

Abstract
Rationale: Recent genetic studies have implicated integrins in asthma and atopy susceptibility. We therefore evaluated the integrin-β3 gene (ITGB3), an integrin gene within an asthma linkage peak on chromosome 17, as a candidate for susceptibility to asthma- and atopy-related phenotypes. Methods and Measurements: We genotyped and performed association tests on 19 single nucleotide polymorphisms in ITGB3 in the Hutterites, a founder population, and in three outbred replication populations. Main Results: Variation in ITGB3 was strongly associated with susceptibility to bronchial hyperresponsiveness and protection from allergic sensitization to mold allergens in this population. Three independent case-control populations representing Caucasians and African Americans were used to replicate this finding, also revealing ITGB3 alleles that are associated with asthma susceptibility and protection from mold allergen sensitization. Conclusions: This study provides evidence that ITGB3 plays a role in the pathogenesis of asthma and sensitization to mold allergens.
Keywords: association, asthma, genetics, hypersensitivity, integrins
Asthma is a complex disease characterized by bronchial hyperresponsiveness (BHR) and reversible airway obstruction, often occurring with atopy (IgE-mediated allergic sensitization). Asthma affects 155 million individuals worldwide and is both clinically and genetically heterogeneous (1). Although over 200 candidate gene studies for asthma and atopy have been published (reviewed in Reference 2), most have focused on genes related to immune function or therapeutic target pathways. However, recent observations suggest that genes in additional pathways may be involved in the development of asthma and atopy. In particular, integrins and related proteins have been implicated in disease susceptibility and pathogenesis. First, ADAM33, a disintegrin and matrix metalloprotease, was identified as influencing susceptibility to BHR in a positional cloning study (3). The membrane-bound proteins in this family have zinc metalloprotease activity and mediate cell–cell adhesion. One of the potential mechanisms of ADAM33 involvement with BHR is through airway remodeling involving integrins (4). Second, we recently reported a significant association between an amino acid polymorphism in the P-selectin gene (SELP Val640Leu) and atopy in the Hutterites (5). P-selectin is an integrin stored in platelet and endothelial cell granules. P-selectin is released by activated cells to mediate interaction with leukocytes and may thus influence the inflammatory response in asthma.
Several genomewide screens of asthma and atopy susceptibility loci identified a linkage peak on chromosome 17, spanning 62 to 100 cM from p-ter (6–8), and a quantitative trait locus (QTL) screen in mice identified linkage to BHR in the syntenic region (9). Recent interest in our laboratory in the gene encoding integrin β3 (ITGB3) (10), which is located on 17q, combined with studies describing integrin associations with similar phenotypes, led us to study this gene as an asthma and atopy susceptibility candidate. ITGB3 encodes a β-integrin that comprises part of the platelet- and monocyte-specific heterodimeric receptor for fibrinogen and the widely expressed receptor for vitronectin. Fibrinogen receptors regulate platelet aggregation, hemostasis, and thrombosis, and they interact with P-selectin to mediate the adhesion of platelets to neutrophils (11). Vitronectin receptors have roles in leukocyte adhesion and extravasation (12), are important in neutrophil braking during transit through the extracellular matrix (13), and signal mast cell degranulation (14). In addition, vitronectin has been shown to downregulate the expression of α-smooth muscle actin, implying an important role in remodeling during lung development or injury response (15). The combined positional and functional evidence suggests ITGB3 as a candidate for asthma and atopy susceptibility.
This study investigated the role of variation in the ITGB3 gene in asthma and atopy susceptibility. We report significant associations of synonymous and noncoding single nucleotide polymorphisms (SNPs) in ITGB3 with asthma and sensitization to mold allergen in four populations, and we suggest that genes in integrin pathways may be important contributors to asthma and sensitization to mold allergens.
METHODS
Hutterites
Our studies of asthma genetics in the Hutterites have been described (16). Briefly, 638 Hutterites were evaluated for asthma and atopy using a modified version of the Collaborative Study on the Genetics of Asthma protocol (8), as reported (17). In this sample, 71 individuals had asthma, 156 had BHR to methacholine, 311 had a positive skin-prick test (SPT) to at least one of 14 airborne allergens. Of the 540 Hutterites in our genotyped case-control sample, 145 had BHR and 68 had a positive SPT to mold allergen. We used BHR as the primary asthma phenotype because, during field trips, we identified undiagnosed asthma, and most Hutterites did not reliably recall past symptoms.
Outbred Chicago Replication Samples
Unrelated asthma cases were recruited in Chicago as part of the Collaborative Study on the Genetics of Asthma and met the same criteria for asthma as the Hutterites (8, 18). Atopy was assessed in the Chicago subjects using the same protocol and a nearly identical panel of allergens as used in the Hutterites (see Reference 18 for details). Cases and control subjects recruited in Chicago reported either at least three grandparents of African American ancestry or of European ancestry. Control subjects were adults reporting a negative personal and first-degree family history for asthma. No phenotyping was performed on control subjects. These samples included 184 African Americans and 108 Caucasians with asthma and 175 African Americans and 186 white control subjects. Of the subjects with asthma, 60 African Americans and 42 Caucasians had a positive SPT to mold allergen.
Outbred Madison Replication Sample
The Childhood Onset of Asthma Study recruited couples in Madison, Wisconsin, before the birth of a child. At least one parent had asthma or allergies (19). In these parents, asthma status was based on a physician's diagnosis and atopy was determined by positive SPT. Our sample included unrelated white parents, of which 335 were atopic (147 with a positive SPT to mold) and 114 were both atopic and asthmatic; 89 parents who were neither atopic nor asthmatic were used as control subjects in our study.
Genotyping
Most SNPs were chosen from dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) to achieve an average spacing of 5 kb, spanning from 5 kb upstream to 5 kb downstream of the gene. All but two SNPs were genotyped in all samples by DNAprint, Inc. (Sarasota, FL) (http://www.dnaprint.com/). The Leu33Pro polymorphism in the Hutterites was genotyped in a linear array panel (Roche Molecular Systems, Inc., Alameda, CA) (20), one SNP (rs15908) was genotyped by an Assay-on-Demand (Applied Biosystems, Foster City, CA) in the Chicago and Madison populations, and one SNP (rs884696) was not typed in the Madison samples because of a limited amount of DNA.
Mendelian error checking was performed for all Hutterite and Madison samples and for Chicago cases with parental data using PedCheck (21). In addition, 12 Hutterite samples were resequenced for the coding regions of ITGB3 and were compared for 9 of 19 sites that fell within sequencing amplicons. The sequence data agreed with genotype data in every case. In the Chicago samples, 65 individuals were genotyped in duplicate and compared as part of genotyping quality control. All genotyping was performed blind to case/control status, and cases and control subjects were genotyped at the same time on the same trays.
Statistical Analysis
Case-control tests in the Hutterites were performed using two association methods designed for related individuals with a known pedigree (5). These tests may have optimal power to detect associations under different genetic models and are therefore complementary tests in the Hutterites. Pairwise haplotypes in the Hutterites were determined by direct observation of alleles segregating in families. Single SNP association tests in the Chicago and Madison samples were performed using χ2 tests for allele and genotype counts. Pairwise haplotypes were inferred and analyzed in the outbred cases and control subjects using FamHap (22, 23).
We did not correct p values for multiple testing because many of the SNPs are correlated, making it impossible to determine the number of independent tests. Instead, we report results in the Hutterites with p values of less than 0.01 and use three replication samples to reduce the likelihood of type 1 error. For the Hutterite case-control tests (corrected χ2 and quasi-likelihood) and replication sample case-control tests (genotype and allele), we present the result with the lower p value.
RESULTS
Allele frequencies at all SNPs in the Hutterite population and three replication samples are shown in Table 1. The numbers of cases and control subjects genotyped for each SNP are reported in Table E2 in the online supplement. Genotypes for these SNPs are in Hardy-Weinberg equilibrium (HWE) in the Hutterites and Chicago and Madison control samples (p> 0.01), except for one 5′ SNP (rs2317385) in the African American sample (deficiency of heterozygotes, HWE p = 0.0026) and one intron 8 SNP (rs2292864) in the Madison sample (deficiency of heterozygotes, HWE p = 0.0045). This is unlikely to be due to genotyping error, as the cases and control subjects were genotyped together and these SNPs were genotyped by the same method in all four populations, the other three of which were all in HWE (p> 0.3) for both SNPs. In addition, a SNP in intron 1 (rs1009312) that is in strong linkage disequilibrium with rs2317385 (African American r2 = 0.94) but genotyped at a different time also showed a slight deficiency of heterozygotes in the African American control subjects (HWE p = 0.040). For both of these SNPs, there was a trend toward excess heterozygotes in African American cases (HWE p = 0.17 and 0.059, respectively).
TABLE 1.
Single nucleotide polymorphisms in ITGB3
| Frequency of Minor Allele
|
|||||||
|---|---|---|---|---|---|---|---|
| rs No. | Location in Gene | Position in Contig NT_035424 |
Minor Allele |
Hutterite | Madison | Chicago White |
Chicago African American |
| rs2317385 | 5′ region | 841140G->A | A | 0.20D | 0.20 | 0.19 | 0.38*† |
| rs1009312 | Intron 1 | 843598G->A | A | 0.20D | 0.18 | 0.20 | 0.39† |
| rs3892085 | Intron 1 | 851938A->G | G | 0.14D | 0.13 | 0.12 | 0.04† |
| rs884696 | Intron 1 | 852702C->A | A | 0.22U | NA | 0.19 | 0.34† |
| rs2015729 | Intron 2 | 865951G->A | A | 0.39A | 0.44 | 0.42 | 0.48 |
| rs1000232 | Intron 2 | 868019T->C | C | 0.37A | 0.36 | 0.36 | 0.48† |
| rs2292867 | Intron 2 | 868947C->T | T | 0.14D | 0.13 | 0.14 | 0.17 |
| rs5918 | Exon 3 | 872188T->C (L33P) | C | 0.21D | 0.14 | 0.15 | 0.10‡ |
| rs5919 | Exon 6 | 875998T->C | C | 0.02A | 0.08 | 0.08 | 0.16† |
| rs2292864 | Intron 8 | 879139C->T | T | 0.14D | 0.12* | 0.12 | 0.14 |
| rs15908 | Exon 9 | 879795A->C | C | 0.38A | 0.41 | 0.38 | 0.42 |
| rs4634 | Exon 10 | 881247G->A | A | 0.36A | 0.30 | 0.31 | 0.30 |
| rs999323 | Intron 10 | 883061T->C | C | 0.36U | 0.33 | 0.33 | 0.31 |
| rs11870252 | Intron 11 | 889283T->C | C | 0.10P | 0.09 | 0.08 | 0.08 |
| rs3760372 | Intron 12 | 891460T->C | T | 0.39A | 0.31 | 0.34 | 0.25‡ |
| rs3809863 | Intron 14 | 896470C->T | T | 0.10D | 0.09 | 0.09 | 0.08 |
| rs17225109 | 3′UTR | 899667G->A | A | 0.07D | 0.04 | 0.04 | 0.01† |
| rs2317676 | 3′UTR | 899741A->G | G | 0.08D | 0.09 | 0.09 | 0.13 |
| rs2317677 | 3′ Region | 904865G->A | A | 0.22D | 0.34 | 0.35 | 0.34 |
Definition of Abbreviation: UTR = untranslated region.
Single nucleotide polymorphisms (SNPs) included in this study are identified by rs number (dbSNP). The SNPs are shown as base change from consensus sequence in contig NT_035424. The frequency of the minor allele in the Hutterite population and outbred control subjects is shown, with a superscript indicating whether this is the ancestral allele (A), derived allele (D), unknown in primates (U), or polymorphic across primates (P) based on sequence and database information.
Not in Hardy-Weinberg equilbrium in this population.
Allele frequency differs between Chicago Caucasian subjects and African American control subjects (p < 0.01).
Allele frequency differs between Chicago Caucasian subjects and African American control subjects (0.05> p> 0.01).
Allele frequencies in the Hutterites were similar to the Chicago and Madison replication samples as seen for alleles at other loci (20). Not surprisingly, the frequencies at 7 of 19 SNPs differed significantly (p < 0.01) between the Chicago African American and white control subjects (Table 1). Pairwise linkage disequilibrium values (r2) for all pairs of SNPs are given in Table E1.
The results of the association studies in the Hutterites and three replication samples are shown in Figure 1. In the Hutterites, the minor alleles at five SNPs in ITGB3 were associated with BHR (p < 0.01; red squares in Figure 1). These include synonymous coding SNPs in exons 9 and 10 and SNPs in introns 2, 10, and 12, but not the nonsynonymous Leu33Pro polymorphism. An additional SNP in intron 1 showed several significant haplotype associations (Figure 2). Seven SNPs were associated with allergic sensitization to mold allergens in the Hutterites (p < 0.01; blue triangles in Figure 1). These include synonymous SNPs in exons 9 and 10 and SNPs in introns 1, 2, 10, and 12. Additional SNPs in exon 6 and the 3′ region showed significant haplotype associations (Figure 2). Interestingly, although five of the significant associations overlap with the BHR-associated SNPs, the alleles overrepresented in BHR cases were underrepresented in positive SPT to mold cases, such that a susceptibility genotype for BHR would be a protective genotype for sensitization to molds and vice versa. Positive SPT to other allergens was not associated with ITGB3 variation in the Hutterites (p> 0.01).
Figure 1.
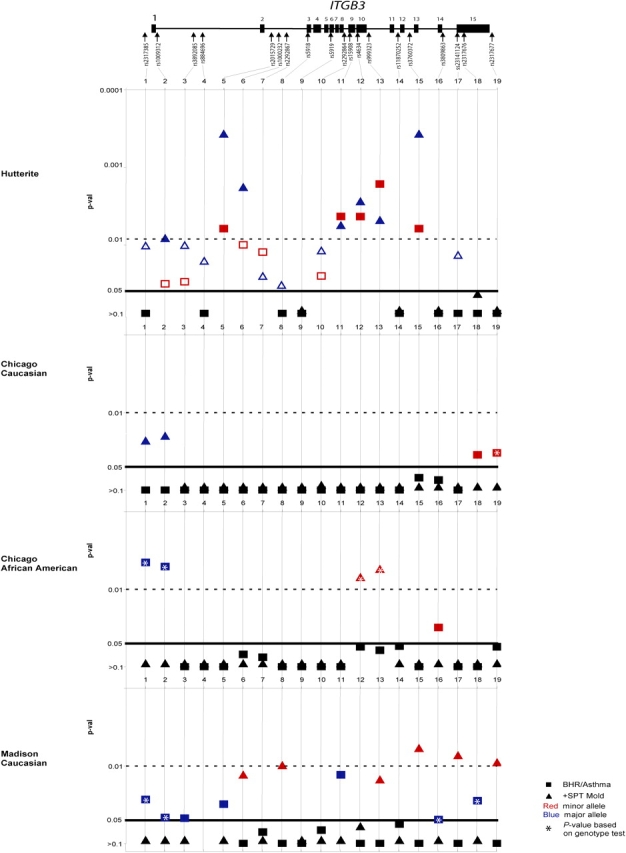
Association of variation in ITGB3 with asthma and atopy phenotypes. The genomic structure of ITGB3 is shown (not to scale), with exons depicted as boxes and arrows identified by rs number in the approximate locations of the variation genotyped. Association signals in the four samples are shown in graphical form. p values are on the Y-axis, with a dashed line showing the threshold for p = 0.01 and a solid line for p = 0.05. Single nucleotide polymorphism (SNP) number in ITGB3 is plotted on the X-axis. Associations with bronchial hyperresponsiveness (BHR) (Hutterites) or asthma (replication samples) are depicted with squares, and associations with allergic sensitization to mold allergen are depicted with triangles. For associations with p < 0.05, associations with the minor allele are in red and associations with the major allele are in blue. p values between 0.05 and 0.01 are shown as open symbols in the Hutterites. The SNP with no result in the Madison sample was not genotyped because of limited DNA. +SPT = positive skin-prick test.
Figure 2.
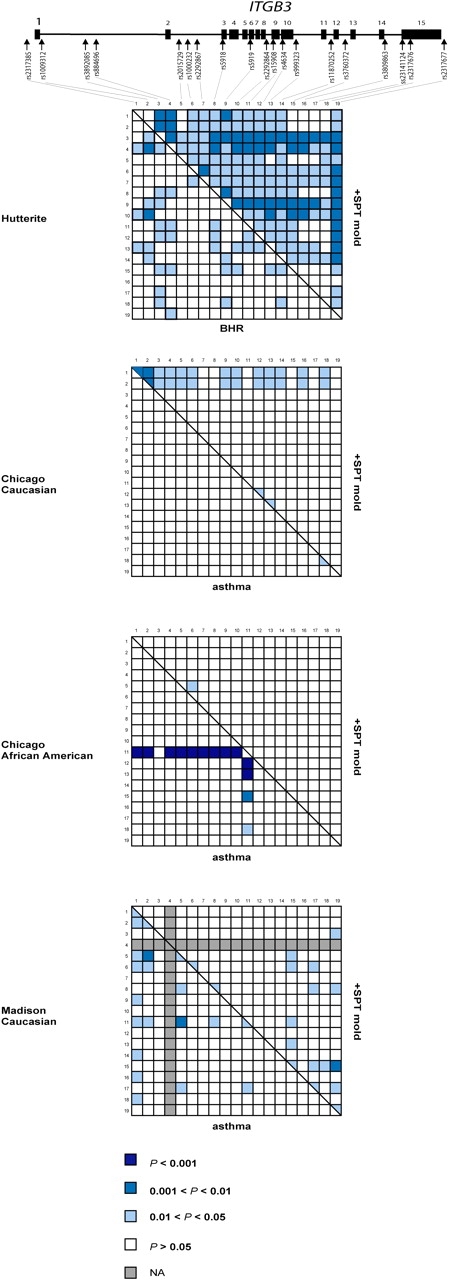
Pairwise haplotype association of variation in ITGB3 with asthma and atopy phenotypes. The genomic structure of ITGB3 is shown (not to scale), with exons depicted as boxes and arrows identified by rs number in the approximate locations of the variation genotyped. Association signals for pairs of SNPs in the four samples are shown in graphical form. For each population, every box in the left triangle represents the association results for a particular pair of SNPs with BHR/asthma, and the right triangle with +SPT mold. p values are indicated by the shading of each square, with darker blue representing more significant results, white representing nonsignificant results, and gray representing tests not performed. NA = tests not performed (not available).
To replicate our findings and further characterize the pattern of association with ITGB3 SNPs, we next studied three outbred populations that had been evaluated for asthma and atopy phenotypes. Because none of the most strongly associated variation in the Hutterites was obviously functional and because we could not predict how similar the patterns of linkage disequilibrium would be across populations, we tested all of the ITGB3 SNPs in the replication samples. In the Chicago Caucasian replication sample, two SNPs at the 3′ end of the gene showed modest association with asthma, and two SNPs at the 5′ end of the gene (and many two-SNP haplotypes including one of these SNPs), showed modest association with sensitization to molds (0.01 < p < 0.05; Figures 1 and 2). Similar to the results in the Hutterites, the minor allele at these SNPs was associated with asthma susceptibility (Figure 1, red squares) and the common allele was associated with a positive SPT to molds (Figure 1, blue triangles).
In the Chicago African American replication sample, two SNPs at the 5′ end of the gene were strongly associated with asthma (p < 0.01), including the SNP that was not in HWE in the control subjects (rs2317385). This suggests that either the genotype association is an artifact caused by chance Hardy-Weinberg disequilibrium in the control subjects, or that the Hardy-Weinberg disequilibrium is a result of the strong association of this variant with asthma. An additional rare SNP in intron 14 was more modestly associated with asthma. Many two-SNP haplotypes including the silent SNP in exon 9 were very strongly associated with asthma (p < 0.001; Figure 2). Two SNPs in exon and intron 10 were associated with sensitization to molds. These overlapped with the SNPs associated with BHR and sensitization to molds in the Hutterites. Consistent with results in the Hutterites and Chicago Caucasians, there were opposite associations between the major or minor alleles with asthma and sensitivity to molds. However, in this sample, the common alleles were associated with asthma (Figure 1, blue squares) and the minor alleles were associated with positive SPT to molds (Figure 1, red triangles).
In the Madison replication sample, six SNPs across the gene were modestly associated with asthma (0.01 < p < 0.05), two of which overlapped with the significant associations in the Hutterites. Many two-SNP haplotypes showed significant association (Figure 2). One SNP was strongly associated with positive SPT to molds (p < 0.01) and overlapped with an associated SNP in the Hutterites, and five were more modestly associated (0.01 < p < 0.05). The Madison sample shows a pattern of association that is the opposite of that observed in the Hutterites and Chicago Caucasians, but similar to the Chicago African American sample: the common alleles were associated with asthma (Figure 1, blue squares) and the minor alleles were associated with positive SPT to mold (Figure 1, red triangles).
DISCUSSION
We examined 19 SNPs spanning the ITGB3 gene for association with phenotypes related to asthma and atopy and identified multiple significant associations with BHR and multiple significant associations of the opposite allele with sensitization to mold allergen in the Hutterites. Associations between ITGB3 alleles and asthma and positive SPT to mold were also identified in three independent replication samples representing Caucasians and African Americans, and in each case, when the common alleles were associated with risk for asthma, the minor alleles were associated with sensitization to mold allergens, and vice versa.
The unexpected pattern of associations between the study samples, and with respect to asthma/BHR susceptibility and allergic sensitization to mold allergens, could reflect the clinical and genetic heterogeneity that characterize these complex phenotypes. For example, the diagnosis of asthma differed between the samples included in this study. Although in the Chicago samples the definition of asthma included both BHR and a doctor's diagnosis of asthma, in the Hutterites it included only BHR and in the Madison sample it included only a doctor's diagnosis. However, this explanation seems unlikely to account for our results because the opposite alleles were associated with each phenotype in the two Chicago samples, despite identical diagnostic criteria and ascertainment. Underlying racial differences in modifying genes or haplotype structure also seem unlikely because the Chicago African Americans and the Madison subjects showed similar patterns of association, which were the opposite of those observed in the Hutterites and the Chicago Caucasians.
Environmental exposures, which are important in both triggering the onset of asthma and influencing exacerbations, also differ between the groups. The Hutterites live on communal farms and in general have mild asthma that seldom requires oral steroid use or hospitalizations. Only half of the Hutterites with asthma or BHR are sensitized to one or more allergen, whereas 70 to 80% of the individuals with asthma in the Chicago samples and all of the Madison individuals with asthma were sensitized to one or more allergen, using roughly the same panel of allergens. The Chicago participants were largely from the inner city, which is in itself a risk factor for asthma prevalence and severity (24), whereas the environmental exposures in the Madison cohort may be intermediate between the rural farming lifestyle of the Hutterites and the urban lifestyle of the Chicago samples with respect to many of the known environmental risk factors. Thus, there are no obvious environmental exposures that would account for the shared associated ITGB3 allele in the Chicago African Americans and Madison sample and between the Chicago Caucasians and Hutterites. However, given the potentially large effect of environmental exposures in early life on the subsequent development of asthma and allergic disease (25–27), it remains possible that the different patterns of association between the samples reflect differences (or similarities) in early-life exposures. Overall, given the heterogeneity between the study samples concerning clinical diagnoses and environmental exposures, it is quite remarkable that we observed associations between variation in the ITGB3 gene and asthma/BHR, and that the asthma-susceptibility allele class was associated with protection against allergic sensitization to mold allergens in four populations. Regardless of the pattern of association, these results implicate another integrin in asthma/atopy susceptibility and raise the possibility that variation in ITGB3 may contribute to some of the linkage signals on chromosome 17 in genomewide screens of asthma- and atopy-related phenotypes in both Caucasians and African Americans (6–8).
The polymorphisms in ITGB3 that showed the most evidence for association in all the samples are silent or noncoding. To date, no other coding variation in ITGB3 has been found in the Hutterites (data not shown), and no coding variation at a frequency more than 0.03 was found in at least two other populations that have been studied (28, 29). The only coding variation (Leu33Pro), which encodes the antigenic platelet polymorphism PlA1/PlA2, did not show the strongest evidence for association in any population, so perhaps the biologically relevant variation (whether or not it was among the set we genotyped) encodes subtle differences in splicing efficiency or expression levels. Our association results may not converge on a single polymorphism because combinations of alleles or genotypes actually influence function, and our methods therefore lack precision. Observing different, inconsistent, or functionally ambiguous allelic associations in replication studies has been particularly characteristic of asthma genetic research (2). In fact, the challenge of replication in the study of complex disease as a whole led a recent review to suggest that the gene is the more appropriate unit for replication than hypothesized susceptibility allele(s) (30), as demonstrated in our study. There are several advantages to testing as much variation as possible in replication samples, as opposed to only the specific SNPs showing association in the original population. First, the gene is truly the unit of interest in the case where there is not a single well-characterized functional polymorphism. Second, a gene is highly consistent in position, sequence, and function across diverse human populations, which is not necessarily the case for any particular SNP or haplotype.
The associations between ITGB3 and asthma are also consistent with previous research implicating platelets and thrombotic factors in asthma and atopy. Increased thrombin (which has been associated with the Pro33 allele [31–33]) has been noted in the airways of patients with asthma as compared with control subjects (34), and enhanced thrombin activity has been hypothesized to contribute to airway remodeling in asthma and allergic inflammation (35). Furthermore, activation of the IgE receptor on platelets (FcεR1) results in the release of serotonin and the cytokine, Regulated on Activation, Normal T-cell Expressed and Secreted, or RANTES, which is important in allergic inflammation (36). Platelets have also been shown to strongly inhibit arachodonic acid 5-lipoxygenation in neutrophils through the fibrinogen receptor (11), and the leukotrienes created by this mechanism are one of the major drug targets in asthma. Together, these previous results and those presented in this study suggest that platelet function, possibly influenced by ITGB3 genotype, may play a direct role in asthma and atopy.
Interestingly, allergic sensitization to molds was consistently associated with variation in ITGB3, whereas other allergens showed little or no association in the Hutterites. Bromley and Donaldson (37) undertook a study on the binding of Aspergillus fumigatus spores to lung epithelial cells and basement membrane proteins because of the particular susceptibility of subjects with asthma to this pathogen. It was found that the Arg-Gly-Asp (RGD) sequence of substrates like fibrinogen and fibronectin may be involved in spore binding to some extracellular matrix proteins, and free fibrinogen may protect against binding of spores to pulmonary epithelium. Notably, the RGD peptide is also the specific ligand for the integrin receptors that contain the β3 subunit. This suggests that there could be direct competition between integrin receptors and mold spores for ligand, and potentially explains the specific association we observed between ITGB3 and sensitivity to mold. One could additionally hypothesize that lower expression or activity of ITGB3-containing receptors would be protective for sensitization to mold allergens by allowing more free fibrinogen. The same reduction in ITGB3 amount or function, however, could increase susceptibility to asthma by decreasing inhibition of leukotriene synthesis, for example. This would be one potential explanation for the direct effects of the same allele in opposite directions for allergic sensitization to mold allergens and asthma susceptibility.
In conclusion, we report evidence for association of ITGB3 with asthma and allergic sensitization to molds in four independent populations. These results, however, pose as many biological questions as they may answer: questions about the environmental exposures and other population-specific parameters that influence the relationship between genotype and phenotype, the genetic variation that determines individual susceptibility, and whether the gene product is directly or indirectly involved in disease pathogenesis. In general, the results presented here, together with the results of studies of other integrin-related genes, suggest that integrins and disintegrins should be considered as functional candidates for asthma and atopy phenotypes in future studies.
Supplementary Material
Acknowledgments
The authors thank Rebecca Anderson and Natasha Phillips for assistance with data collection, and anonymous reviewers for helpful comments. They thank the Hutterites, Childhood Onset of Asthma (COAST) families, and Collaborative Study on the Genetics of Asthma subjects for their participation. The support and participation of the following hospitals and clinics have been key to the success of the COAST project: the obstetric nursing staff at Meriter Hospital; St. Mary's Medical Center; Fort Atkinson Memorial Health Services, Inc.; St. Clare Hospital; Reedsburg Area Medical Center; and Sauk Prairie Memorial Hospital. In addition, they thank the clinic staff from Physicians Plus, Associated Physicians, Dean Medical Center, and Group Health in the Madison area.
Supported by the National Institutes of Health grants HL56399, HL66533, HL72414, and HL70831 (C.O.), and M01 RR00055 to the University of Chicago Clinical Research Center. L.A.W. is supported by a National Science Foundation graduate research fellowship.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Conflict of Interest Statement: L.A.W. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; L.A.L. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; J.E.G. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; R.L.W. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; R.P. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; R.F.L. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; J.S. received $240,151 in support of research from GlaxoSmithKline (GSK), from 2001 to 2004. He received $3,000 in honorarium from GSK, and $2,500 in consulting fees each from Wyeth and AstraZeneca. He also received financial support for community, physician, and patient education/outreach activities from Merck ($6,000), GSK ($23,600), and AstraZeneca ($4,000); C.O. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Beasley R. The burden of asthma with specific reference to the United States. J Allergy Clin Immunol 2002;109(Suppl):S482–S489. [DOI] [PubMed] [Google Scholar]
- 2.Hoffjan S, Nicolae D, Ober C. Association studies for asthma and atopic diseases: a comprehensive review of the literature. Respir Res 2003;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, Torrey D, Pandit S, McKenny J, Braunschweiger K, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 2002;418:426–430. [DOI] [PubMed] [Google Scholar]
- 4.Davies DE, Wicks J, Powell RM, Puddicombe SM, Holgate ST. Airway remodeling in asthma: new insights. J Allergy Clin Immunol 2003;111(2):215–225. [DOI] [PubMed] [Google Scholar]
- 5.Bourgain C, Hoffjan S, Nicolae R, Newman D, Steiner L, Walker K, Reynolds R, Ober C, McPeek MS. Novel case-control test in a founder population identifies P-selectin as an atopy-susceptibility locus. Am J Hum Genet 2003;73:612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koppelman GH, Stine OC, Xu J, Howard TD, Zheng SL, Kauffman HF, Bleecker ER, Meyers DA, Postma DS. Genome-wide search for atopy susceptibility genes in Dutch families with asthma. J Allergy Clin Immunol 2002;109:498–506. [DOI] [PubMed] [Google Scholar]
- 7.Dizier MH, Besse-Schmittler C, Guilloud-Bataille M, Annesi-Maesano I, Boussaha M, Bousquet J, Charpin D, Degioanni A, Gormand F, Grimfeld A, et al. Genome screen for asthma and related phenotypes in the French EGEA study. Am J Respir Crit Care Med 2000;162:1812–1818. [DOI] [PubMed] [Google Scholar]
- 8.Collaborative Study on the Genetics of Asthma. A genome-wide search for asthma susceptibility loci in ethnically diverse populations. Nat Genet 1997;15:389–392. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Lefort J, Kearsey V, Lapa e Silva JR, Cookson WO, Vargaftig BB. A genome-wide screen for asthma-associated quantitative trait loci in a mouse model of allergic asthma. Hum Mol Genet 1999;8:601–605. [DOI] [PubMed] [Google Scholar]
- 10.Weiss LA, Veenstra-Vanderweele J, Newman DL, Kim SJ, Dytch H, McPeek MS, Cheng S, Ober C, Cook EH, Abney M. Genome-wide association study identifies ITGB3 as a QTL for whole blood serotonin. Eur J Hum Genet 2004;12:949–954. [DOI] [PubMed] [Google Scholar]
- 11.Chabannes B, Moliere P, Merhi-Soussi F, Poubelle PE, Lagarde M. Platelets may inhibit leucotriene biosynthesis by human neutrophils at the integrin level. Br J Haematol 2003;121:341–348. [DOI] [PubMed] [Google Scholar]
- 12.Thompson RD, Wakelin MW, Larbi KY, Dewar A, Asimakopoulos G, Horton MA, Nakada MT, Nourshargh S. Divergent effects of platelet-endothelial cell adhesion molecule-1 and beta 3 integrin blockade on leukocyte transmigration in vivo. J Immunol 2000;165:426–434. [DOI] [PubMed] [Google Scholar]
- 13.Bruyninckx WJ, Comerford KM, Lawrence DW, Colgan SP. Phosphoinositide 3-kinase modulation of beta(3)-integrin represents an endogenous “braking” mechanism during neutrophil transmatrix migration. Blood 2001;97:3251–3258. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharyya SP, Mekori YA, Hoh D, Paolini R, Metcalfe DD, Bianchine PJ. Both adhesion to immobilized vitronectin and FcepsilonRI cross-linking cause enhanced focal adhesion kinase phosphorylation in murine mast cells. Immunology 1999;98:357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scaffidi AK, Moodley YP, Weichselbaum M, Thompson PJ, Knight DA. Regulation of human lung fibroblast phenotype and function by vitronectin and vitronectin integrins. J Cell Sci 2001;114:3507–3516. [DOI] [PubMed] [Google Scholar]
- 16.Ober C, Cox NJ. The genetics of asthma: mapping genes for complex traits in founder populations. Clin Exp Allergy 1998;28(Suppl):101–105; discussion 108–110. [DOI] [PubMed] [Google Scholar]
- 17.Ober C, Tsalenko A, Parry R, Cox NJ. A second-generation genomewide screen for asthma-susceptibility alleles in a founder population. Am J Hum Genet 2000;67:1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lester LA, Rich SS, Blumenthal MN, Togias A, Murphy S, Malveaux F, Miller ME, Dunston GM, Solway J, Wolf RL, et al. Ethnic differences in asthma and associated phenotypes: collaborative study on the genetics of asthma. J Allergy Clin Immunol 2001;108:357–362. [DOI] [PubMed] [Google Scholar]
- 19.Lemanske RF Jr. The childhood origins of asthma (COAST) study. Pediatr Allergy Immunol 2002;13:38–43. [DOI] [PubMed] [Google Scholar]
- 20.Newman DL, Hoffjan S, Bourgain C, Abney M, Nicolae RI, Profits ET, Grow MA, Walker K, Steiner L, Parry R, et al. Are common disease susceptibility alleles the same in outbred and founder populations? Eur J Hum Genet 2004;12:584–590. [DOI] [PubMed] [Google Scholar]
- 21.O'Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 1998;63:259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becker T, Knapp M. A powerful strategy to account for multiple testing in the context of haplotype analysis. Am J Hum Genet 2004;75:561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becker T, Knapp M. Maximum-likelihood estimation of haplotype frequencies in nuclear families. Genet Epidemiol 2004;27:21–32. [DOI] [PubMed] [Google Scholar]
- 24.Shapiro GG, Stout JW. Childhood asthma in the United States: urban issues. Pediatr Pulmonol 2002;33:47–55. [DOI] [PubMed] [Google Scholar]
- 25.Holt PG. The role of genetic and environmental factors in the development of T-cell mediated allergic disease in early life. Paediatr Respir Rev 2004;5(Suppl A):S27–S30. [DOI] [PubMed] [Google Scholar]
- 26.Stein RT, Martinez FD. Asthma phenotypes in childhood: lessons from an epidemiological approach. Paediatr Respir Rev 2004;5:155–161. [DOI] [PubMed] [Google Scholar]
- 27.Hoffjan S, Nicolae D, Ostrovnaya I, Roberg K, Evans M, Mirel DB, Steiner L, Walker K, Shult P, Gangnon RE, et al. Gene-environment interaction effects on the development of immune responses in the 1st year of life. Am J Hum Genet 2005;76(4):696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genomics and proteomics of cell injury and inflammation. NHLBI Program for Genomic Applications (grant ID 5U01HL6688002) [Internet]. Dallas, TX: University of Texas Southwestern Medical Center; [September 2003]. Available from: http://pga.swmed.edu/Data/SNPs/PGA/ITGB3.html.
- 29.Sherry ST, Ward M, Sirotkin K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 1999;9:677–679. [PubMed] [Google Scholar]
- 30.Neale BM, Sham PC. The future of association studies: gene-based analysis and replication. Am J Hum Genet 2004;75:353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng D, Lindpaintner K, Larson MG, Rao VS, O'Donnell CJ, Lipinska I, Schmitz C, Sutherland PA, Silbershatz H, D'Agostino RB, et al. Increased platelet aggregability associated with platelet GPIIIa PlA2 polymorphism: the Framingham Offspring Study. Arterioscler Thromb Vasc Biol 1999;19:1142–1147. [DOI] [PubMed] [Google Scholar]
- 32.Vijayan KV, Goldschmidt-Clermont PJ, Roos C, Bray PF. The Pl(A2) polymorphism of integrin beta(3) enhances outside-in signaling and adhesive functions. J Clin Invest 2000;105:793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Undas A, Brummel K, Musial J, Mann KG, Szczeklik A. Pl(A2) polymorphism of beta(3) integrins is associated with enhanced thrombin generation and impaired antithrombotic action of aspirin at the site of microvascular injury. Circulation 2001;104:2666–2672. [DOI] [PubMed] [Google Scholar]
- 34.Gabazza EC, Taguchi O, Tamaki S, Takeya H, Kobayashi H, Yasui H, Kobayashi T, Hataji O, Urano H, Zhou H, et al. Thrombin in the airways of asthmatic patients. Lung 1999;177:253–262. [DOI] [PubMed] [Google Scholar]
- 35.Terada M, Kelly EA, Jarjour NN. Increased thrombin activity after allergen challenge: a potential link to airway remodeling? Am J Respir Crit Care Med 2004;169:373–377. [DOI] [PubMed] [Google Scholar]
- 36.Hasegawa S, Pawankar R, Suzuki K, Nakahata T, Furukawa S, Okumura K, Ra C. Functional expression of the high affinity receptor for IgE (FcepsilonRI) in human platelets and its intracellular expression in human megakaryocytes. Blood 1999;93:2543–2551. [PubMed] [Google Scholar]
- 37.Bromley IM, Donaldson K. Binding of Aspergillus fumigatus spores to lung epithelial cells and basement membrane proteins: relevance to the asthmatic lung. Thorax 1996;51:1203–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.