

Abstract
We recently found that low-molecular-weight hyaluronan was induced by cyclic stretch in lung fibroblasts and accumulated in lungs from animals with ventilator-induced lung injury. The low-molecular-weight hyaluronan produced by stretch increased interleukin-8 production in epithelial cells, and was accompanied by an upregulation of hyaluronan synthase–3 mRNA. We hypothesized that low-molecular-weight hyaluronan induced by high VT was dependent on hyaluronan synthase 3, and was associated with ventilator-induced lung injury. Effects of high VT ventilation in C57BL/6 wild-type and hyaluronan synthase–3 knockout mice were compared. Significantly increased neutrophil infiltration, macrophage inflammatory protein–2 production, and lung microvascular leak were found in wild-type animals ventilated with high VT. These reactions were significantly reduced in hyaluronan synthase–3 knockout mice, except the capillary leak. Wild-type mice ventilated with high VT were found to have increased low-molecular-weight hyaluronan in lung tissues and concomitant increased expression of hyaluronan synthase–3 mRNA, neither of which was found in hyaluronan synthase–3 knockout mice. We conclude that high VT induced low-molecular-weight hyaluronan production is dependent on de novo synthesis through hyaluronan synthase 3, and plays a role in the inflammatory response of ventilator-induced lung injury.
Keywords: hyaluronic acid, knockout mice, mechanical ventilation, tidal volume
The management of acute lung injury and acute respiratory distress syndrome (ARDS) requires the use of positive-pressure ventilation to provide adequate oxygenation. When ARDS develops, the lungs are affected nonhomogeneously, which leads to areas with different compliance. As the low compliant areas increase, the uneven distribution delivered by traditional or even smaller tidal volumes (Vt) will result in overdistension of those normal, compliant areas. A clinical trial by the ARDS Network has documented that mechanical ventilation with a smaller Vt (6 m/kg) decreased the mortality rate in patients with ARDS (1), suggesting the potential role of conventional Vt in lung injury. Ventilator-induced lung injury (VILI) has been studied in different animal models with high Vt ventilation (2–4). It has been characterized by neutrophil sequestration, increased vascular permeability, and elevated levels of chemoattractant cytokines, particularly macrophage inflammatory protein 2 (MIP-2), the rodent equivalent for human interleukin 8 (IL-8) (5–10). Furthermore, MIP-2 receptor knockout mice have been shown to have less VILI than wild-type mice (9). However, the mechanisms of VILI during large Vt ventilation are not fully understood.
Several studies of fetal lung cells have documented that cyclic stretch led to increased extracellular matrix synthesis and secretion (11–14). Increased synthesis of lung extracellular matrix components has been found in animal models of VILI (15, 16). These findings suggested that extracellular matrix may be involved in the pathogenesis of VILI.
Hyaluronan (HA) is a common component of extracellular matrix. In addition to being an essential structural molecule for tissue architecture, different forms of HA have various biological functions. High-molecular-weight (HMW) HA exists in most healthy tissues, whereas low-molecular-weight (LMW) HA accumulates at sites of inflammation. LMW HA acts as a potent signaling molecule, triggering the production of chemokines, cytokines, and growth factors by many cell types (17–19). HA is synthesized by HA synthase (Has) located at the inner face of the plasma membrane (20). Three isoforms of Has have been identified in mammals. Each isoform is encoded by a separate gene (21). In vitro Has1 and Has2 produce HMW HA, whereas Has3 produces LMW HA (21, 22). This relationship has not been confirmed to date in any in vivo model.
In our previous studies, LMW HA was produced in stretched fibroblasts and in rats ventilated with high Vt, both of which were accompanied with increased expression of Has3 mRNA (23). Furthermore, LMW HA was demonstrated to enhance the production of IL-8 in stretched epithelial cells (23). On the basis of these results, we hypothesized that the production of LMW HA via Has3 plays a role in VILI. To investigate this hypothesis, we used Has3 knockout and wild-type mice in an in vivo model of VILI. We found that Has3 was necessary for the high Vt–induced LMW HA and that the resultant neutrophil infiltration was partly dependent on Has3.
METHODS
Creation of Has3-deficient Mice by Gene Targeting
The genomic organization of the mouse Has3 gene has been previously described (21). A replacement type that targets vector was designed and created from a P1 genomic clone derived from the 129Sv/J strain. Germ-line transmission was monitored through backcrossing of male chimeras to C57BL/6J dams. As expected, 50% of the agouti pups were heterozygous for the targeted Has3 allele. These animals were progressively backcrossed against C57BL/6J to create a congenic line, and were also intercrossed to derive homozygous Has3-deficient animals.
Animal Preparation and Ventilator Protocol
Male and female Has3−/−, Has3+/−, and Has3+/+ mice were used. These animals were between N6 and N9 on the backcross series to C57BL/6J. Wild-type C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). Both wild-type and knockout mice were subjected to the same ventilator protocol. Animals were randomized into three groups: (1) a high Vt group, ventilated with a Vt of 30 ml/kg for 5 hours; (2) a low Vt group, ventilated with a Vt of 6 ml/kg for 5 hours; and (3) a control group, without mechanical ventilation. No positive end-expiratory pressure was used, and the peak inspiratory pressure at the beginning of ventilation was 12.9 ± 0.9 cm H2O for the low Vt group and 32.8 ± 1.2 cm H2O for the high Vt group.
We used an established rodent model of VILI as previously described (8, 24, 25). In the same VILI model, we have previously shown that mice with different ventilation strategies did not show any statistical difference in arterial blood gases, mean arterial pressures, and peak inspiratory pressures (25).
Measurement and Size Fractionation of HA
Total HA deposition in lung tissues was localized by using an avidin-biotin-peroxidase histochemical technique with a biotinylated fragment of HA binding protein (Seikagaku Corp., Tokyo, Japan) (23). An ELISA method based on biotinylated fragment of HA binding protein was used to quantify total HA in bronchoalveolar lavage (BAL) and lung tissues (BAL, n = 4/group; lung tissue, two sets of animals, n = 5/set).
Sepharose CL-4B size exclusion chromatography was used to determine the molecular size of HA (n = 5/group). The column was calibrated in the following manner: void volume (Vo) was measured using Blue Dextran 2000 (Pharmacia Fine Chemicals AB, Uppsala, Sweden); total volume(Vt) was measured using uronic acid. HA sizes were determined by relative elution volume (Kav) compared with three HA standards of known molecular weight (Sigma Chemical Co. and Biomatrix, Inc., Ridgefield, NJ). Kav was calculated from the following equation: Kav = (elution volume − Vo)/(Vt − Vo). The three HA standards, 1,600 kD HMW HA, 370 kD LMW HA, and 178 kD LMW HA, had a Kav of 0.17, 0.33, and 0.8, respectively. HA peaks were detected using HA ELISA as described previously.
Statistical Analysis
The data were compared by analysis of variance and then subsequent multiple comparisons by the Tukey-Kramer (Statview 5.0; Abacus Concepts, Inc., SAS Institute, Inc., Cary, NC). By F test for homogenous variances, only the data on Evans blue dye had inhomogeneous variances between groups. For these data, a Box-Cox transform analysis was performed to identify the optimal transformation. The assumption of equal variance was met by Bartlett's test, confirming that the transformation was effective. Then, the data were analyzed by analysis of variance followed by multiple comparisons with the Tukey test (Stata statistical software, release 8.0; Stata Corp., College Station, TX). Statistical significance was defined as a p value less than 0.05. All values were expressed as mean ± SEM.
RESULTS
Generation of Has3-deficient Mouse Lines
Has3-deficient mice were created using homologous recombination in mouse ES cells. The targeted Has3 allele was designed such that a large portion of the fourth exon, encoding 100 amino acids from within the catalytic region of the polypeptide, would be deleted, effectively creating a functionally null Has3 allele (Figure 1). A similar strategy has been successfully used by us to create a functionally null Has2 allele (26). Correctly targeted clones were obtained at a frequency of 1 in 30 geneticin-resistant clones. Two clones were expanded and microinjected into blastocysts to yield germline chimeras at high frequency. Heterozygous Has3+/− animals were obtained at the expected frequency and appeared normal in all respects. Heterozygous intercrosses yielded the expected mendelian frequency (25%) of Has3−/− animals, all of which appeared normal, and had normal fertility and life spans. These animals did not exhibit any age-related changes and exhibited normal motor activity (data not shown). The static and dynamic compliances for wild-type and Has3−/− mice were not significantly different (static, 0.61 ± 0.03 vs. 0.66 ± 0.09 μl/cm H2O/g, p = 0.44, and dynamic, 0.47 ± 0.03 vs. 0.54 ± 0.02 μl/cm H2O/g, p = 0.12, respectively).
Figure 1.

Structure of the Has3 gene locus before and after gene targeting by homologous recombination. Exons 2–4 are shown. The extent of the open-reading frame is indicated by the black-filled boxes and the ATG (start) and TGA (stop) codons. The extent of the restriction fragments that were used as the 5′ and 3′ arms of homology in the targeting vector are indicated by the thick black lines above the Has3 wild-type (WT) gene locus illustration. Correctly targeted clones were identified by Southern analyses using the flanking probe indicated and BamHI restriction endonuclease digests. The flanking probe detected a diagnostic 2.5-kb pair restriction fragment indicative of homologous recombination at the Has3 locus. The relative positions of polymerase chain reaction (PCR) primers that were routinely used to genotype animals are indicated. PGKneo = neomycin-phospho transfer gene.
High VT–induced Infiltration of Neutrophils
Lung myeloperoxidase activity, neutrophil count of BAL, peribronchiolar neutrophil count, MIP-2 mRNA expression in lung, and MIP-2 protein level in BAL were used to assess the neutrophil infiltration in the lungs.
In wild-type mice, the expression of MIP-2 mRNA in lung (Figure 2A), levels of MIP-2 protein in BAL (Figure 2B), and lung myeloperoxidase activity (Figure 2C) were significantly elevated as compared with low Vt and control groups. The BAL neutrophil count (Figure 2D) and the average number of peribronchiolar neutrophils (Figure 2E) were significantly higher in the high Vt group as compared with control groups. No significant difference of BAL MIP-2 level and lung myeloperoxidase activity was found between low Vt and control groups (Figures 2B and 2C). The lavage recovery was equivalent in all groups (average, 1.2 cc).
Figure 2.
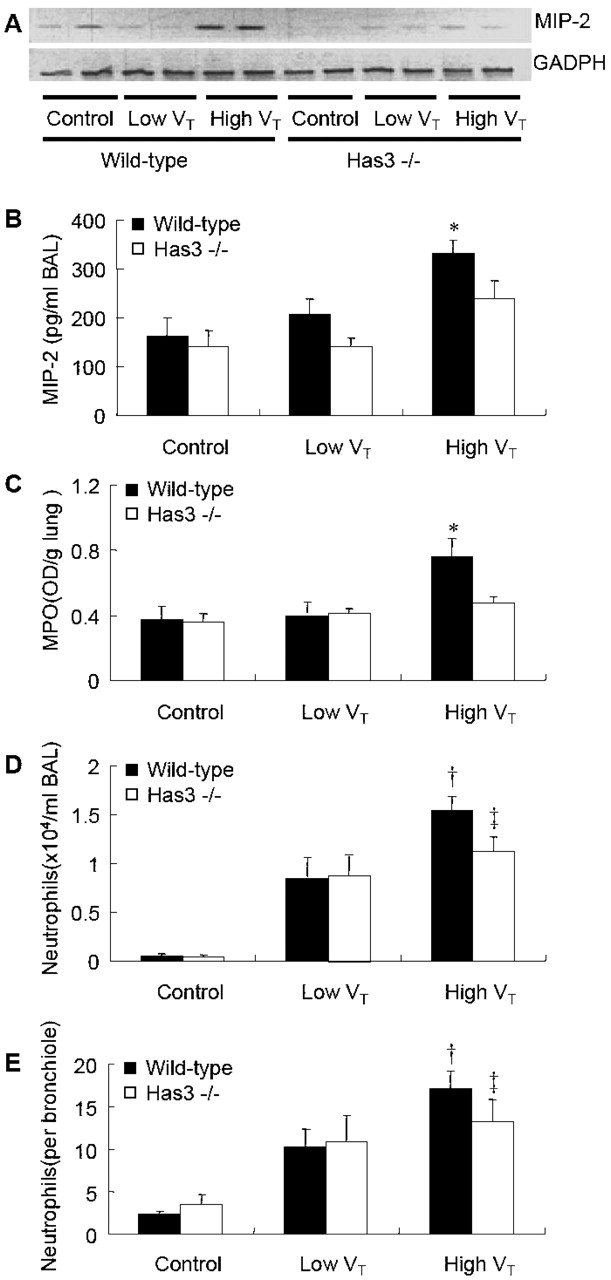
High VT–ventilation-induced lung neutrophil infiltrations were reduced in Has3−/−mice. (A) reverse transcription (RT)–PCR results of the expression of macrophage inflammatory protein 2 (MIP-2) mRNA, which was upregulated in the high VT group of wild-type mice. The expression of high VT–induced MIP-2 mRNA was reduced in Has3−/− mice (n = 2/group). Bronchoalveolar lavage (BAL) MIP-2 level (B) and neutrophil counts (D) were significantly increased in wild-type mice ventilated with high VT. Both were significantly reduced in Has3−/− mice (n = 5 in control and low VT groups, n = 7 in Has3−/−, and n = 10 in the wild-type high VT group). Lung myeloperoxidase (MPO) activity (C) was significantly lower in Has3−/− mice with high VT ventilation (n = 5 in control and low VT groups, n = 6 in Has3−/−, and n = 9 in the wild-type high VT group). Peribronchiolar neutrophil infiltration (E) was lower in Has3−/− mice ventilated with high VT, but did not reach significant difference (n = 5 in control and low VT groups, n = 5 in Has3−/−, and n = 7 in the wild-type high VT group). *p < 0.05 versus all control and low VT groups; †p < 0.05 versus wild-type and Has3−/− control and wild-type low VT groups; ‡p < 0.05 versus control groups. GADPH = glyceraldehyde-3-phosphate dehydrogenase.
High VT–induced Infiltration of Neutrophils Was Reduced in Has3−/− Mice
In contrast to the wild-type animals, the Has3−/− mice subjected to the high Vt regimen did not show any significant increases in MIP-2 mRNA expression, BAL MIP-2 protein level, and lung myeloperoxidase activity as compared with the low Vt and control groups (Figures 2A–2C). The BAL content of neutrophils and the peribronchiolar neutrophil infiltration were significantly higher in the Has3−/− high Vt group when compared with the control group, but were not significantly different from that of the low Vt group (Figures 2D and 2E).
High VT–induced Microvascular Leak Was Not Different between Has3−/− and Wild-Type Mice
Extravasation of Evans blue dye (Figure 3A) and concentration of BAL total protein (Figure 3B) were used to evaluate the extent of microvascular leak of VILI. Both were significantly elevated in wild-type and Has3−/− mice ventilated with high Vt compared with control groups, signifying increased microvascular leak after high Vt ventilation. There was no significant difference in these two values between low Vt and control groups of wild-type mice.
Figure 3.

High VT–ventilation-induced microvascular leakage was not significantly different between wild-type and Has3−/− mice. Evans blue dye (EBD) extravasation (A) was significantly increased in the high VT ventilation group (n = 5 in control and low VT groups, n = 5 in Has3−/−, and n = 8 in the wild-type high VT group). Total protein level in BAL (B) was significantly higher in mice ventilated with high VT (n = 5/group). There was no significant difference of EBD extravasation and BAL total protein level between wild-type and Has3−/− mice of the high VT group. *p < 0.05 versus all control and low VT groups; †p < 0.05 versus all control groups.
In Has3−/− mice, the Evans blue dye extravasation was significantly higher in the low Vt group as compared with control groups (Figure 3A). In groups ventilated with high Vt, the degrees of Evans blue dye extravasation and BAL protein concentration were not significantly different between wild-type and Has3−/− mice (Figure 3).
High VT–induced Elevation of Total HA
In lung tissue of wild-type mice, increased deposition of total HA was noted in the high Vt group as compared with that of control group (Figures 4A and 4B). The HA accumulated in large airways and within lung parenchyma. For further quantitative measurement, we determined the total HA levels in lung tissue (Figure 5A) and BAL (Figure 5B) of wild-type mice. There were significantly higher levels of total HA in the high Vt group as compared with low Vt and control groups. No significant difference in total HA level was found between the low Vt and control groups of wild-type mice.
Figure 4.

Histochemical staining for hyaluronan (HA) in wild-type and Has3−/− mice. Histochemical staining of total HA in lungs of (A) the control group of wild-type, (B) the high VT group of wild-type, (C) the control group of Has3−/−, and (D) the high VT group of Has3−/− mice. There was an increase of total HA accumulation in the large airways and parenchyma of lung in wild-type mice with high VT ventilation.
Figure 5.

High VT–ventilation-induced lung HA accumulation was reduced in Has3−/− mice. Measurement of total HA in lung (A) and BAL (B) showed significantly higher levels in wild-type mice with high VT ventilation, as compared with control, low VT groups, and the high VT group of Has3−/− mice. In Has3−/− mice, the total HA of the high VT group was significantly higher than that of the control group, but not different from the low VT group (two sets, and n = 5/group in each set for lung HA measurement, n = 4/group for BAL HA measurement). *p < 0.05 versus all control and low VT groups, and high VT Has3−/−; †p < 0.05 versus all control and low VT groups, and the high VT wild-type.
Reduced High VT–induced Elevation of Total HA in Has3−/− Mice
The histochemical staining of total HA did not show any difference between high Vt and control groups of Has3−/− mice (Figures 4C and 4D). Although total HA levels in lung tissue of the Has3−/− high Vt group were significantly higher than those of the control group, the levels were not significantly different from those of the low Vt group (Figure 5A). There was no significant increase in total HA in BAL of Has3−/− mice with low or high Vt ventilation. As compared with the high Vt group of wild-type mice, Has3−/− mice ventilated with high Vt had less deposition of total HA by biotinylated hyaluronic acid binding protein staining (Figures 4B and 4D).
High VT–induced Elevation of LMW HA in Wild-Type, but Not in Has3−/− Mice
For further evaluation of the molecular weight of HA induced by Vt, we used Sepharose CL-4B size exclusion chromatography (Amersham Pharmacia, Little Chalfont, Bucks, UK) to analyze the total HA extracted from lungs (Figure 6). We found that both HMW HA (1,600 kD) and LMW HA (< 370 kD) were present in the lungs of wild-type mice after high Vt ventilation, but only HMW HA (1,600 kD) was found in the lungs of the other groups, including the high Vt group of Has3−/− mice.
Figure 6.

High VT–ventilation-induced low-molecular-weight (LMW) HA in lung (n = 5/group). HA from lung tissues was run on Sepharose CL-4B (150 × 2 cm; total volume [Vt] 120 ml, void volume [Vo] 30-ml column) for chromatography. Three standard HA: HMW HA (1,600 kD) had a relative elution volume (Kav) of 0.17, LMW HA (370 kD) had a Kav of 0.33, and LMW HA (178 kD) had a Kav of 0.8. The HA in wild-type mice ventilated with high VT (closed squares) had a Kav of 0.17 and a Kav from 0.55 to 0.77. The HA of wild-type control (closed diamonds), wild-type low VT (closed triangles), Has3−/− control (open diamonds), Has3−/− low VT (open triangles), and Has3−/− high VT (open squares) had a Kav of 0.17.
High VT–induced Expression of Has3 mRNA
To explore the role of Has3 in high Vt–induced LMW HA production, we performed reverse transcription–polymerase chain reaction to detect the expression of the respective Has isoforms. There was an increased expression of Has3 mRNA in the high Vt group of wild-type mice as compared with the other groups (Figure 7A). The concomitant upregulation of Has3 mRNA and LMW HA was only found in the high Vt group wild-type, but not in Has3−/− mice. The expression of Has2 and Has1 was increased in the high Vt group of Has3−/− mice (Figures 5 and 7A).
Figure 7.

High VT ventilation increased the expression of lung Has mRNA. Total RNA from mice lung was isolated for analysis of Has isoforms by RT-PCR. (A) The expression of Has3 mRNA was increased in wild-type mice ventilated with high VT as compared with other groups. (B) Has3 mRNA in Has3+/− mice was only mildly increased with high VT ventilation (n = 2/group).
It should be noted that the Has3+/− and wild-type littermates of the Has3−/− mice demonstrated the same responses to high and low Vt ventilation as C57BL/6 wild-type animals purchased from Charles River Laboratories (data not shown). The Has3 mRNA expression was only mildly elevated in Has3+/− mice with high Vt ventilation (Figure 7B).
DISCUSSION
By using a mouse model of VILI, we demonstrated that high Vt ventilation induced the appearance of LMW HA and resulted in neutrophil infiltration in the lungs. In Has3−/− animals with high Vt ventilation, LMW HA was not induced and a reduced inflammatory response was observed. These results indicate that high Vt–induced LMW HA production was dependent on Has3, and LMW HA played an important role in VILI.
HA is an integral and critical component of the extracellular matrix, where it can act as the backbone around which large space-filling matrices can be assembled. HA has been associated with many forms of pulmonary disease, including acute lung injury and ARDS (27, 28). Hallgren and colleagues (29) found that the median BAL HA level was six times higher in patients with ARDS and the median serum HA level was 20 times higher than those of control patients. A high BAL HA level in ARDS was also found by Modig and Hallgren (30). In patients with ARDS treated with an extracorporeal CO2 removal device, Kropf and colleagues (31) found that there was a sustained increase of serum HA level in those who did not respond to therapy. However, the molecular weight of HA was not measured in these experiments, and the exact role of different-sized HA remains unknown.
HA acts as a potent signaling molecule in many biological contexts (32–34). The interaction of HA with HA receptors, primarily CD44 and RHAMM, has been shown to stimulate multiple intracellular signaling cascades (34, 35). Significantly, the signaling potency of HA is dependent on its molecular mass (17, 18, 33–36). For instance, in human alveolar macrophages, McKee and coworkers (17) have found that LMW HA, but not HMW HA, induced IL-8 mRNA expression. Noble and colleagues (18) showed that LMW HA activated the transcriptional factor nuclear factor (NF)-κB in mouse macrophages, whereas HMW HA had no such effect. Fitzgerald and associates (37) further demonstrated that the LMW HA activation of NF-κB was dependent on CD44 binding and protein kinase C pathway activation. These findings suggested that LMW HA is able to induce chemokine gene expression and to stimulate proinflammatory pathways, which may contribute to the development or maintenance of inflammation. Moreover, both the activation of NF-κB and the production of IL-8 (or MIP-2) have been demonstrated to be involved in the pathogenesis of VILI (6, 25, 38–40).
Using our in vitro model, which simulates cyclic stretch of lung cells during ventilator use, we have demonstrated that LMW HA was produced by stretched human fibroblasts, whereas only HMW HA was found in the supernatant of unstretched control cultures (23). We further showed that the LMW HA produced by stretched fibroblasts was able to induce IL-8 production in nonstretched cells, and enhance the production of IL-8 from cyclic stretched epithelial cells. There was no increase of IL-8 when these epithelial cells were treated with HMW HA. We have now confirmed these findings in vivo. Wild-type mice were found to produce LMW HA after high Vt ventilation. This group had a higher degree of MIP-2 production and neutrophil infiltration as compared with those groups that only produced HMW HA (Figures 2, 5, and 6). These results support our hypothesis that LMW HA plays a role in the inflammatory response of VILI.
The synthesis of HA is dependent on HA synthases, which are located at the plasma membrane and expressed widely in human tissues. The three identified Has isoforms have distinct enzymatic properties (21, 22). In vitro, Has3 catalyzed the biosynthesis of LMW HA, whereas Has1 and Has2 activity resulted in HMW HA (21, 22). Although this result has remained intriguing, the possible role of Has3 in the in vivo biosynthesis of LMW HA had not been determined. In our present study, Has3 expression and function was necessary for the LMW HA (178–370 kD) induced by high Vt ventilation. HA in this size range was similar to that synthesized by Has3 in previous in vitro experiments (21, 22). Furthermore, HA within this size range has been demonstrated to activate transcriptional factors and induce chemokine release (17–19, 37, 41). Our results differ from those found in chondrocytes exposed to mechanical stretch (42). In chondrocytes, mechanical stretch increased the expression of Has2 but not Has3 mRNA, suggesting the response to stretch is cell-specific.
LMW HA may be produced by Has3 or by cleavage of HMW HA to LMW forms. The expression of Has3 mRNA and production of LMW HA were only increased in wild-type mice ventilated with high Vt, whereas neither was found in Has3−/− mice (Figures 6 and 7A). These results suggest that the LMW HA produced by high Vt ventilation is dependent on de novo synthesis via Has3. It has been shown, however, that LMW HA can be generated from HMW HA through chemical attack, particularly by reactive oxygen species. We cannot absolutely rule out that breakdown of HMW HA by reactive oxygen species may also play a role in the production of LMW HA related to VILI. The ventilator-induced inflammation was only partially blocked in the Has3−/− mice, suggesting other pathways may also be involved. A critical role for c-Jun N-terminal kinase (JNK) activation in VILI has been demonstrated in previous studies (25, 38–40). In our cell stretch model, the IL-8 production induced by cyclic stretch of A549 cells was shown to be dependent on JNK activation (40). Using the same in vivo model of VILI as this study, we have shown that both JNK knockout and wild-type mice pretreated with specific JNK inhibitor had significantly reduced ventilation-induced neutrophil infiltration and MIP-2 production (25). We have also found that LMW HA–enhanced IL-8 production was dependent on the JNK activation (unpublished data). These findings suggested that LMW HA may augment stretch-induced JNK activation and inflammation, but may not be the only mechanism of JNK activation.
The expressions of Has1 and Has2 were upregulated in Has3−/− animals ventilated with high Vt, which may account for the significantly increased total HA as compared with the control group (Figures 5 and 7A). It is possible that there is a feedback mechanism where cells are able to sense that relative amount of HA that is present on the cell surface or in the immediate surrounding matrix. More production of HA has been shown in certain cells treated with hyaluronidase, in which response appeared to be mediated both through resident Has and by modest upregulation of expression (A.P.S., unpublished data). As the three Has genes are located on three separate autosomes, it would not be highly unlikely for the targeted mutation of Has3 to have a direct effect on any regulatory elements within the Has1 and/or Has2 genes.
High Vt ventilation in the normal lung of the mouse led to moderate, but real changes in lung neutrophil infiltration and MIP-2 production (Figure 2) of the same magnitude as found by other investigators (9). The changes in MIP-2 can be variable between animals as demonstrated by others (10). We have found that mice are more resistant to the effects of high Vt ventilation than rats (8) and show a lower magnitude of difference in cytokines and neutrophil infiltration. However, the mouse model allows the use of knockouts.
With histochemical staining in both our rat model (23) and this mouse model, we found the largest amounts of HA accumulation around the airways. We have tested individual lung cell types in vitro, and found that pulmonary artery endothelial cells (unpublished data), bronchial epithelial cells (unpublished data), and fibroblasts (23) all produce HA when exposed to stretch. However, fibroblasts made the largest amounts of HA in vitro (data not shown).
Though HA has been associated with the process of pulmonary edema in bleomycin-induced pulmonary fibrosis (43), we did not find the same phenomenon in the current study. The microvascular leak induced by high Vt ventilation was not significantly decreased in Has3−/− mice, suggesting that at least LMW HA was not involved in the process of pulmonary edema in VILI (Figure 3).
In conclusion, ventilator-induced inflammation is, at least in part, dependent on the production of LMW HA by Has3. This is the first in vivo demonstration of a role for Has3 in LMW HA production. Furthermore, the results from this study may provide a molecular target at which we can aim novel therapeutic strategies for the treatment of respiratory disease and damage.
Supplementary Material
Acknowledgments
The authors thank Susannah Wood for her generous support and encouragement, and Danielle Higgins, Li-Fu Li, Olga Syrkina, Bin Ouyang, and John Beagle for their expert technical assistance.
Supported by the National Institutes of Health grants HL03920, 2T32HL07874, and funds from the Texas A&M University System Health Science Center to A.P.S.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Conflict of Interest Statement: K.-J.B. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; A.P.S. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; M.M.M. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; L.Y. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; C.D.O. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; H.G.G. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; D.A.Q. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 2000;342:1301–1308. [DOI] [PubMed] [Google Scholar]
- 2.American Thoracic Society. International consensus conferences in intensive care medicine: ventilator-associated lung injury in ARDS. Am J Respir Crit Care Med 1999;160:2118–2124. [DOI] [PubMed] [Google Scholar]
- 3.Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 1998;157:294–323. [DOI] [PubMed] [Google Scholar]
- 4.Slutsky AS. Lung injury caused by mechanical ventilation. Chest 1999;116:9S–15S. [DOI] [PubMed] [Google Scholar]
- 5.Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest 1997;99:944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Held HD, Boettcher S, Hamann L, Uhlig S. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med 2001;163:711–716. [DOI] [PubMed] [Google Scholar]
- 7.Ricard JD, Dreyfuss D, Saumon G. Production of inflammatory cytokines in ventilator-induced lung injury: a reappraisal. Am J Respir Crit Care Med 2001;163:1176–1180. [DOI] [PubMed] [Google Scholar]
- 8.Quinn DA, Moufarrej RK, Volokhov A, Hales CA. Interactions of lung stretch, hyperoxia, and MIP-2 production in ventilator-induced lung injury. J Appl Physiol 2002;93:517–525. [DOI] [PubMed] [Google Scholar]
- 9.Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, Phillips RJ, Strieter RM. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest 2002;110:1703–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson MR, Choudhury S, Goddard ME, O'Dea KP, Nicholson AG, Takata M. High tidal volume upregulates intrapulmonary cytokines in an in vivo mouse model of ventilator-induced lung injury. J Appl Physiol 2003;95:1385–1393. [DOI] [PubMed] [Google Scholar]
- 11.Xu J, Liu M, Liu J, Caniggia I, Post M. Mechanical strain induces constitutive and regulated secretion of glycosaminoglycans and proteoglycans in fetal lung cells. J Cell Sci 1996;109:1605–1613. [DOI] [PubMed] [Google Scholar]
- 12.Xu J, Liu M, Post M. Differential regulation of extracellular matrix molecules by mechanical strain of fetal lung cells. Am J Physiol 1999;276:L728–L735. [DOI] [PubMed] [Google Scholar]
- 13.Mourgeon E, Xu J, Tanswell AK, Liu M, Post M. Mechanical strain-induced posttranscriptional regulation of fibronectin production in fetal lung cells. Am J Physiol 1999;277:L142–L149. [DOI] [PubMed] [Google Scholar]
- 14.Breen EC. Mechanical strain increases type I collagen expression in pulmonary fibroblasts in vitro. J Appl Physiol 2000;88:203–209. [DOI] [PubMed] [Google Scholar]
- 15.Parker JC, Breen EC, West JB. High vascular and airway pressures increase interstitial protein mRNA expression in isolated rat lungs. J Appl Physiol 1997;83:1697–1705. [DOI] [PubMed] [Google Scholar]
- 16.Al Jamal R, Ludwig MS. Changes in proteoglycans and lung tissue mechanics during excessive mechanical ventilation in rats. Am J Physiol Lung Cell Mol Physiol 2001;281:L1078–L1087. [DOI] [PubMed] [Google Scholar]
- 17.McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, Noble PW. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages: the role of HA size and CD44. J Clin Invest 1996;98:2403–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noble PW, McKee CM, Cowman M, Shin HS. Hyaluronan fragments activate an NF-kappa B/I-kappa B alpha autoregulatory loop in murine macrophages. J Exp Med 1996;183:2373–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horton MR, McKee CM, Bao C, Liao F, Farber JM, Hodge-DuFour J, Pure E, Oliver BL, Wright TM, Noble PW. Hyaluronan fragments synergize with interferon-gamma to induce the C-X-C chemokines mig and interferon-inducible protein-10 in mouse macrophages. J Biol Chem 1998;273:35088–35094. [DOI] [PubMed] [Google Scholar]
- 20.Philipson LH, Schwartz NB. Subcellular localization of hyaluronate synthetase in oligodendroglioma cells. J Biol Chem 1984;259:5017–5023. [PubMed] [Google Scholar]
- 21.Spicer AP, McDonald JA. Characterization and molecular evolution of a vertebrate hyaluronan synthase gene family. J Biol Chem 1998;273:1923–1932. [DOI] [PubMed] [Google Scholar]
- 22.Itano N, Sawai T, Yoshida M, Lenas P, Yamada Y, Imagawa M, Shinomura T, Hamaguchi M, Yoshida Y, Ohnuki Y, et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J Biol Chem 1999;274:25085–25092. [DOI] [PubMed] [Google Scholar]
- 23.Mascarenhas MM, Day RM, Ochoa CD, Choi WI, Yu L, Ouyang B, Garg HG, Hales CA, Quinn DA. Low molecular weight hyaluronan from stretched lung enhances interleukin-8 expression. Am J Respir Cell Mol Biol 2004;30:51–60. [DOI] [PubMed] [Google Scholar]
- 24.Hales CA, Du HK, Volokhov A, Mourfarrej R, Quinn DA. Aquaporin channels may modulate ventilator-induced lung injury. Respir Physiol 2001;124:159–166. [DOI] [PubMed] [Google Scholar]
- 25.Li LF, Yu L, Quinn DA. Ventilation-induced neutrophil infiltration depends on c-Jun N-terminal kinase. Am J Respir Crit Care Med 2004;169:518–524. [DOI] [PubMed] [Google Scholar]
- 26.Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest 2000;106:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savani RC, DeLisser HM. Hyaluronan and its receptors in lung health and disease. In: Garg HG, Roughley PJ, Hales CA, editors. Proteoglycans in lung disease. New York: Marcel Dekker; 2002. pp. 23–36.
- 28.Turino GM, Cantor JO. Hyaluronan in respiratory injury and repair. Am J Respir Crit Care Med 2003;167:1169–1175. [DOI] [PubMed] [Google Scholar]
- 29.Hallgren R, Samuelsson T, Laurent TC, Modig J. Accumulation of hyaluronan (hyaluronic acid) in the lung in adult respiratory distress syndrome. Am Rev Respir Dis 1989;139:682–687. [DOI] [PubMed] [Google Scholar]
- 30.Modig J, Hallgren R. Increased hyaluronic acid production in lung–a possible important factor in interstitial and alveolar edema during general anesthesia and in adult respiratory distress syndrome. Resuscitation 1989;17:223–231. [DOI] [PubMed] [Google Scholar]
- 31.Kropf J, Grobe E, Knoch M, Lammers M, Gressner AM, Lennartz H. The prognostic value of extracellular matrix component concentrations in serum during treatment of adult respiratory distress syndrome with extracorporeal CO2 removal. Eur J Clin Chem Clin Biochem 1991;29:805–812. [DOI] [PubMed] [Google Scholar]
- 32.Bourguignon LY, Lokeshwar VB, Chen X, Kerrick WG. Hyaluronic acid-induced lymphocyte signal transduction and HA receptor (GP85/CD44)-cytoskeleton interaction. J Immunol 1993;151:6634–6644. [PubMed] [Google Scholar]
- 33.Lee JY, Spicer AP. Hyaluronan: a multifunctional, megaDalton, stealth molecule. Curr Opin Cell Biol 2000;12:581–586. [DOI] [PubMed] [Google Scholar]
- 34.Day RM, Mascarenhas M. Signal transduction associated with hyaluronan. In: Garg HG, Hales CA, editors. Chemistry and biology of hyaluronan. Amsterdam: Elsevier; 2004.
- 35.Turley EA, Noble PW, Bourguignon LY. Signaling properties of hyaluronan receptors. J Biol Chem 2002;277:4589–4592. [DOI] [PubMed] [Google Scholar]
- 36.Neumann A, Schinzel R, Palm D, Riederer P, Munch G. High molecular weight hyaluronic acid inhibits advanced glycation endproduct-induced NF-kappaB activation and cytokine expression. FEBS Lett 1999;453:283–287. [DOI] [PubMed] [Google Scholar]
- 37.Fitzgerald KA, Bowie AG, Skeffington BS, O'Neill LA. Ras, protein kinase C zeta, and I kappa B kinases 1 and 2 are downstream effectors of CD44 during the activation of NF-kappa B by hyaluronic acid fragments in T-24 carcinoma cells. J Immunol 2000;164:2053–2063. [DOI] [PubMed] [Google Scholar]
- 38.Oudin S, Pugin J. Role of MAP kinase activation in interleukin-8 production by human BEAS-2B bronchial epithelial cells submitted to cyclic stretch. Am J Respir Cell Mol Biol 2002;27:107–114. [DOI] [PubMed] [Google Scholar]
- 39.Uhlig U, Haitsma JJ, Goldmann T, Poelma DL, Lachmann B, Uhlig S. Ventilation-induced activation of the mitogen-activated protein kinase pathway. Eur Respir J 2002;20:946–956. [DOI] [PubMed] [Google Scholar]
- 40.Li LF, Ouyang B, Choukroun G, Matyal R, Mascarenhas M, Jafari B, Bonventre JV, Force T, Quinn DA. Stretch-induced IL-8 depends on c-Jun NH2-terminal and nuclear factor-kappaB-inducing kinases. Am J Physiol Lung Cell Mol Physiol 2003;285:L464–L475. [DOI] [PubMed] [Google Scholar]
- 41.McKee CM, Lowenstein CJ, Horton MR, Wu J, Bao C, Chin BY, Choi AM, Noble PW. Hyaluronan fragments induce nitric-oxide synthase in murine macrophages through a nuclear factor kappaB-dependent mechanism. J Biol Chem 1997;272:8013–8018. [DOI] [PubMed] [Google Scholar]
- 42.Yamazaki K, Fukuda K, Matsukawa M, Hara F, Matsushita T, Yamamoto N, Yoshida K, Munakata H, Hamanishi C. Cyclic tensile stretch loaded on bovine chondrocytes causes depolymerization of hyaluronan: involvement of reactive oxygen species. Arthritis Rheum 2003;48:3151–3158. [DOI] [PubMed] [Google Scholar]
- 43.Nettelbladt O, Tengblad A, Hallgren R. Lung accumulation of hyaluronan parallels pulmonary edema in experimental alveolitis. Am J Physiol 1989;257:L379–L384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.