (Figure 7A) as previously reported (29, 35), and did not differ between wild-type and STAT6-/- mice. After 4 OVA challenges there was significantly more Th2 cell recruitment in the mice that received wild-type dendritic cells compared to mice that received STAT6-/- dendritic cells or DT treatment without dendritic cell transfer (Figure 7B). These data demonstrate that the CD11c+/CD11b+ cell population in the lung is sufficient for Th2 cell recruitment and also demonstrate that the Th2 cell recruitment defect was not due to a non-specific effect of DT treatment.
Figure 7. Intratracheal transfer of bone marrow-derived dendritic cells restores Th2 cell recruitment in CD11b+ myeloid cell depleted CD11b-DTR mice.

A) Bone marrow cells from wild-type or STAT6-/- mice were cultured for 10 days in media supplemented with GM-CSF. The non-adherent cells were harvested and stained with antibodies to CD11b, CD11c, Gr-1, and MHC II. B) Number of OVA-specific Th2 cells in the BAL following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges in CD11b-DTR mice which had been injected with DT and had received intratracheal transfer of wild-type or STAT6-/- bone marrow derived dendritic cells (n = 10 mice per group, from 2 experiments). CD11b-DTR mice treated with DT or PBS following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 8 mice per group, from 2 experiments) served as controls.
It is possible that depletion of the CD11b+ myeloid cells in the lung may have also impaired antigen presentation of OVA to the injected Th2 cells. If this was occurring, we would expect to see comparatively lower numbers of OVA-specific Th2 cells in the thoracic lymph nodes of DT treated mice compared to PBS treated mice due to a decrease in antigen-induced proliferation of the Th2 cells (14). When we measured the accumulation of transferred OVA-specific Th2 cells in the draining thoracic lymph nodes following OVA challenge, however, we saw a similar number of OVA-specific Th2 cells in the lymph nodes of the PBS and DT treated mice (Figure 8A). These data demonstrate that the transferred Th2 cells proliferated in DT-treated mice, consistent with at least some effective antigen presentation. Despite these data, we think that it is possible that there is still a potential defect in antigen presentation in the DT treated mice, but this does not rule out a simultaneous role for these cells in Th2 cell specific chemokine production. To definitively demonstrate that a CD11b+ myeloid cell is the critical cell that controls Th2-cell-active chemokine production, we treated CD11b-DTR mice following DT or PBS injection with IL-13 administration in the lung. This allowed us to measure effector functions of these cells independent of antigen presentation. We then analyzed RNA from the lungs of these mice for chemokine production. The levels of CCL17, CCL22 and CCL24 RNA were all significantly reduced in the DT-treated mice compared to PBS-treated mice (Figure 8B). CCL11 levels were not different between the two groups, consistent with a non-CD11b+ cellular source for this chemokine. This may be due to the strong stimulation of other cells by the high levels of IL-13 introduced into the airways. Overall, these data confirm that a CD11b+ population is critical for Th2 cell-active chemokine production.
Figure 8. DT administration to CD11b-DTR mice does not impair Th2 cell accumulation in the lymph node but does reduce IL-13 induced chemokine production.
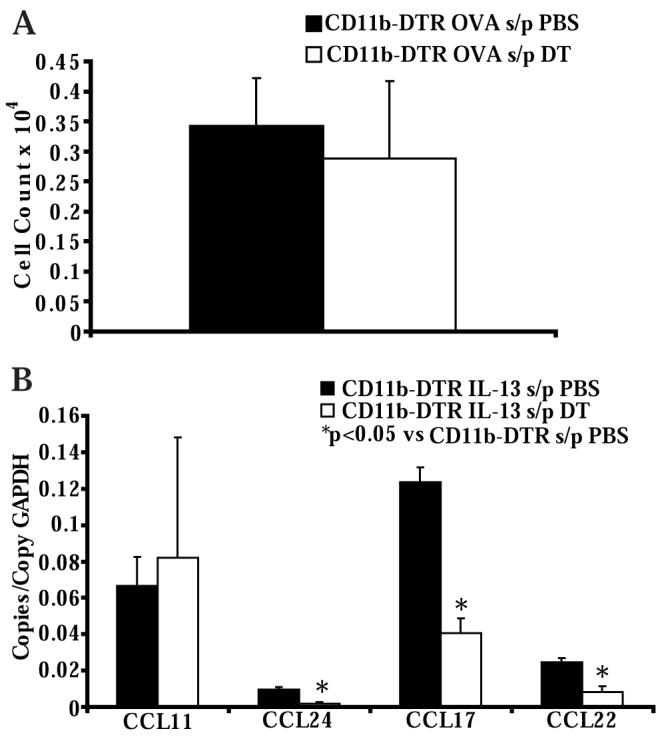
A) Numbers of OVA-specific Th2 cells in the thoracic lymph nodes of CD11b-DTR mice after treatment with PBS or DT and following adoptive transfer of OVA-specific in vitro polarized Th2 lymphocytes and 4 OVA challenges (n = 5 mice per group, from 2 experiments). B) Lung chemokine RNA copies normalized to copies of GAPDH RNA in CD11b-DTR mice after treatment with PBS or DT and 24 hours following intranasal IL-13 administration (n = 6 mice per group, from 2 experiments).
Discussion
The transcription factor STAT6 has been shown to be an important regulator of Th2-type inflammation making it a potential target for therapy in allergic inflammatory disorders such as asthma (38). Among the many functions of STAT6 is the regulation of chemokine production in the lung during the establishment of allergic airway inflammation (23). The goal of the current study was to define the critical cellular mediators of STAT6-dependent Th2 cell-active chemokine secretion in the lung during allergic inflammation. Although there are multiple potential cellular sources for these chemokines in the lung (39-56), we hypothesized that myeloid-derived cells in the lung, such as pulmonary dendritic cells and/or macrophages, were the primary mediators of chemokine production and Th2 cell recruitment in this model of asthma. Our data suggest that these cells are bone marrow-derived CD11b+ myeloid cells. These cells have already been demonstrated to be critical for initiation of the adaptive immune response in asthma (35, 37); however our data reveal a novel role for these cells in controlling the effector phase of allergic inflammation by mediating Th2 cell recruitment.
We have previously demonstrated that following adoptive transfer, recruitment of Th2 cells into the airways following antigen exposure occurs via both STAT6-dependent and -independent mechanisms (16, 23, 57). This is based on the fact that STAT6-/- mice have reduced Th2 cell recruitment into the airways compared to wild-type mice, but these mice maintain some minimal Th2 cell recruitment that is fully eliminated when chemoattractant receptor signaling is disrupted with pertussis treatment of the Th2 cells (16, 23). However, STAT6 is clearly necessary for the full dramatic accumulation of Th2 cells in the airways that develops in response to antigen challenge (16, 23, 27). Since Th2 cells in the lung and airway do not proliferate extensively in response to antigen (14), the increase in the number of Th2 cells, as well as eosinophils, is dependent on cellular recruitment. These data suggest that in response to IL-4 and IL-13 (produced by the Th2 cells recruited into the lung by STAT6-independent mechanisms) resident lung cells are stimulated via STAT6 to produce chemokines, such as CCL17, CCL22, CCL11, and CCL24. These chemokines then amplify allergic inflammation by increasing the recruitment of Th2 cells and initiating the recruitment of eosinophils into the airways. Consistent with this, studies on humans with asthma have demonstrated increased levels of the Th2-active chemokines CCL17 and CCL22 in the airways (58, 59), and inhibition of the activity of these chemokines has been shown to reduce allergic inflammation in murine models of asthma (18, 52). Similarly, the deletion of the eosinophil-active chemokines CCL11 or CCL24 reduced lung eosinophil accumulation in murine models of asthma (44, 53, 56). These data suggest that STAT6 is a master regulator of cellular recruitment in asthma, and thus may be an effective target to reduce Th2 cell and eosinophil recruitment into the lung. Given the potential therapeutic utility of targeting of STAT6, a more cell-specific approach would be ideal. Thus, we used a strategy that targeted specific cell types in the lung looking for a cell or cells that are necessary and sufficient to lead to Th2 lymphocyte recruitment.
In our experiments, we used the adoptive transfer model of allergic airway inflammation (16, 23). This model was chosen because the T cell polarization step is performed in vitro, thus we could eliminate effects on T cell priming/activation and focus on effects on recruitment. In addition, mast cells are not activated in this model, simplifying the analysis of our findings. Previously, we had shown in this model that there was significantly reduced wild-type OVA-specific Th2 cell recruitment into the airways of STAT6-/- recipient mice compared to wild-type recipient mice following OVA challenge. These results differ from data recently published by King et al., which did not demonstrate a Th2 cell recruitment defect in STAT6-/- mice following naïve T cell transfer and N. brasiliensis infection with OVA sensitization (60). However, their model involved T cell priming/activation in vivo and intact IgE-mast cell responses, so is not completely analogous to our model of allergic inflammation. Furthermore, even though the authors report that there was not a Th2 cell recruitment defect, the percentage of OVA-specific T cells recruited into the BAL in their model was 50% less in STAT6-/- mice compared to wild-type recipients.
In our first set of experiments, we used mice engineered to express STAT6 under control of the CC10 promoter (31) to test the role of airway lining cells in the recruitment of Th2 cells and eosinophils. Expression of STAT6 in these cells restored mucus production in the model but did not lead to increased Th2 cell recruitment or eosinophil recruitment compared to STAT6-/- mice. The EpiSTAT6 mice as well as STAT6-/- mice had reduced levels of CCL17, CCL11, and CCL24 RNA in the lung. Interestingly, RNA levels of CCL22 were restored but there was no detectable protein in the BAL. It may be that CCL22 is not secreted or remains bound to cellular surfaces as seen with other chemokines (61). The protein levels of CCL17 and CCL24 were reduced in the BAL of EpiSTAT6 and STAT6-/- mice compared to wild-type mice, but we were unable to detect significant amounts of CCL11 in the BAL in wild-type, STAT6-/- mice or the EpiSTAT6 mice. Our experiments demonstrate that expression of STAT6 in CC10 expressing airway lining cells is not sufficient to restore Th2 cell and eosinophil trafficking into the lung in this model. We cannot rule out a role for non-CC10 expressing airway epithelia, however, as these cells would remain STAT6-/- in the EpiSTAT6 mouse.
We wondered if the eosinophil trafficking defect seen in the STAT6-/- mice and EpiSTAT6 mice was secondary to the Th2 cell trafficking defect or if there was also a coexisting defect in eosinophil trafficking that resulted directly from STAT6-deficiency. In our experiments we bypassed the need for Th2 cell recruitment by transferring the cells directly into the airways via an intratracheal injection. In these experiments, transfer of wild-type OVA-specific Th2 cells directly into the lung of STAT6-/- mice did not restore eosinophil recruitment or the production in the lung of the eosinophil-active chemokines. These data demonstrate that the eosinophil recruitment in this model is also dependent on STAT6-induced chemokine production in the lung.
Voehringer et al have demonstrated that STAT6 expression in a bone marrow derived myeloid cell is necessary for Th2 cell and eosinophil trafficking into the lung in a model of pulmonary Th2 inflammation induced by N. brasiliensis infection (62, 63). However, these experiments did not identify whether the critical cellular mediator was a lung dendritic cell or macrophage, nor did the studies directly address Th2 cell recruitment in an asthma model. We sought to determine if a similar mechanism was involved in Th2 cell and eosinophil recruitment in a model of allergic airway inflammation. For these experiments, we reconstituted STAT6-/- mice with wild-type, STAT6-/- or RAG1-/- bone marrow. Wild-type or RAG1-/- bone marrow was able to fully restore the recruitment of Th2 cells and eosinophils in the model. In these experiments, the expression of the chemokines CCL17, CCL22, and CCL24 was restored with wild-type or RAG1-/- bone marrow transplant into STAT6-/- mice. Interestingly, CCL11 was not expressed in large amounts at either the RNA or the protein level. Given that STAT6-/- mice reconstituted with RAG1-/- bone marrow would only have STAT6 restored in myeloid cells, these data demonstrate that expression of STAT6 in bone marrow derived myeloid cells is sufficient to restore Th2 cell and eosinophil trafficking in a model of allergic airway inflammation.
The bone marrow reconstitution experiments do not fully define the cellular mediator of STAT6-dependent recruitment in this model. For this reason, we wanted to use a system that allowed us to selectively deplete myeloid cell populations in the lung. The identification of dendritic cells and macrophages using cell-type specific markers is different in the lung than in other organs (32). Specifically, the dendritic cell marker CD11c has been shown to be expressed on both pulmonary macrophages and dendritic cells, and the myeloid marker CD11b, is seen at high levels on pulmonary dendritic cells and at low levels on pulmonary macrophages (32, 36). Thus, we decided to utilize a transgenic mouse that allowed us to deplete the CD11b+ subpopulation of myeloid cells in the lung using DT, which should have its primary effect on dendritic cells with minimal effects on macrophages. Careful phenotyping of cell types in the lungs of mice after DT administration indicated a very specific reduction in the number of CD11b+/CD11c+/MHC II+/Gr-1- myeloid cells in the lung, and minimal effects on other CD11b+ or CD11c+ populations. In these experiments, the reduction in CD11b+ cells significantly attenuated the recruitment of Th2 cells and eosinophils into the lung following adoptive transfer of wild-type Th2 cells and OVA challenge. Furthermore, add back of wild-type bone-marrow derived dendritic cells was able to restore the recruitment of Th2 cells in these mice, demonstrating that these cells were sufficient for Th2 cell recruitment and that the recruitment defect in the DT treated CD11b-DTR mice was due to the deletion of these myeloid cells. In addition, there was a significant reduction in the expression of CCL17, CCL22, CCL11, and CCL24 RNA in the lungs of the CD11b-DTR mice treated with DT compared to those treated with PBS. These data are consistent with the RNA expression profile we generated by isolating these cells from the lung using a cell sorter as well as with data published by others (37, 64). It is interesting that there is such a dramatic decrease in Th2 cell recruitment and chemokine expression with a 3-fold reduction in this cell population; however, we speculate that we may be depleting the most active CD11b+ cells with DT treatment. Alternatively, we may be reducing the numbers of a subpopulation of CD11b+/CD11c+/MHC II+/Gr-1- myeloid cells below a critical threshold that significantly impairs chemokine production and Th2 cell recruitment. Our results differs somewhat from data published by Beaty et al. which demonstrated that both CD11c+/CD11bhigh dendritic cells and CD11c+/CD11blow/CD103+ dendritic cells in the lung can produce high levels of the chemokines CCL17 and CCL22 in a murine model of asthma (33). However, they used a different model of asthma for their experiments. In addition, our findings are in agreement with findings by van Rijt et al. (35), which demonstrated that DT-induced depletion of CD11c+ cells in the lungs prevented the development of allergic airway inflammation. This study utilized the OVA immunization model to demonstrate the importance of dendritic cells in the development of allergic airway inflammation. While their study largely focused on the early role of these cells in antigen presentation to T cells, they also examined the effects of CD11c+ depletion on allergic inflammation in the adoptive transfer model. However, in those experiments deletion of CD11c+ cells depleted pulmonary macrophages (CD11c+/CD11b-) as well as myeloid dendritic cells (CD11c+/CD11b+), and thus they were not able to define the critical cellular mediator of Th2 cell recruitment in the adoptive transfer model of asthma. In our experiments, we were able to delete only the CD11c+/CD11b+ myeloid cell population and therefore we have extended their observations by more specifically defining the critical cellular mediator of Th2 cell recruitment.
An alternative explanation for our findings is that STAT6 deficiency in CD11b+ myeloid cells affects antigen presentation to Th2 cells, thus the decreased accumulation of Th2 cells in the lung is secondary to a failure to reactivate primed Th2 cells. Recent data has demonstrated that IL-4 and IL-13 can influence the maturation of dendritic cells in the lung (presumably via STAT6 signaling) and can affect their ability to regulate cytokine expression by memory CD4+ T cells (65). Although this mechanism may be partially responsible for our findings, we believe our findings demonstrate that CD11b+ myeloid cells in the lung have an additional role in directing Th2 cell recruitment into the airways via STAT6-dependent chemokine production. To evaluate the possibility that the phenotype we observed was also secondary to effects on antigen presentation, we examined at the accumulation of the transferred Th2 cells in the draining thoracic lymph nodes. Previous studies had demonstrated that transferred Th2 cells will proliferate in the lymph node only following presentation of inhaled antigen by dendritic cells (14). The fact that the numbers of transferred Th2 cells in the draining thoracic lymph node were similar in mice following DT-induced depletion of CD11b+ myeloid cells compared to PBS control mice, suggests that antigen presentation was at least partially intact. In addition, we did not see a difference in blood eosinophilia in the two groups of mice suggesting that IL-5 secretion from Th2 cells following CD11b+ cell depletion was not affected. As further evidence, we used IL-13 to stimulate chemokine production (thus bypassing antigen presentation) in CD11b-DTR mice and demonstrate that Th2-active chemokine production was significantly impaired after treatment with DT compared to mice treated with PBS. Thus, we believe these data definitively demonstrate that CD11b+ myeloid cells have a critical role mediating Th2 cell recruitment independent of their role in antigen presentation.
Our data confirm that the recruitment of eosinophils in this model is also dependent on STAT6 expression in a resident lung cell (23, 53, 56, 62, 63). Consistent with prior data from others (53, 56), we demonstrate that CD11c+/CD11b+ cells are a source of CCL24 but so are CD11c+/CD11b- cells (alveolar macrophages), and neither cell makes large amounts of CCL11. In addition to effects on cellular recruitment, STAT6 also is necessary for mucus hypersecretion in this model (23). In our experiments, we observed that following adoptive transfer and OVA challenge, EpiSTAT6 mice develop mucus hypersecretion similar to wild-type mice, but reconstitution of STAT6 expression in myeloid cells did not restore this endpoint of allergic inflammation. These data are similar to those of Kuperman et al., which demonstrated that mucus producing cells are derived from Clara cells in the airways (66). Our data also suggest that the multiple endpoints of allergic inflammation can be anatomically compartmentalized with different cell-types controlling different STAT6-dependent processes.
In conclusion, our data demonstrate that STAT6 expression in a CD11b+ myeloid cell in the lung is necessary for effective Th2 cell recruitment into the airways in a murine model of allergic airway inflammation. We propose that following STAT6 independent recruitment of Th2 cells into the airway, CD11b+ cells in the lung are stimulated by IL-4 and IL-13 to produce CCL17 and CCL22 via STAT6. These chemokines then help orchestrate the amplification of Th2 cell recruitment. Our studies illuminate a novel link between the innate and adaptive immune systems and suggest that a therapeutic strategy targeting STAT6 in CD11b+ cells may provide a novel means of reducing allergic airway inflammation.
Acknowledgements
The authors would like to thank Andrew Carafone, Ryan Jackobek, Francisco Zincone, and Carol Leary for technical support with this research.
Footnotes
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
Grant Support: This work was supported by the National Institutes of Health grants K08 HL072775 to B.D. Medoff and R01 AI40618 to A.D. Luster.
- AHR
- airway hyperresponsiveness
- BAL
- bronchoalveolar lavage
- PAS
- Periodic-Acid-Schiff
- TCR
- T cell receptor
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Busse WW, Lemanske RF., Jr. Asthma. N Engl J Med. 2001;344:350–362. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- 2.Larche M, Robinson DS, Kay AB. The role of T lymphocytes in the pathogenesis of asthma. J Allergy Clin Immunol. 2003;111:450–463. doi: 10.1067/mai.2003.169. quiz 464. [DOI] [PubMed] [Google Scholar]
- 3.Woodfolk JA. T-cell responses to allergens. J Allergy Clin Immunol. 2007;119:280–294. doi: 10.1016/j.jaci.2006.11.008. quiz 295-286. [DOI] [PubMed] [Google Scholar]
- 4.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 5.Del Prete GF, De Carli M, D’Elios MM, Maestrelli P, Ricci M, Fabbri L, Romagnani S. Allergen exposure induces the activation of allergen-specific Th2 cells in the airway mucosa of patients with allergic respiratory disorders. Eur J Immunol. 1993;23:1445–1449. doi: 10.1002/eji.1830230707. [DOI] [PubMed] [Google Scholar]
- 6.Walker C, Bode E, Boer L, Hansel TT, Blaser K, Virchow JC., Jr. Allergic and nonallergic asthmatics have distinct patterns of T-cell activation and cytokine production in peripheral blood and bronchoalveolar lavage. Am Rev Respir Dis. 1992;146:109–115. doi: 10.1164/ajrccm/146.1.109. [DOI] [PubMed] [Google Scholar]
- 7.Brusselle G, Kips J, Joos G, Bluethmann H, Pauwels R. Allergen-induced airway inflammation and bronchial responsiveness in wild-type and interleukin-4-deficient mice. American Journal of Respiratory Cell & Molecular Biology. 1995;12:254–259. doi: 10.1165/ajrcmb.12.3.7873190. [DOI] [PubMed] [Google Scholar]
- 8.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. Journal of Experimental Medicine. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, Corry DB. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 11.Cohn L, Homer RJ, Marinov A, Rankin J, Bottomly K. Induction of airway mucus production By T helper 2 (Th2) cells: a critical role for interleukin 4 in cell recruitment but not mucus production. Journal of Experimental Medicine. 1997;186:1737–1747. doi: 10.1084/jem.186.10.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li XM, Schofield BH, Wang QF, Kim KH, Huang SK. Induction of pulmonary allergic responses by antigen-specific Th2 cells. J Immunol. 1998;160:1378–1384. [PubMed] [Google Scholar]
- 13.Li L, Xia Y, Nguyen A, Feng L, Lo D. Th2-induced eotaxin expression and eosinophilia coexist with Th1 responses at the effector stage of lung inflammation. J Immunol. 1998;161:3128–3135. [PubMed] [Google Scholar]
- 14.Harris NL, Watt V, Ronchese F, Le Gros G. Differential T cell function and fate in lymph node and nonlymphoid tissues. J Exp Med. 2002;195:317–326. doi: 10.1084/jem.20011558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Idzko M, Hammad H, van Nimwegen M, Kool M, Muller T, Soullie T, Willart MA, Hijdra D, Hoogsteden HC, Lambrecht BN. Local application of FTY720 to the lung abrogates experimental asthma by altering dendritic cell function. J Clin Invest. 2006;116:2935–2944. doi: 10.1172/JCI28295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathew A, Medoff BD, Carafone AD, Luster AD. Cutting edge: th2 cell trafficking into the allergic lung is dependent on chemoattractant receptor signaling. J Immunol. 2002;169:651–655. doi: 10.4049/jimmunol.169.2.651. [DOI] [PubMed] [Google Scholar]
- 17.Chvatchko Y, Hoogewerf AJ, Meyer A, Alouani S, Juillard P, Buser R, Conquet F, Proudfoot AE, Wells TN, Power CA. A key role for CC chemokine receptor 4 in lipopolysaccharide-induced endotoxic shock. J Exp Med. 2000;191:1755–1764. doi: 10.1084/jem.191.10.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalo JA, Pan Y, Lloyd CM, Jia GQ, Yu G, Dussault B, Powers CA, Proudfoot AE, Coyle AJ, Gearing D, Gutierrez-Ramos JC. Mouse monocyte-derived chemokine is involved in airway hyperreactivity and lung inflammation. J Immunol. 1999;163:403–411. [PubMed] [Google Scholar]
- 19.Lloyd CM, Delaney T, Nguyen T, Tian J, Martinez AC, Coyle AJ, Gutierrez-Ramos JC. CC chemokine receptor (CCR)3/eotaxin is followed by CCR4/monocyte-derived chemokine in mediating pulmonary T helper lymphocyte type 2 recruitment after serial antigen challenge in vivo. J Exp Med. 2000;191:265–274. doi: 10.1084/jem.191.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothenberg M, MacLean J, Pearlman E, Leder P. Targeted disruption of the chemokine eotaxin partially reduces peripheral blood and antigen induced tissue eosinophilia. J. Exp. Med. 1997;185:785–790. doi: 10.1084/jem.185.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goya I, Villares R, Zaballos A, Gutierrez J, Kremer L, Gonzalo JA, Varona R, Carramolino L, Serrano A, Pallares P, Criado LM, Kolbeck R, Torres M, Coyle AJ, Gutierrez-Ramos JC, Martinez AC, Marquez G. Absence of CCR8 does not impair the response to ovalbumin-induced allergic airway disease. J Immunol. 2003;170:2138–2146. doi: 10.4049/jimmunol.170.4.2138. [DOI] [PubMed] [Google Scholar]
- 22.Chung CD, Kuo F, Kumer J, Motani AS, Lawrence CE, Henderson WR, Jr., Venkataraman C. CCR8 is not essential for the development of inflammation in a mouse model of allergic airway disease. J Immunol. 2003;170:581–587. doi: 10.4049/jimmunol.170.1.581. [DOI] [PubMed] [Google Scholar]
- 23.Mathew A, MacLean JA, DeHaan E, Tager AM, Green FH, Luster AD. Signal transducer and activator of transcription 6 controls chemokine production and T helper cell type 2 cell trafficking in allergic pulmonary inflammation. J Exp Med. 2001;193:1087–1096. doi: 10.1084/jem.193.9.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoshino A, Tsuji T, Matsuzaki J, Jinushi T, Ashino S, Teramura T, Chamoto K, Tanaka Y, Asakura Y, Sakurai T, Mita Y, Takaoka A, Nakaike S, Takeshima T, Ikeda H, Nishimura T. STAT6-mediated signaling in Th2-dependent allergic asthma: critical role for the development of eosinophilia, airway hyper-responsiveness and mucus hypersecretion, distinct from its role in Th2 differentiation. Int Immunol. 2004;16:1497–1505. doi: 10.1093/intimm/dxh151. [DOI] [PubMed] [Google Scholar]
- 25.Tomkinson A, Duez C, Lahn M, Gelfand EW. Adoptive transfer of T cells induces airway hyperresponsiveness independently of airway eosinophilia but in a signal transducer and activator of transcription 6-dependent manner. J Allergy Clin Immunol. 2002;109:810–816. doi: 10.1067/mai.2002.123531. [DOI] [PubMed] [Google Scholar]
- 26.Medoff BD, Tager AM, Jackobek R, Means TK, Wang L, Luster AD. Antibody-antigen interaction in the airway drives early granulocyte recruitment through BLT1. Am J Physiol Lung Cell Mol Physiol. 2006;290:L170–178. doi: 10.1152/ajplung.00212.2005. [DOI] [PubMed] [Google Scholar]
- 27.Tager AM, Bromley SK, Medoff BD, Islam SA, Bercury SD, Friedrich EB, Carafone AD, Gerszten RE, Luster AD. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. 2003;4:982–990. doi: 10.1038/ni970. [DOI] [PubMed] [Google Scholar]
- 28.Mikhak Z, Fleming CM, Medoff BD, Thomas SY, Tager AM, Campanella GS, Luster AD. STAT1 in peripheral tissue differentially regulates homing of antigen-specific Th1 and Th2 cells. J Immunol. 2006;176:4959–4967. doi: 10.4049/jimmunol.176.8.4959. [DOI] [PubMed] [Google Scholar]
- 29.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 30.Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, Bennett M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res. 2007;100:884–893. doi: 10.1161/01.RES.0000260802.75766.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, Erle DJ. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8:885–889. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- 32.Vermaelen K, Pauwels R. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A. 2004;61:170–177. doi: 10.1002/cyto.a.20064. [DOI] [PubMed] [Google Scholar]
- 33.Beaty SR, Rose CE, Jr., Sung SS. Diverse and potent chemokine production by lung CD11bhigh dendritic cells in homeostasis and in allergic lung inflammation. J Immunol. 2007;178:1882–1895. doi: 10.4049/jimmunol.178.3.1882. [DOI] [PubMed] [Google Scholar]
- 34.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, Hoogsteden HC, Lambrecht BN. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med. 2005;201:981–991. doi: 10.1084/jem.20042311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jakubzick C, Tacke F, Llodra J, van Rooijen N, Randolph GJ. Modulation of Dendritic Cell Trafficking to and from the Airways. J Immunol. 2006;176:3578–3584. doi: 10.4049/jimmunol.176.6.3578. [DOI] [PubMed] [Google Scholar]
- 37.Kohl J, Baelder R, Lewkowich IP, Pandey MK, Hawlisch H, Wang L, Best J, Herman NS, Sproles AA, Zwirner J, Whitsett JA, Gerard C, Sfyroera G, Lambris JD, Wills-Karp M. A regulatory role for the C5a anaphylatoxin in type 2 immunity in asthma. J Clin Invest. 2006;116:783–796. doi: 10.1172/JCI26582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foster PS. STAT6: an intracellular target for the inhibition of allergic disease. Clin Exp Allergy. 1999;29:12–16. doi: 10.1046/j.1365-2222.1999.00476.x. [DOI] [PubMed] [Google Scholar]
- 39.Ying S, Meng Q, Zeibecoglou K, Robinson DS, Macfarlane A, Humbert M, Kay AB. Eosinophil chemotactic chemokines (eotaxin, eotaxin-2, RANTES, monocyte chemoattractant protein-3 (MCP-3), and MCP-4), and C-C chemokine receptor 3 expression in bronchial biopsies from atopic and nonatopic (Intrinsic) asthmatics. J Immunol. 1999;163:6321–6329. [PubMed] [Google Scholar]
- 40.Sekiya T, Miyamasu M, Imanishi M, Yamada H, Nakajima T, Yamaguchi M, Fujisawa T, Pawankar R, Sano Y, Ohta K, Ishii A, Morita Y, Yamamoto K, Matsushima K, Yoshie O, Hirai K. Inducible expression of a Th2-type CC chemokine thymus- and activation-regulated chemokine by human bronchial epithelial cells. J Immunol. 2000;165:2205–2213. doi: 10.4049/jimmunol.165.4.2205. [DOI] [PubMed] [Google Scholar]
- 41.Li L, Xia Y, Nguyen A, Lai YH, Feng L, Mosmann TR, Lo D. Effects of Th2 cytokines on chemokine expression in the lung: IL-13 potently induces eotaxin expression by airway epithelial cells. J Immunol. 1999;162:2477–2487. [PubMed] [Google Scholar]
- 42.Lilly CM, Nakamura H, Kesselman H, Nagler-Anderson C, Asano K, Garcia-Zepeda EA, Rothenberg ME, Drazen JM, Luster AD. Expression of eotaxin by human lung epithelial cells: induction by cytokines and inhibition by glucocorticoids. J Clin Invest. 1997;99:1767–1773. doi: 10.1172/JCI119341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsukura S, Stellato C, Plitt JR, Bickel C, Miura K, Georas SN, Casolaro V, Schleimer RP. Activation of eotaxin gene transcription by NF-kappa B and STAT6 in human airway epithelial cells. J Immunol. 1999;163:6876–6883. [PubMed] [Google Scholar]
- 44.Rothenberg ME, Luster AD, Leder P. Murine eotaxin: an eosinophil chemoattractant inducible in endothelial cells and in interleukin 4-induced tumor suppression. Proc Natl Acad Sci U S A. 1995;92:8960–8964. doi: 10.1073/pnas.92.19.8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gounni AS, Hamid Q, Rahman SM, Hoeck J, Yang J, Shan L. IL-9-mediated induction of eotaxin1/CCL11 in human airway smooth muscle cells. J Immunol. 2004;173:2771–2779. doi: 10.4049/jimmunol.173.4.2771. [DOI] [PubMed] [Google Scholar]
- 46.Shore SA. Direct effects of Th2 cytokines on airway smooth muscle. Current opinion in pharmacology. 2004;4:235–240. doi: 10.1016/j.coph.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 47.Miyamasu M, Misaki Y, Yamaguchi M, Yamamoto K, Morita Y, Matsushima K, Nakajima T, Hirai K. Regulation of human eotaxin generation by Th1-/Th2-derived cytokines. Int Arch Allergy Immunol. 2000;122(Suppl 1):54–58. doi: 10.1159/000053634. [DOI] [PubMed] [Google Scholar]
- 48.Kumagai N, Fukuda K, Nishida T. Synergistic effect of TNF-alpha and IL-4 on the expression of thymus- and activation-regulated chemokine in human corneal fibroblasts. Biochem Biophys Res Commun. 2000;279:1–5. doi: 10.1006/bbrc.2000.3890. [DOI] [PubMed] [Google Scholar]
- 49.Andrew DP, Chang MS, McNinch J, Wathen ST, Rihanek M, Tseng J, Spellberg JP, Elias CG., 3rd STCP-1 (MDC) CC chemokine acts specifically on chronically activated Th2 lymphocytes and is produced by monocytes on stimulation with Th2 cytokines IL-4 and IL-13. J Immunol. 1998;161:5027–5038. [PubMed] [Google Scholar]
- 50.Rodenburg RJ, Brinkhuis RF, Peek R, Westphal JR, Van Den Hoogen FH, van Venrooij WJ, van de Putte LB. Expression of macrophage-derived chemokine (MDC) mRNA in macrophages is enhanced by interleukin-1beta, tumor necrosis factor alpha, and lipopolysaccharide. J Leukoc Biol. 1998;63:606–611. doi: 10.1002/jlb.63.5.606. [DOI] [PubMed] [Google Scholar]
- 51.Bonecchi R, Sozzani S, Stine JT, Luini W, D’Amico G, Allavena P, Chantry D, Mantovani A. Divergent effects of interleukin-4 and interferon-gamma on macrophage-derived chemokine production: an amplification circuit of polarized T helper 2 responses. Blood. 1998;92:2668–2671. [PubMed] [Google Scholar]
- 52.Kawasaki S, Takizawa H, Yoneyama H, Nakayama T, Fujisawa R, Izumizaki M, Imai T, Yoshie O, Homma I, Yamamoto K, Matsushima K. Intervention of thymus and activation-regulated chemokine attenuates the development of allergic airway inflammation and hyperresponsiveness in mice. J Immunol. 2001;166:2055–2062. doi: 10.4049/jimmunol.166.3.2055. [DOI] [PubMed] [Google Scholar]
- 53.Pope SM, Zimmermann N, Stringer KF, Karow ML, Rothenberg ME. The Eotaxin Chemokines and CCR3 Are Fundamental Regulators of Allergen-Induced Pulmonary Eosinophilia. J Immunol. 2005;175:5341–5350. doi: 10.4049/jimmunol.175.8.5341. [DOI] [PubMed] [Google Scholar]
- 54.Oliveira SH, Lukacs NW. Stem cell factor and igE-stimulated murine mast cells produce chemokines (CCL2, CCL17, CCL22) and express chemokine receptors. Inflamm Res. 2001;50:168–174. doi: 10.1007/s000110050741. [DOI] [PubMed] [Google Scholar]
- 55.Wirnsberger G, Hebenstreit D, Posselt G, Horejs-Hoeck J, Duschl A. IL-4 induces expression of TARC/CCL17 via two STAT6 binding sites. Eur J Immunol. 2006;36:1882–1891. doi: 10.1002/eji.200635972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pope SM, Fulkerson PC, Blanchard C, Akei HS, Nikolaidis NM, Zimmermann N, Molkentin JD, Rothenberg ME. Identification of a cooperative mechanism involving interleukin-13 and eotaxin-2 in experimental allergic lung inflammation. J Biol Chem. 2005;280:13952–13961. doi: 10.1074/jbc.M406037200. [DOI] [PubMed] [Google Scholar]
- 57.Medoff BD, Thomas SY, Luster AD. T Cell Trafficking in Allergic Asthma: The Ins and Outs. Annu Rev Immunol. 2008;26:205–232. doi: 10.1146/annurev.immunol.26.021607.090312. [DOI] [PubMed] [Google Scholar]
- 58.Pilette C, Francis JN, Till SJ, Durham SR. CCR4 ligands are up-regulated in the airways of atopic asthmatics after segmental allergen challenge. Eur Respir J. 2004;23:876–884. doi: 10.1183/09031936.04.00102504. [DOI] [PubMed] [Google Scholar]
- 59.Ying S, O’Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, Robinson D, Zhang G, Zhao J, Lee TH, Corrigan C. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. 2005;174:8183–8190. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- 60.King SB, Knorn AM, Ohnmacht C, Voehringer D. Accumulation of effector CD4 T cells during type 2 immune responses is negatively regulated by Stat6. J Immunol. 2008;180:754–763. doi: 10.4049/jimmunol.180.2.754. [DOI] [PubMed] [Google Scholar]
- 61.Yuan Q, Campanella GS, Colvin RA, Hamilos DL, Jones KJ, Mathew A, Means TK, Luster AD. Membrane-bound eotaxin-3 mediates eosinophil transepithelial migration in IL-4-stimulated epithelial cells. Eur J Immunol. 2006;36:2700–2714. doi: 10.1002/eji.200636112. [DOI] [PubMed] [Google Scholar]
- 62.Voehringer D, Shinkai K, Locksley RM. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 2004;20:267–277. doi: 10.1016/s1074-7613(04)00026-3. [DOI] [PubMed] [Google Scholar]
- 63.Voehringer D, van Rooijen N, Locksley RM. Eosinophils develop in distinct stages and are recruited to peripheral sites by alternatively activated macrophages. J Leukoc Biol. 2007;81:1434–1444. doi: 10.1189/jlb.1106686. [DOI] [PubMed] [Google Scholar]
- 64.Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med. 2006;203:269–273. doi: 10.1084/jem.20051745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Webb DC, Cai Y, Matthaei KI, Foster PS. Comparative roles of IL-4, IL-13, and IL-4Ralpha in dendritic cell maturation and CD4+ Th2 cell function. J Immunol. 2007;178:219–227. doi: 10.4049/jimmunol.178.1.219. [DOI] [PubMed] [Google Scholar]
- 66.Kuperman DA, Huang X, Nguyenvu L, Holscher C, Brombacher F, Erle DJ. IL-4 receptor signaling in Clara cells is required for allergen-induced mucus production. J Immunol. 2005;175:3746–3752. doi: 10.4049/jimmunol.175.6.3746. [DOI] [PubMed] [Google Scholar]