

Abstract
Rationale: Airway infection with Haemophilus influenzae causes airway inflammation, and isolation of new strains of this bacteria is associated with increased risk of exacerbations in patients with chronic obstructive pulmonary disease (COPD). Objective: To determine whether strains of H. influenzae associated with exacerbations cause more inflammation than strains that colonize the airways of patients with COPD. Methods: Exacerbation strains of H. influenzae were isolated from patients during exacerbation of clinical symptoms with subsequent development of a homologous serum antibody response and were compared with colonization strains that were not associated with symptom worsening or an antibody response. Bacterial strains were compared using an in vivo mouse model of airway infection and in vitro cell culture model of bacterial adherence and defense gene and signaling pathway activation in primary human airway epithelial cells. Results: H. influenzae associated with exacerbations caused more airway neutrophil recruitment compared with colonization strains in the mouse model of airway bacterial infection. Furthermore, exacerbation strains adhered to epithelial cells in significantly higher numbers and induced more interleukin-8 release after interaction with airway epithelial cells. This effect was likely mediated by increased activation of the nuclear factor-κB and p38 mitogen-activated protein kinase signaling pathways. Conclusions: The results indicate that H. influenzae strains isolated from patients during COPD exacerbations often induce more airway inflammation and likely have differences in virulence compared with colonizing strains. These findings support the concept that bacteria infecting the airway during COPD exacerbations mediate increased airway inflammation and contribute to decreased airway function.
Keywords: bacterial adhesion, interleukin-8, NF-κB, neutrophil infiltration, p38 mitogen-activated protein kinases
Acute pulmonary exacerbations are an important contributor to the health care costs, quality of life, morbidity, and mortality of patients with chronic obstructive pulmonary disease (COPD) (1–4). COPD exacerbations are associated with increased markers of inflammation in the airway, leading to the hypothesis that factors that promote airway inflammation lead to worsening airway function (5–7). Although infection clearly has the ability to induce airway inflammation, the role that bacteria play in COPD exacerbations remains controversial (8, 9). Bacteria commonly colonize the airways of patients with COPD, making the presence of bacteria insufficient to explain worsening airway function (10–12). However, COPD exacerbation could be precipitated by increase in bacterial number, change in bacterial location in the airway, or acquisition of a new, more virulent, or more proinflammatory bacterial species or strain (8, 13, 14). Indeed, previous reports have revealed a correlation between the level of airway inflammation and bacterial presence, number, and pathogenicity (7, 12, 15). Furthermore, COPD exacerbations are associated with acquisition of new strains of bacteria (14). Patients with COPD exacerbations recover more rapidly with antibiotic therapy, supporting the possibility that bacteria modulate airway inflammation and function (16).
Nontypeable Haemophilus influenzae is the bacterial species most commonly isolated from airway samples during COPD exacerbations (14, 17). This respiratory pathogen is a pleomorphic gram-negative bacilli that fails to agglutinate with typing antisera against known capsular structures, and multiple strains from human airways have been identified using molecular typing (18). These bacteria frequently colonize human respiratory mucosa and can produce respiratory tract disease including otitis media, sinusitis, bronchitis, and pneumonia, particularly in patients with underlying airway diseases such as COPD, bronchiectasis, and cystic fibrosis. When constitutive innate defense mechanisms in respiratory epithelium (e.g., mucociliary clearance, antibacterial molecules) are overwhelmed by H. influenzae, an inflammatory response is activated that is characterized by recruitment of leukocytes, particularly neutrophils, to sites of infection (12, 19, 20). The ability of H. influenzae to cause intense airway inflammation and the association of exacerbations with development of humoral immune responses to H. influenzae acquired for the first time supports the role of infection by this organism in causing COPD exacerbations (18).
Airway epithelial cells actively participate in neutrophil recruitment, retention, and activation in response to H. influenzae by expression of several airway defense genes, including the leukocyte adhesion glycoprotein intercellular adhesion molecule-1 (ICAM-1) and neutrophil chemoattractant and activating factors such as the chemokine interleukin (IL)-8. Both ICAM-1 and IL-8 appear important for neutrophilic inflammation in COPD (21, 22). Inflammatory gene expression by epithelial cells in response to bacterial infection may be mediated both indirectly through communication with other cells types by soluble mediators and directly by epithelial cell interaction with bacteria in the airway. Direct contact between H. influenzae and airway epithelial cells occurs during airway infection, and attachment to epithelial cells is mediated by specific bacterial surface molecules (23, 24). H. influenzae may also reside in the airway lumen or in the mucosa between, below, or inside epithelial cells (13, 25). Interestingly, strains of H. influenzae vary in their ability to induce inflammatory responses after interaction with epithelial cells, suggesting that different strains might have different effects on airway function (26).
In this study, we questioned whether strains of H. influenzae associated with exacerbations cause more inflammation than strains that colonize the airways of patients with COPD. Bacterial strains were obtained from a cohort of patients with COPD that underwent serial assessment of clinical status, sputum microbiology, and serum antibody production against homologous strains of sputum bacterial isolates (14, 18, 27). Bacteria were tested in a mouse model of airway inflammation and using a primary cell culture model of airway epithelial cell inflammatory responses. We show that H. influenzae isolated for the first time during COPD exacerbations induce more leukocyte recruitment in our mouse model and more airway epithelial cell activation, leading to increased mediator release in our isolated airway epithelial cell model. Our results support the concept that bacterial infection of the airway during a COPD exacerbation mediates increased airway inflammation and contributes to decreased airway function.
Some of the results of these studies have been previously reported in abstract form (28).
METHODS
Patients with COPD
The prospective, longitudinal study of patients with COPD at the Buffalo Veterans Affairs Medical Center (Buffalo, NY) has been described previously (14, 18, 27). All participants gave written informed consent under a protocol approved by the Human Studies Subcommittee of the Veterans Affairs Western New York Healthcare System.
Bacterial Isolation
Sputum samples were spontaneously expectorated the morning of the clinic visit and were homogenized, diluted, and plated for identification and quantitation as described previously (14, 18). For this study, exacerbation strains were defined by the following criteria: (1) the patient experienced clinical exacerbation symptoms at the time the strain was isolated from sputum; (2) the strain was isolated for the first time at exacerbation symptom onset; and (3) serum obtained 1 month after the exacerbation contained new bactericidal antibodies to the homologous infecting strain compared with serum obtained 1 month before exacerbation. Colonization strains were defined by the following criteria: (1) the patient had no clinical signs of exacerbation at the time the strain was isolated from sputum; (2) the strain was newly acquired; and (3) there was absence of a new serum antibody response to the homologous strain. H. influenzae strain 12 is a well characterized, nontypeable isolate that has been described previously (19).
Bacterial Preparation
Aerated, log-phase cultures of H. influenzae were prepared and quantitated as described previously (19, 29), and infection with equivalent bacterial inoculums was verified in each experiment.
Mouse Airway Infection Model
Mouse airway infection with H. influenzae with assessment of airway leukocyte recruitment, lung chemokines expression, and bacterial load was performed under a protocol approved by the University of Iowa Institutional Animal Care and Use Committee as described previously (19, 20).
Biofilm Formation Assay
Biofilm formation of H. influenzae isolates were assayed by the ability of the bacteria to adhere to the walls of a 96-well microplate as described previously (30).
Airway Epithelial Cell Infection Model
Human tracheobronchial epithelial (hTBE) cells were obtained under a protocol approved by the University of Iowa Institutional Review Board (29, 31). Assays for epithelial cell bacterial adherence, ICAM-1 expression, IL-8 release, nuclear factor (NF)-κB activation, and mitogen-activated protein (MAP) kinase phosphorylation have been described previously (19, 29, 32–34).
Signaling Pathway Inhibition
Inhibition of NF-κB–dependent signaling was accomplished by infection of hTBE cells with a recombinant adenoviral vector that expresses a dominant-negative mutant form of IκBα. Adenoviral vector expression of green fluorescence protein was used both as a control and to assess the level of epithelial cell transgene expression. Inhibition of p38 MAP kinase was accomplished using the inhibitor SB203580.
Statistical Analysis
Experimental results involving comparison of two conditions were analyzed for statistical significance using two-tailed, unpaired Student's t tests, with p < 0.05 considered significant. Experimental results with multiple comparisons were analyzed using one-way analysis of variance (ANOVA) for a factorial experimental design. The multicomparison significance level for the one-way ANOVA was 0.05. If significance was achieved by one-way analysis, post-ANOVA comparison of means was performed using Scheffe F tests (35).
Additional details on patients with COPD, bacterial isolation and preparation, mouse airway infection model, biofilm formation assay, airway epithelial cell model, signaling pathway inhibition, and analysis of epithelial cell bacterial adherence, ICAM-1 and IL-8 expression, and NF-κB and MAP kinase activation are provided in the online supplement.
RESULTS
H. influenzae from Patients with COPD with Exacerbation Induce More Airway Inflammation
Previous reports of epithelial responses to H. influenzae primarily tested single bacterial isolates, often laboratory strains or from sources other than patients with COPD. Therefore, our experiments were directed at testing inflammatory responses to 17 H. influenzae strains isolated from 15 individuals with COPD (Table 1). H. influenzae for these experiments were divided into isolates cultured from sputum when the patient was having a clinical exacerbation (n = 10) versus those associated with stable respiratory status (n = 7). Infection of mice with one of three or four bacterial isolates from each of these groups confirmed that H. influenzae associated with both exacerbation and colonization in patients with COPD induced high levels of neutrophil recruitment into the airway 24 hours after inoculation (Figure 1A). Because of difficulty in precisely equalizing the inoculum of each bacterial isolate used to infect animals, we divided the number of neutrophils in bronchoalveolar lavage (BAL) by the bacterial load detected in lung homogenates at the time of specimen acquisition to normalize for differences in bacterial number and thus the stimulus for leukocyte recruitment. We observed significantly higher numbers of airway neutrophils per bacteria in animals infected with H. influenzae from patients with COPD exacerbation versus isolates associated with colonization. We also measured lung levels of the mouse CXC chemokines keratinocyte–derived chemokine or keratinocyte chemoattractant and macrophage-inflammatory protein-2, which are potent neutrophil chemoattractants and functional mouse homologs for IL-8 that are often elevated during pulmonary bacterial infection (19). Although we observed a higher mean level of these mediators in the lungs of mice infected with H. influenzae associated with COPD exacerbation (Figure 1B), because of variability among animals infected with different isolates, results did not reach statistical significance (keratinocyte chemoattractant: p = 0.08; macrophage-inflammatory protein-2, p = 0.07). These results suggest that H. influenzae isolated from patients with COPD exacerbation induce more airway neutrophil recruitment than those that colonize their airway.
TABLE 1.
Characteristics of patients with chronic obstructive pulmonary disease with sputum isolation of haemophilusinfluenzae
| Characteristic | Value (n = 15) |
|---|---|
| Age, yr | 64.8 ± 2.0 |
| Sex, female/male | 1/14 |
| Pack-years of smoking | 91.8 ± 6.4 |
| Years since diagnosis | 11.3 ± 2.9 |
| Baseline FEV1, L | 1.53 ± 0.12 |
| Baseline FEV1, % predicted | 44.0 ± 3.0 |
| Exacerbations per year | 2.1 ± 0.4 |
| Visits with H. influenzae isolated from sputum, % | 28.7 ± 5.8 |
Values are expressed as mean ± SEM or as a ratio.
Figure 1.

Haemophilus influenzae from patients with chronic obstructive pulmonary disease (COPD) exacerbation induce more airway inflammation. Bronchoalveolar lavage (BAL) neutrophil numbers (A) and lung keratinocyte chemoattractant (KC) and macrophage-inflammatory protein-2 (MIP-2) levels (B) were determined in C57BL/6J mice after tracheobronchial injection of agar particles suspended with H. influenzae isolates from patients with COPD. At 24 hours after inoculation, the left lung underwent bronchoalveolar lavage with quantification of neutrophils, and the right lung was homogenized for chemokine assay and bacterial quantitation. (A, B) Values are expressed as mean neutrophil number or chemokine concentration per bacterial number ± SEM (n = 3–4 bacterial isolates per condition, with each isolate used to infect two to three individual mice), and a significant difference in levels between animals infected with H. influenzae associated with colonization (Col) versus exacerbation (Exac) is indicated by an asterisk.
H. influenzae from Patients with COPD with Exacerbation Adhere More to Epithelial Cells
Although bacterial capacity to induce inflammation may affect airway function in COPD, we were also interested in other differences in virulence between bacterial groups that could affect H. influenzae ability to infect the airway. Our preliminary results did not detect a significant difference in bacterial growth rates between H. influenzae isolates, both in vitro and in our mouse model (results not shown). Furthermore, although H. influenzae form biofilms that are likely important in pathogenesis (36, 37), no significant difference in capacity for biofilm formation was observed (Figure 2A). However, H. influenzae from patients with COPD with exacerbation adhered in greater numbers to human airway epithelial cells (Figure 2B). Although adherence has been shown to correlate poorly with H. influenzae capacity to induce inflammatory mediator expression in airway epithelial cells (19), differences in bacterial adherence likely affect bacterial pathogenicity (38).
Figure 2.

H. influenzae from patients with COPD with exacerbation adhere more to airway epithelial cells. (A) Bacterial biofilm formation was assessed after adherence of H. influenzae isolates from patients with COPD to polystyrene for 96 hours. Biofilms were stained with crystal violet, nonadherent bacteria were removed by washing, biofilms were dissolved in ethanol, and absorbance at 570 nm was determined. (B) Bacterial adherence to human tracheobronchial epithelial cell monolayers was assessed after incubation of cells for 30 minutes with H. influenzae isolates from patients with COPD. Nonadherent bacteria were removed by washing, cells and bacteria were released by saponin treatment and scraping, and numbers of adherent bacteria were determined by quantitative culture. (A, B) Values are expressed as mean ± SEM (n = 7–10), and a significant difference in bacterial adherence between H. influenzae associated with colonization versus exacerbation is indicated by an asterisk.
H. influenzae from Patients with COPD with Exacerbation Induce More IL-8
Based on results from experiments using our animal model of airway infection, we questioned whether differences in inflammation induced by H. influenzae isolates in vivo might correlate with epithelial cell inflammatory mediator expression in response to these bacterial strains using a cell culture model of primary airway epithelial cells. In experiments using this in vitro model, H. influenzae from patients with COPD with exacerbation induced slightly more ICAM-1 expression compared with equivalent inoculums of colonizing strains after epithelial cell interaction with bacteria for 24 hours, but this difference did not reach statistical significance (Figure 3A). The lack of statistical significance may relate to the fact that these bacterial isolates induced relatively low levels of epithelial cell ICAM-1, thereby making differences harder to detect. In contrast, bacteria are potent inducers of epithelial cell IL-8 release, making the ability to detect differences more likely. We observed that H. influenzae isolates from patients with COPD with exacerbation induced significantly higher levels of IL-8 compared with colonizers (Figure 3B). These results support the concept that differences in bacterial effects on epithelial cell inflammatory mediators could account for differences in inflammation.
Figure 3.

H. influenzae from patients with COPD with exacerbation induce more interleukin (IL)-8. Intercellular adhesion molecule-1 (ICAM-1) protein expression on the cell surface (A) and IL-8 secretion into the culture media (B) were determined using enzyme-linked immunoassays with hTBE cell monolayers that were incubated for 24 hours without or with equivalent inoculums of H. influenzae isolates from patients with COPD. (A, B) Values are expressed as mean ± SEM (n = 7–10), and a significant difference in IL-8 release induced by H. influenzae associated with colonization versus exacerbation is indicated by an asterisk.
H. influenzae Induction of Epithelial Cell ICAM-1 and IL-8 requires NF-κB and p38
The transcription factor NF-κB is important in the regulation of inflammatory gene expression in response to bacteria, including H. influenzae (39). In addition, other cell signaling pathways appear to modulate multiple steps in inflammatory gene activation. Important examples include members of the MAP kinase family of evolutionarily conserved enzymes that are activated by phosphorylation of specific threonine and tyrosine residues in response to cell surface events (40). There are three subfamilies of MAP kinases that have been well characterized: (1) extracellular signal–regulated kinases (ERK1 and ERK2); (2) c-Jun NH2-terminal kinases (JNK1, JNK2, and JNK3); and (3) p38 kinases (p38α, p38β, p38γ, and p38δ). Through phosphorylation of nuclear and cytoplasmic proteins, these signaling molecules regulate cellular gene expression by modulating transcription factor or basal transcription complex function, stabilizing mRNA, or enhancing translation. Interaction between MAP kinases and NF-κB–dependent gene expression has been identified in the regulation of several genes that mediate inflammation (41). To confirm that members of these two signaling pathway families are involved in ICAM-1 and IL-8 expression in epithelial cells, a well characterized strain of H. influenzae (nontypeable strain 12) was allowed to interact with primary human airway epithelial cells. However, we first infected epithelial cells with an adenoviral vector that expressed either a control transgene or a mutant of IκBα that binds NF-κB in its inactivated form, but cannot be phosphorylated to allow for release of activated NF-κB. We found that epithelial expression of the dominant-negative IκBα strongly inhibited expression of ICAM-1 and IL-8 in hTBE cells incubated with H. influenzae (Figure 4A) under conditions that resulted in transgene expression in the majority of cells in the sample (Figure 4B). Decreased ICAM-1 and IL-8 expression with uninfected cells expressing dominant-negative IκBα suggests that constitutive levels of these genes are also regulated by low-level activation of NF-κB. We also pretreated airway epithelial cells with a small-molecule inhibitor of the kinase function of p38 and found that this inhibitor caused a significant and dose-dependent inhibition of increases in ICAM-1 and IL-8 levels that occurred in response to airway epithelial cell interaction with H. influenzae (Figure 4C). Taken together, these results indicate that NF-κB and p38 MAP kinases participate in regulation of epithelial cell ICAM-1 and IL-8 expression in response to H. influenzae.
Figure 4.
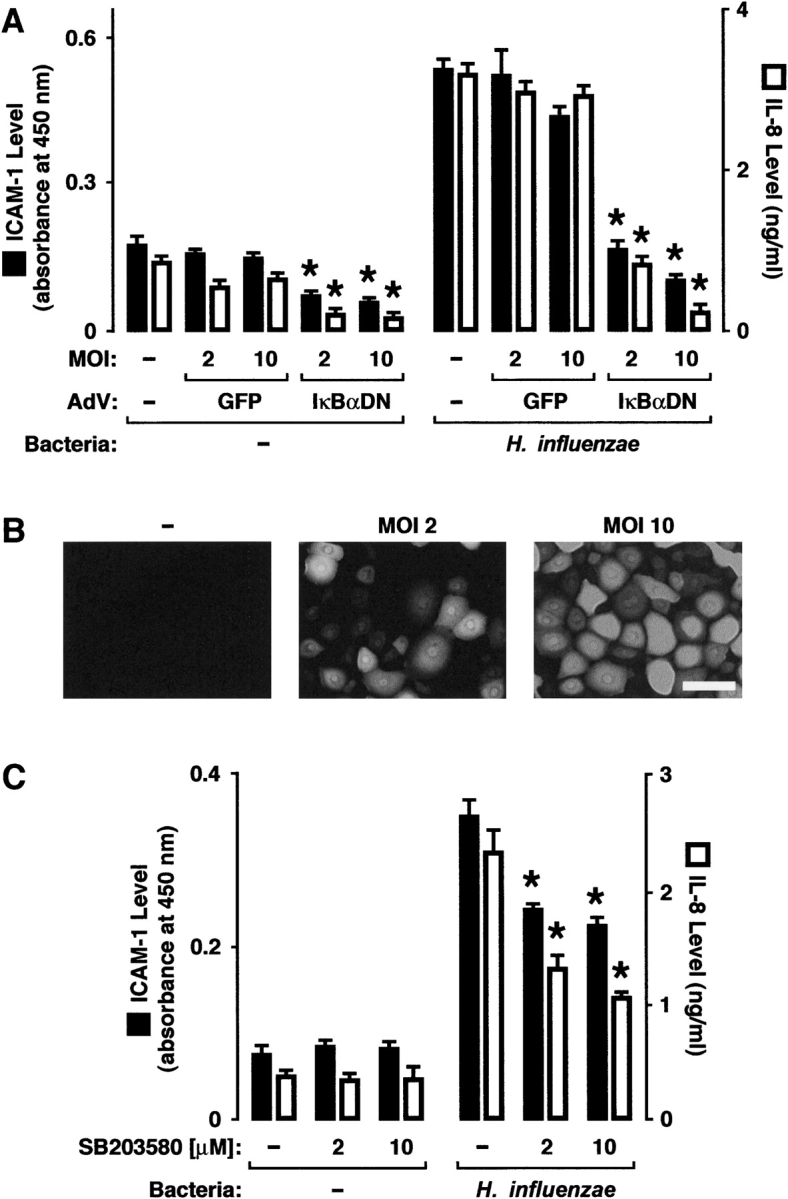
H. influenzae induction of epithelial cell ICAM-1 and IL-8 requires nuclear factor κB and p38. (A) ICAM-1 protein expression on the cell surface and IL-8 secretion into the culture media were determined using enzyme-linked immunoassays with hTBE cell monolayers that were left uninfected or were infected with adenoviral vectors expressing control green fluorescence protein (GFP) transgene or dominant-negative IκBα. Cells were then incubated for 24 hours without or with nontypeable H. influenzae strain 12. (B) The proportion of cells expressing transgene protein at each level of infection in (A) was assessed by epifluorescence photomicroscopy with hTBE cell monolayers that were left uninfected or were infected with an adenoviral vector expressing GFP at the indicated MOI. Scale bar = 30 μm. (C) ICAM-1 and IL-8 protein levels were determined using enzyme-linked immunoassays with hTBE cell monolayers that were pretreated with carrier control or a small molecule inhibitor of p38 at the indicated concentrations. Cells were then incubated for 24 hours without or with H. influenzae strain 12. (A,C) Values are expressed as mean ± SEM (n = 3–6), and a significant difference in ICAM-1 expression or IL-8 secretion compared with cells treated with the control is indicated by an asterisk.
H. influenzae from Patients with COPD with Exacerbation Activate More NF-κB and p38
To determine if differences in inflammatory responses to H. influenzae isolates correlated with activation of signal transduction pathways important for expression of ICAM-1 and IL-8, NF-κB and p38 activation after bacterial interaction with hTBE cells was assessed. H. influenzae from patients with COPD exacerbation induced higher levels of NF-κB activation compared with colonizing bacteria (Figure 5A). For p38 activation, there was variability in the response to bacterial isolates (Figure 5B), but when evaluated as a group the mean activation of this MAP kinase was higher in epithelial cells exposed to H. influenzae from patients with COPD exacerbation versus colonization (Figure 5C). We have previously found that ERK members of this family of kinases do not have a direct effect on airway epithelial cell ICAM-1 and IL-8 expression in response to H. influenzae (results not shown). Assessment of the activation of ERK revealed that there was high basal activation in hTBE cells with no significant activation in response to bacteria. We also found no significant overall difference in ERK activation after exposure to the two H. influenzae isolate groups, indicating that differences between bacteria regulated specific epithelial cell signaling pathways.
Figure 5.

H. influenzae from patients with COPD with exacerbation activate more NF-κB and p38. (A) NF-κB activation was determined using luciferase assays with hTBE cell monolayers that were initially infected for 24 hours with an adenoviral vector expressing a luciferase gene driven by four tandem NF-κB sites. Cells were then incubated for an additional 24 hours without or with equivalent inoculum of H. influenzae isolates from patients with COPD. (B) Phosphorylation of p38 and extracellular signal–regulated kinases (ERK) mitogen-activated protein (MAP) kinase were assessed using immunoblot analysis of extracts from hTBE cell monolayers that were left uninfected or were infected for 1 hour with equivalent inoculum of H. influenzae isolates from patients with COPD. The positions of phosphorylated and total p38 and ERK are indicated by arrows. (C) Phosphorylation of p38 and ERK that was detected in (B) were quantified using densitometry. (A, C) Values are expressed as mean relative luciferase or phosphorylated/total protein level ± SEM (n = 7–10), and a significant difference in NF-κB or MAP kinase activation induced by H. influenzae associated with colonization versus exacerbation is indicated by an asterisk.
DISCUSSION
Acute pulmonary exacerbation of COPD can be precipitated by multiple factors that include environmental exposures (air pollution, dust, temperature) and infection (bacterial or viral). Although the role that bacteria play in COPD is not completely defined, it is clear that bacterial infection can cause inflammation in the airway and markers of inflammation are increased in patients with COPD exacerbations (5–7). Increased airway inflammation in patients with COPD could be mediated by increase in bacterial number, change in the airway compartment that bacteria are located, or acquisition of a new, more virulent, or more proinflammatory bacteria (8, 13, 14). Strains of nontypeable H. influenzae that persist in the airways of patients with chronic bronchitis have been reported to induce less IL-6 and IL-8 release from a respiratory epithelial cell line compared with nonpersisting isolates, providing precedent for the possibility that bacteria isolated under different clinical circumstances might have different effects on airway inflammation and function (26). Our report addresses the possibility that isolates of H. influenzae might have different effects on airway inflammation and these effects might correlate with COPD exacerbations and subsequent development of a humoral antibacterial immune response. We demonstrate that H. influenzae strains isolated for the first time from patients with COPD during exacerbations caused more airway neutrophil recruitment in a mouse model of bacterial infection and more inflammatory signaling pathway activation and IL-8 expression in cultures of primary human airway epithelial cells when compared with bacterial strains isolated from patients with respiratory symptoms at baseline. These findings support the concept that bacteria infecting the airway during COPD exacerbations can mediate increased airway inflammation and contribute to decreased airway function.
It is intuitive that higher bacterial loads provide more stimulation for inflammation in the airway (12). Therefore, in our experiments, it was important to ensure that differences in airway and cellular responses to bacterial isolates were not the result of variation in bacterial numbers. No difference between groups in the growth rate of bacteria was observed either in vitro or in vivo (results not shown), but bacterial numbers were monitored in experiments to address potential differences. In our mouse model of airway infection by H. influenzae, markers of airway inflammation were divided by lung bacterial load in each animal to control for differences in bacterial number that would drive inflammation. In our in vitro studies, experiments were designed to ensure equivalent bacterial inoculum for each strain, thereby excluding the possibility that different bacterial numbers accounted for the results. Although these methods of normalization assume that there is a direct relationship between the level of inflammation and number of bacteria, the lack of a completely linear relationship does not invalidate the results we observed.
The capacity of bacterial strains to induce inflammation was assessed using multiple markers relevant to airway inflammation in COPD. Because of variation in assay conditions, it was not possible to correlate the effects of a single bacterial strain across all assays. However, it is interesting to note the variability within a single assay of inflammatory markers. For example, some H. influenzae strains associated with COPD exacerbations and stimulation of other inflammatory markers were poor inducers of p38 activation, whereas the group as a whole was a strong inducer. The relatively small number of bacterial isolates tested in our study and the variability of responses limits the ability to generalize some findings. Testing isolates from patients with COPD in other clinical situations may help clarify associations between bacterial factors and airway responses. It is likely that multiple bacterial factors determine the capacity of a single strain to induce airway inflammation, and differences in only a few factors may be required to make a strain a strong inducer of inflammation.
Regulation of airway defense involves detection of multiple bacterial macromolecules by host receptors that mediate inflammation in the airway. Many pathogen-associated molecular patterns are recognized by members of the toll-like receptor family of surface proteins on cells in the airway (42). One important example is detection of a component of the outer membrane of gram-negative bacteria, called lipopolysaccharide, by host cell toll-like receptor 4, resulting in inflammatory gene activation. Haemophilus species synthesize a form of lipopolysaccharide, referred to as lipo-oligosaccharide, which contains an oligosaccharide linked to lipid A without repeating subunit O-antigen polysaccharide chains. However, airway epithelial cells are poorly responsive to lipopolysaccharide and lipo-oligosaccharide, suggesting that these bacterial molecules are not responsible for differences in epithelial defense gene activation between strains of H. influenzae (19). The presence or level of other H. influenzae factors that could account for strain differences in epithelial cell activation include the P6 outer membrane lipoprotein, lipo-oligosaccharide glycoforms containing phosphorylcholine that bind to the platelet-activating factor receptor, and small cytoplasmic molecules that activate multiple epithelial cell signaling pathways (39, 43, 44). In addition, several H. influenzae proteins that promote adherence to epithelial cells have been identified, and expression of these adhesins depends on the isolate (45). These include the Hia adhesin, high-molecular-weight adhesins HMW1 and HMW2, and the Hap serine protease. A small subset of nontypeable H. influenzae express hemagglutinating pili, which are polymeric adhesive structures expressed on many gram-negative bacteria, and components of pili from other bacteria have been shown to induce epithelial cell IL-8 (46, 47). Although strains of H. influenzae associated with exacerbation adhered in greater numbers to airway epithelial cells compared with colonizing strains, we have previously reported that these bacterial adhesive molecules are not required for H. influenzae induction of epithelial cell inflammatory genes (19). The critical role for adhesins in the pathogenesis of airway infection is likely by providing adherence to epithelial cell and other surfaces allowing resistance to airway clearance mechanisms.
The finding that new isolates of H. influenzae that mediate increased airway inflammation and activate a humoral immune response are associated with exacerbation of COPD raises interesting questions. These results support the possibility that acquisition of a new, more virulent or more proinflammatory bacterial species or strain may be one pathway for induction of COPD exacerbations. This assumes that a temporal and causal relationship exists between acquiring a proinflammatory isolate and developing worse COPD symptoms. However, environmental factors have the capacity to change bacterial behavior and can also result in permanent changes in the bacterial genome itself (48). Therefore, another possibility is that the airway environment for these bacteria during an exacerbation changes their behavior into a more proinflammatory phenotype. This possibility seems counterintuitive because of the assumption that airway inflammation and a humoral immune response make the airway environment more hostile for bacterial survival. Determining the macromolecular interactions that mediate airway inflammation and vary between isolates will provide a better understanding of the effects that new bacteria acquisition has in COPD exacerbation and may uncover new therapeutic strategies. It seems likely that manipulating bacterial capacity to affect the airway inflammatory and immune response could improve airway function in patients with COPD.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge D. Brenner, P. McCray, and the University of Iowa Center for Gene Therapy for generous gifts of cells and reagents, and thank M. Apicella and G. Hunninghake for helpful discussion.
Supported by grants from the National Institutes of Health.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Conflict of Interest Statement: C.L.C. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; L.J.M. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; E.E.L. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; A.L.H. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; L.S. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; T.D.S. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; G.M.D. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; T.F.M. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; S.S. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; D.C.L. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Burrows B, Earle RH. Course and prognosis of chronic obstructive lung disease: a prospective study of 200 patients. N Engl J Med 1969;280:397–404. [DOI] [PubMed] [Google Scholar]
- 2.Connors AFJ, Dawson NV, Thomas C, Harrell FEJ, Desbiens N, Fulkerson WJ, Kussin P, Bellamy P, Goldman L, Knaus WA. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments). Am J Respir Crit Care Med 1996;154:959–967. [DOI] [PubMed] [Google Scholar]
- 3.Seemungal TAR, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;157:1418–1422. [DOI] [PubMed] [Google Scholar]
- 4.Donaldson GC, Seemungal TAR, Bhowmik A, Wedzicha JA. The relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002;57:847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax 2000;55:114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aaron SD, Angel JB, Lunau M, Wright K, Fex C, Le Saux N, Dales RE. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001;163:349–355. [DOI] [PubMed] [Google Scholar]
- 7.Qiu Y, Zhu J, Bandi V, Atmar RL, Hattotuwa K, Guntupalli KK, Jeffery PK. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003;168:968–975. [DOI] [PubMed] [Google Scholar]
- 8.Hirschmann JV. Do bacteria cause exacerbation of COPD? Chest 2000;118:193–203. [DOI] [PubMed] [Google Scholar]
- 9.Murphy TF, Sethi S, Niederman MS. The role of bacteria in exacerbations of COPD—a constructive view. Chest 2000;118:204–209. [DOI] [PubMed] [Google Scholar]
- 10.Cabello H, Torres A, Celis R, El-Ebiary M, Puig de la Bellacasa J, Xaubet A, Gonzalez J, Agusti C, Soler N. Bacterial colonization of distal airways in healthy subjects and chronic lung disease: a bronchoscopic study. Eur Respir J 1997;10:1137–1144. [DOI] [PubMed] [Google Scholar]
- 11.Soler N, Ewig S, Torres A, Filella X, Gonzalez J, Zaubet A. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur Respir J 1999;14:1015–1022. [DOI] [PubMed] [Google Scholar]
- 12.Hill AT, Campbell EJ, Hill SL, Bayley DL, Stockley RA. Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am J Med 2000;109:288–295. [DOI] [PubMed] [Google Scholar]
- 13.Bandi V, Apicella MA, Mason E, Murphy TF, Siddiqi A, Atmar RL, Greenberg SB. Nontypeable Haemophilus influenzae in the lower respiratory tract of patients with chronic bronchitis. Am J Respir Crit Care Med 2001;164:2114–2119. [DOI] [PubMed] [Google Scholar]
- 14.Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med 2002;347:465–471. [DOI] [PubMed] [Google Scholar]
- 15.Bresser P, Out TA, van Alphen L, Jansen HM, Lutter R. Airway inflammation in nonobstructive and obstructive chronic bronchitis with chronic Haemophilus influenzae airway infection: comparison with noninfected patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000;162:947–952. [DOI] [PubMed] [Google Scholar]
- 16.Saint S, Bent S, Vittinghoff E, Grady D. Antibiotics in chronic obstructive pulmonary disease exacerbations: a meta-analysis. JAMA 1995;273:957–960. [PubMed] [Google Scholar]
- 17.Soler N, Torres A, Ewig S, Gonzalez J, Celis R, El-Ebiary M, Hernandez C, Rodriguez-Roisin R. Bronchial microbial patterns in severe exacerbations of chronic obstructive pulmonary disease (COPD) requiring mechanical ventilation. Am J Respir Crit Care Med 1998;157:1498–1505. [DOI] [PubMed] [Google Scholar]
- 18.Sethi S, Wrona C, Grant BJ, Murphy TF. Strain-specific immune response to Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004;169:448–453. [DOI] [PubMed] [Google Scholar]
- 19.Frick AG, Joseph TD, Pang L, Rabe AM, St. Geme JW III, Look DC. Haemophilus influenzae stimulates ICAM-1 expression on respiratory epithelial cells. J Immunol 2000;164:4185–4196. [DOI] [PubMed] [Google Scholar]
- 20.Humlicek AL, Pang L, Look DC. Modulation of airway inflammation and bacterial clearance by epithelial cell ICAM-1. Am J Physiol Lung Cell Mol Physiol 2004;287:L598–L607. [DOI] [PubMed] [Google Scholar]
- 21.Di Stefano A, Maestrelli P, Roggeri A, Turato G, Calabro S, Potena A, Mapp CE, Ciaccia A, Covacev L, Fabbri LM, et al. Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. Am J Respir Crit Care Med 1994;149:803–810. [DOI] [PubMed] [Google Scholar]
- 22.Beeh KM, Kornmann O, Buhl R, Culpitt SV, Giembycz MA, Barnes PJ. Neutrophil chemotactic activity of sputum from patients with COPD: role of interleukin-8 and leukotriene B4. Chest 2003;123:1240–1247. [DOI] [PubMed] [Google Scholar]
- 23.Hers JFP, Mulder J. The mucosal epithelium of the respiratory tract in muco-purulent bronchitis caused by Haemophilus influenzae. J Pathol Bacteriol 1953;56:103–108. [DOI] [PubMed] [Google Scholar]
- 24.St. Geme JW III. Molecular determinants of the interaction between Haemophilus influenzae and human cells. Am J Respir Crit Care Med 1996;154:S192–S196. [DOI] [PubMed] [Google Scholar]
- 25.Ketterer MR, Shao JQ, Hornick DB, Buscher B, Bandi VK, Apicella MA. Infection of primary human bronchial epithelial cells by Haemophilus influenzae: macropinocytosis as a mechanism of airway epithelial cell entry. Infect Immun 1999;67:4161–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bresser P, van Alphen L, Habets FLM, Hart AAM, Dankert J, Jansen HM, Lutter R. Persisting Haemophilus influenzae strains induce lower levels of interleukin-6 and interleukin-8 in H292 lung epithelial cells than nonpersisting strains. Eur Respir J 1997;10:2319–2326. [DOI] [PubMed] [Google Scholar]
- 27.Murphy TF, Brauer AL, Schiffmacher AT, Sethi S. Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004;170:266–272. [DOI] [PubMed] [Google Scholar]
- 28.Manzel LJ, Chin CL, McNaughton EE, Laurie NL, Look DC. Epithelial cell activation pathways for induction of airway defense gene expression by Haemophilus influenzae. Pediatr Pulmonol 2003;273.
- 29.Aldallal N, McNaughton EE, Manzel LJ, Richards AM, Zabner J, Ferkol TW, Look DC. Inflammatory response in airway epithelial cells isolated from patients with cystic fibrosis. Am J Respir Crit Care Med 2002;166:1248–1256. [DOI] [PubMed] [Google Scholar]
- 30.O'Toole GA, Kolter R. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol 1998;28:449–461. [DOI] [PubMed] [Google Scholar]
- 31.Look DC, Keller BT, Rapp SR, Holtzman MJ. Selective induction of intercellular adhesion molecule-1 by interferon-γ in human airway epithelial cells. Am J Physiol 1992;263:L79–L87. [DOI] [PubMed] [Google Scholar]
- 32.Look DC, Roswit WT, Frick AG, Gris-Alevy Y, Dickhaus DM, Walter MJ, Holtzman MJ. Direct suppression of Stat1 function during adenoviral infection. Immunity 1998;9:871–880. [DOI] [PubMed] [Google Scholar]
- 33.Joseph TD, Look DC. Specific inhibition of interferon signal transduction pathways by adenoviral infection. J Biol Chem 2001;276:47136–47142. [DOI] [PubMed] [Google Scholar]
- 34.Sanlioglu S, Williams CM, Samavati L, Butler NS, Want G, McCray PB Jr, Ritchie TC, Hunninghake GW, Zandi E, Engelhardt JF. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-α secretion through IKK regulation of NF-κB. J Biol Chem 2001;276:30188–30198. [DOI] [PubMed] [Google Scholar]
- 35.Zar JH. Biostatistical analysis. Englewood Cliffs, NJ: Prentice-Hall, Inc.; 1984.
- 36.Murphy TF, Kirkham C. Biofilm formation by nontypeable Haemophilus influenzae: strain variability, outer membrane antigen expression and role of pili. BMC Microbiol 2002;2:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greiner LL, Watanabe H, Phillips NJ, Shao J, Morgan A, Zaleski A, Gibson BW, Apicella MA. Nontypeable Haemophilus influenzae strain 2019 produces a biofilm containing N-acetylneuraminic acid that may mimic sialylated O-linked glycans. Infect Immun 2004;72:4249–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rao VK, Krasan GP, Hendrixson DR, Dawid S, St. Geme JW III. Molecular determinants of the pathogenesis of disease due to non-typable Haemophilus influenzae. FEMS Microbiol Rev 1999;23:99–129. [DOI] [PubMed] [Google Scholar]
- 39.Shuto T, Xu H, Wang B, Han J, Kai H, Gu X, Murphy TF, Lim DJ, Li J. Activation of NF-κB by nontypeable Haemophilus influenzae is mediated by toll-like receptor 2–TAK1-dependent NIK-IKKα/β-IκBα and MKK3/6-p38 MAP kinase signaling pathway in epithelial cells. Proc Natl Acad Sci USA 2001;98:8774–8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 2001;81:807–869. [DOI] [PubMed] [Google Scholar]
- 41.Carter AB, Knudtson KL, Monick MM, Hunninghake GW. The p38 mitogen-activated protein kinase is required for NF-kappaB-dependent gene expression: the role of TATA-binding protein (TBP). J Biol Chem 1999;274:30858–30863. [DOI] [PubMed] [Google Scholar]
- 42.Akira S. Toll-like receptor signaling. J Biol Chem 2003;278:38105–38108. [DOI] [PubMed] [Google Scholar]
- 43.Swords WE, Ketterer MR, Shao J, Campbell CA, Weiser JN, Apicella MA. Binding of the non-typeable Haemophilus influenzae lipooligosaccharide to the PAF receptor initiates host cell signalling. Cell Microbiol 2001;3:525–536. [DOI] [PubMed] [Google Scholar]
- 44.Wang B, Cleary PP, Xu H, Li J. Up-regulation of interleukin-8 by novel small cytoplasmic molecules of nontypeable Haemophilus influenzae via p38 and extracellular signal-regulated kinase pathways. Infect Immun 2003;71:5523–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.St. Geme JW III. The pathogenesis of nontypable Haemophilus influenzae otitis media. Vaccine 2000;19:S41–S50. [DOI] [PubMed] [Google Scholar]
- 46.DiMango E, Zar HJ, Bryan R, Prince A. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J Clin Invest 1995;96:2204–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krasan GP, Cutter D, Block SL, St. Geme JW III. Adhesin expression in matched nasopharyngeal and middle ear isolates of nontypeable Haemophilus influenzae from children with acute otitis media. Infect Immun 1999;67:449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boles BR, Thoendel M, Singh PK. Self-generated diversity produces “insurance effects” in biofilm communities. Proc Natl Acad Sci USA 2004;101:16630–16635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.