

Abstract
Rationale: Inducible nitric oxide synthase (iNOS) has been implicated in the development of acute lung injury. Recent studies indicate a role for mechanical stress in iNOS and endothelial NOS (eNOS) regulation. Objectives: This study investigated changes in lung NOS expression and activity in a mouse model of ventilator-induced lung injury. Methods: C57BL/6J (wild-type [WT]) and iNOS-deficient (iNOS−/−) mice received spontaneous ventilation (control) or mechanical ventilation (MV; VT of 7 and 20 ml/kg) for 2 hours, after which NOS gene expression and activity were determined and pulmonary capillary leakage assessed by the Evans blue albumin assay. Results: iNOS mRNA and protein expression was absent in iNOS−/− mice, minimal in WT control mice, but significantly upregulated in response to 2 hours of MV. In contrast, eNOS protein was decreased in WT mice, and nonsignificantly increased in iNOS−/− mice, as compared with control animals. iNOS and eNOS activities followed similar patterns in WT and iNOS−/− mice. MV caused acute lung injury as suggested by cell infiltration and nitrotyrosine accumulation in the lung, and a significant increase in bronchoalveolar lavage cell count in WT mice, findings that were reduced in iNOS−/− mice. Finally, Evans blue albumin accumulation in lungs of WT mice was significant (50 vs. 15% increase in iNOS−/− mice compared with control animals) in response to MV and was prevented by treatment of the animals with the iNOS inhibitor aminoguanidine. Conclusion: Taken together, our results indicate that iNOS gene expression and activity are significantly upregulated and contribute to lung edema in ventilator-induced lung injury.
Keywords: inducible nitric oxide synthase, lung permeability, mechanical ventilation
Nitric oxide (NO) is involved in many physiologic and pathologic conditions, such as blood vessel relaxation, neurotransmission, and host defense. NO also plays a critical role in tissue injury in the context of various inflammatory conditions (1). NO is produced by three isoforms (neuronal, endothelial, and inducible) of NO synthase (NOS). Endothelial NOS (eNOS), which is calcium/calmodulin (Ca2+/CaM)-dependent and activated by agonists (e.g., acetylcholine), produces a low level of NO output, whereas inducible NOS (iNOS) is independent of Ca2+/CaM, is transcriptionally regulated by proinflammatory products (e.g., LPS) and cytokines (interleukin 1β [IL-1β], tumor necrosis factor α [TNF-α], IFN-γ), and results in sustained and elevated release of NO (2). Therefore, overproduction of NO, in particular in the setting of superoxide production (3), leads to oxidative stress and tissue injury in conditions such as endotoxin-induced acute lung injury (ALI) (4).
ALI is characterized by a severe inflammatory process, profound hypoxia, and respiratory failure most often requiring mechanical ventilation (MV) for life support. However, it is now recognized that mechanical stress related to positive-pressure MV may cause or aggravate ALI, in particular when high Vt (HVt) ventilation is applied (5). Furthermore, a systemic inflammatory response may be elicited by MV during recruitment or derecruitment of collapsed lung units and when alveolar regions are overdistended (6). Several investigators, including our group, have demonstrated increased activity of certain signaling pathways in animal models of MV (7–10). A recent study by Frank and colleagues (11) demonstrated increased lung iNOS protein expression and total nitrite levels in bronchoalveolar lavage (BAL) fluid, and a decrease in airspace fluid clearance in rats ventilated with HVt as compared with low Vt (LVt). Inhibition of iNOS prevented the decrease in airspace fluid clearance, suggesting a role for reactive nitrogen species in the pathogenesis of ventilator-associated lung injury (VILI). In contrast, work by Choi and colleagues (12) demonstrated an increase in eNOS protein expression in lungs and kidneys in a rat model of HVt (20 ml/kg for 2 hours) but without detectable iNOS expression in this model. Although these investigators did not measure specific NOS activity in their model, they demonstrated that the nonspecific NOS inhibitor N-nitro-l-arginine methyl ester attenuated lung and kidney microvascular leakage (12).
The aim of this study was to investigate changes in lung NOS expression and activity in a mouse model of VILI. We also assessed the specific role of iNOS in the development of ventilator-induced capillary leakage with the use of iNOS-deficient mice and specific chemical inhibition. Our results indicate that iNOS expression and activity are significantly upregulated by HVt ventilation, are accompanied by nitrotyrosine deposition, mainly in capillary endothelial cells, and correlate with increased lung capillary permeability. Furthermore, iNOS-deficient mice were protected from pulmonary edema in response to MV with HVt. Finally, specific chemical inhibition of iNOS activity with aminoguanidine (AG) abrogated the capillary leakage produced by MV in wild-type animals. Some of the results of these studies have been previously published in abstract form (13).
METHODS
Johns Hopkins University Institutional Animal Care and Use Committee approved all animal protocols. Additional method details are reported in the online supplement.
Animal Preparation
Male C57BL/6 and iNOS knock-out mice, 8 to 10 weeks old, were anesthetized and underwent tracheotomy, and the jugular vein was cannulated. The mice were then exposed to LVt (7 ml/kg, 120 breaths/minute) and HVt (20 ml/kg, 60 breaths/minute) MV for 2 hours. In some experiments, animals were pretreated with AG (15 mg/kg, intraperitoneally), a selective iNOS inhibitor (Sigma, St. Louis, MO), 1 hour before exposure to MV. The adequacy of MV settings on gas exchange was confirmed in preliminary experiments by arterial blood gases analysis (Instrumentation Laboratories, Lexington, MA), which revealed stable levels of arterial oxygen and carbon dioxide.
Immunoblotting
Western blotting was performed as previously described (14, 15) after tissue homogenates were prepared and protein quantified by bicinchoninic acid assay (Pierce, Rockford, IL).
Assessment of Lung Capillary Leakage
Evans blue dye albumin (EBA; 20 mg/kg) was injected into the internal jugular vein 30 minutes before the termination of the experiment to assess vascular leak, as previously described (10, 16, 17).
Determination of BAL Protein, Total Cell Counts, and Inflammatory Cytokines
BAL was performed by intratracheal injection of 1 ml of Hank's balanced salt solution followed by gentle aspiration. The recovered fluid was processed for protein and cell count, as described previously (16), and measurement of cytokines was performed by specific ELISA.
Lung Immunohistochemistry
After removal of paraffin, lung tissue sections were incubated for 1 hour at room temperature with diluted monoclonal antinitrotyrosine (Transduction Laboratories, Lexington, KY) or antineutrophil (Dako Corporation, Carpinteria, CA) primary antibodies. The immunohistochemical reaction was visualized by incubation with 0.05% diaminobenzidine (DAB).
Lung Immunofluorescence
Tissue sections were incubated with anti-iNOS, antinitrotyrosine, anti–surfactant protein C, and anti-CD34 primary antibodies. The samples were then incubated with fluorescein isothiocyanate–labeled secondary antibodies and visualized under fluorescent microscopy.
Lung Morphology
Lungs from animals in each experimental group were inflated under a pressure of 25 cm H2O with 0.5% of low melting agarose (Invitrogen, Carlsbad, CA) for histologic evaluation by hematoxylin and eosin staining as previously described (18, 19).
Measurement of NOS Activity
NOS activity was determined by measuring the conversion of [3H]l-arginine to [3H]l-citrulline using a modification of the Bredt and Snyder procedure (20). Calcium-dependent eNOS activity was determined by the addition of 0.6 mM CaCl2, whereas the addition of 1 mM ethyleneglycol-bis-(β-aminoethyl ether)-N,N′-tetraacetic acid allowed the determination of the calcium-independent iNOS activity. Enzymatic activity is reported as pmol l-citruline/mg protein/minute.
Semiquantitative Reverse Transcription–Polymerase Chain Reaction
Total RNA was isolated and then used for a one-step semiquantitative reverse transcription–polymerase chain reaction (Invitrogen). Primers specific for mouse iNOS were designed on the basis of published cDNA sequences (22). Primers for 18S ribosomal RNA were used as internal positive controls under the same conditions. The steady-state mRNA levels are expressed in arbitrary units as the ratio of iNOS/18S expression (23).
Statistical Analysis
All experiments were repeated at least three times. Representative experiments are shown with values expressed as means ± SE, with n ⩾ 3 for each condition. Data were analyzed by two-way analysis of variance with Bonferroni correction, and significance in all cases was defined at p < 0.05.
RESULTS
Effect of MV on Peak Airway Pressure
We have previously demonstrated that MV with HVt (17 ml/kg) for 2 hours causes significant pulmonary vascular leakage in C57/BL6 mice, an effect that was attenuated by intravenous treatment with the lipid growth factor sphingosine 1-phosphate (10). We used a similar animal model of VILI to examine the effects of MV on NOS gene expression and activity. C57BL/6J and iNOS-deficient (iNOS−/−) mice obtained from the same strain background were exposed to MV for 2 hours at LVt (7 ml/kg) or HVt (20 ml/kg). Peak airway pressure was monitored continuously as detailed in Methods. As shown in Figure 1, peak airway pressures were significantly higher in wild-type and iNOS−/− mice exposed to HVt as compared with LVt. However, in both LVt and HVt groups, there was a small but significant increase in airway pressure (average of 1 and 3 cm H2O for LVt and HVt, respectively) over time (at 60 and 120 minutes) compared with baseline (Time 0) value. There were no trend differences in peak airway pressures between wild-type and iNOS−/− mice (Figure 1).
Figure 1.

Effect of low (LVT) and high (HVT) VT ventilation on peak airway pressure. Peak airway pressure was measured continuously, as detailed in METHODS, in three animals in each exposure group. There were significant differences in peak airway pressures between LVT and HVT exposures but no trend differences between C57BL/6J (wild-type [WT]) and inducible nitric oxide synthase–deficient (iNOS−/−) mice in each exposure group. In both LVT and HVT exposures, there was a small but significant increase in airway pressure (average of 1 and 3 cm H2O for LVT and HVT, respectively) over time compared with baseline value (Time 0). *p < 0.05 versus baseline (Time 0) for the same exposure; +p < 0.05 versus LVT exposure.
Effect of MV with HVT on iNOS Gene Expression
In a separate group of animals, total lung mRNA and protein were collected for semiquantitative polymerase chain reaction and Western blot analysis after exposure to MV. As expected, there was no iNOS mRNA expression in iNOS−/− mice breathing spontaneously or exposed to MV (results not shown). As shown in Figure 2, iNOS mRNA expression in spontaneously breathing (sham-treated) C57BL/6J control mice was minimal but increased significantly (∼ 2.7-fold) in animals exposed to HVt. iNOS protein expression was undetectable in iNOS−/− mice breathing spontaneously or exposed to HVt and minimal in C57BL/6J mice breathing spontaneously (Figure 3A). However, although LVt exposure caused a nonsignificant increase in iNOS protein, HVt exposure produced a 3.5-fold increase (p < 0.05 vs. sham) in this expression in C57BL/6J mice (Figure 3A). Changes in iNOS protein produced over time were examined in a separate group of C57BL/6J mice exposed to HVt MV and were time-dependent (Figure 3B).
Figure 2.

Effect of HVT mechanical ventilation (MV) on iNOS mRNA expression. Upper panel: RNA was extracted from lungs of mice breathing spontaneously (controls) or exposed to HVT MV for 2 hours. A one-step reverse transcription–polymerase chain reaction was performed with specific primers as described in METHODS. Amplification of 18S ribosomal RNA was also performed to confirm equal loading of cDNA samples. As shown in this representative experiment, iNOS expression was minimal in control mice and increased 2.7-fold in animals exposed to HVT. Lower panel: iNOS/18S densitometric measurements from n ⩾ 3 animals for each experimental condition. *p < 0.05 versus control.
Figure 3.


(A) Induction of iNOS protein by MV is dependent on VT. Upper panel: Immunoblots were prepared from extracts of lungs of C57BL/6J (WT) and iNOS−/− mice breathing spontaneously (controls) or exposed to MV with LVT (6 ml/kg) and HVT (20 ml/kg) as described in METHODS. Lower panel: Densitometric assessment of iNOS/actin protein expression (n ⩾ 3 for each condition). iNOS protein expression was undetectable in control iNOS−/− or HVT-exposed iNOS−/− mice. iNOS protein expression was minimal in the WT sham group and increased significantly (3.5-fold) in response to HVT for 2 hours. *p < 0.05 versus sham. (B) Induction of iNOS protein by HVT is time-dependent. C57BL/6J mice were exposed to HVT MV and killed at various time points (three animals/time point) before iNOS immunoblotting. *p < 0.05 versus Time 0. Upper panel: iNOS and actin immunoblots. Lower panel: Densitometric assessment of iNOS/actin protein expression (n = 3 for each condition).
Using immunohistochemistry with antibodies specific for iNOS and endothelial (anti-CD34) and epithelial (anti–surfactant protein C) cells, iNOS immunostaining was prominent in C57BL/6J mice exposed to HVt (Figure 4) and was localized predominantly in pulmonary vessels (Figure 4a) and endothelial cells (Figure 4e), and less markedly in epithelial cells (Figure 4f).
Figure 4.
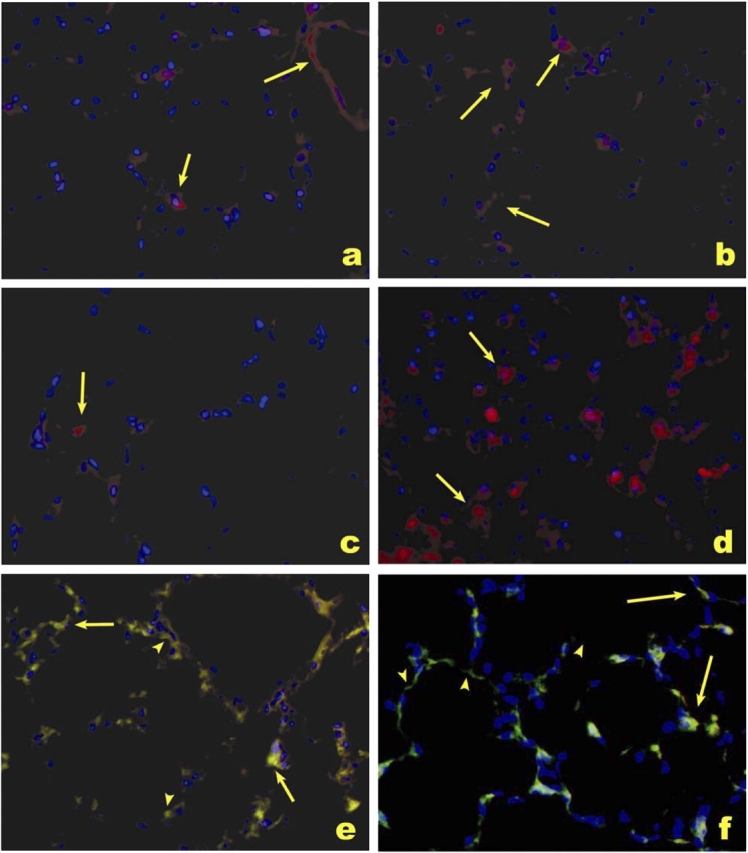
Immunolocalization of iNOS in response to HVT. Alveolar localization of iNOS (red signal) in C57BL/6J mice treated with HVT (a and b), sham ventilation (c), and LPS used as a positive control (d). Note the enhanced expression of iNOS in a pulmonary vessel (a, arrow in right upper corner) and along alveolar septa (a and b, arrows), as compared with the normal lung expression pattern (c). There was colocalization of iNOS expression (red signal) with endothelial cells (CD34, green signal; e) and type II epithelial cells (surfactant protein C, green signal; f) in lungs treated with HVT (e and f). Most iNOS expression was localized in alveolar capillary endothelial cells (e, arrows, yellow signal), with less prominent expression in type II epithelial cells (f, arrows, yellow signal). Lack of expression of iNOS in some endothelial cells (e) or type II cells (f) is highlighted by arrowheads. a–f were counterstained with Dapi (blue signal) for nuclear localization.
Effect of MV on eNOS Protein Expression
The effect of HVt MV on eNOS expression was also assessed in C57BL/6J and iNOS−/− mice. As expected, eNOS protein was constitutively expressed in spontaneously breathing (sham-treated) C57BL/6J and iNOS−/− mice (Figure 5). In comparison, this expression was significantly decreased in C57BL/6J mice, but relatively unchanged in iNOS−/− mice, in response to HVt (Figure 5).
Figure 5.

Effect of MV on endothelial NOS (eNOS) protein expression. Upper panel: Immunoblots were prepared from extracts of lungs of C57BL/6J (WT) and iNOS−/− mice breathing spontaneously (controls) or exposed to MV with HVT (20 ml/kg) as described in METHODS. Lower panel: Densitometric assessment of eNOS/actin protein expression (n ⩾ 3 for each condition). eNOS protein was constitutively expressed in WT and iNOS−/− mice breathing spontaneously. MV with HVT caused a significant decrease in eNOS expression in WT mice and a small, nonsignificant increase in iNOS−/− mice.
Increased iNOS Activity by MV
We also examined lung eNOS and iNOS activities in C57BL/6J and iNOS−/− mice exposed to HVt MV. As shown in Figure 6A, there was a significant increase in lung iNOS activity in C57BL/6J mice in response to HVt as compared with spontaneously breathing control animals. As expected, there was no significant Ca2+-independent lung NOS activity in iNOS−/− mice (Figure 6A) and no change in response to HVt. However, eNOS activity tended to decrease in C57BL/6J mice, but increased significantly in iNOS−/− mice in response to HVt, as compared with control counterparts (Figure 6B).
Figure 6.


Changes in lung NOS activity in response to MV. (A) iNOS activity. There was no detectable iNOS activity in lungs of iNOS−/− mice breathing spontaneously (controls) or exposed to HVT. In contrast, there was minimal iNOS activity in control WT mice, with a significant increase in response to HVT (n ⩾ 4 for each condition). *p < 0.05 versus control animals. (B) eNOS activity. At baseline (spontaneous breathing), lungs of both WT and iNOS−/− mice displayed activity. HVT resulted in a mild decrease and a significant increase in that activity in WT and iNOS−/− mice, respectively. *p < 0.05 versus control animals.
Deficiency of iNOS Protects against Ventilator-induced VILI
We assessed lung injury (by histology and BAL protein and cell count) and pulmonary vascular leakage (by the EBA technique) in a separate group of C57BL/6J and iNOS−/− mice exposed to MV.
There were no striking histologic changes in lungs of C57BL/6J mice exposed to LVt as compared with control animals (results not shown). However, HVt caused ALI as suggested by parenchymal cell infiltration as well as hemorrhage in C57BL/6J mice, findings that were minimal in iNOS−/− mice (Figures 7A and 7B). In addition, there was a significant increase in BAL total cell count in C57BL/6J mice in response to HVt (50 ± 5 vs. 36.4 ± 4 cells/ml of BAL fluid in HVt compared with the sham group, respectively; p < 0.05), but no change in iNOS−/− mice (28.7 ± 3 vs. 30.4 ± 4 cells/ml in HVt compared with the sham group, respectively). Similarly, the number of neutrophils present in the lung parenchyma was significantly increased in C57BL/6J mice, but unchanged in iNOS−/− mice, in response to HVt exposure (Figure 8). Furthermore, there was accumulation of nitrotyrosine product (as assessed by immunohistochemistry [Figure 7C] and digital counting of the immunostaining images [Figure 9]) in the lungs of C57BL/6J, but not in iNOS−/− mice, in response to HVt. As shown in Figure 10, accumulation of nitrotyrosine was predominant in endothelial and epithelial cells, which were sites of increased iNOS expression (Figure 4). However, there was no difference in BAL protein in C57BL/6J and iNOS−/− mice in response to HVt (Figure 11). Finally, EBA accumulation in the lungs of C57BL/6J mice exposed to LVt was trivial, but increased significantly in response to HVt. In sharp contrast, HVt caused no significant EBA leakage in iNOS−/− mice (Figure 12).
Figure 7.

Histologic assessment of the effect of HVT on lung inflammation and injury. (A) Hematoxylin and eosin stain: histologic analysis of lung tissue (40×) obtained from representative WT and iNOS−/− (KO) control mice breathing spontaneously (sham) demonstrated preserved lung parenchymal architecture (a and b). In contrast, exposure of WT mice to HVT for 2 hours (c) produced neutrophil infiltration and occasional alveolar hemorrhage (arrow). These features were absent in iNOS−/− mice exposed to HVT (d). The scale bar represents 25 μm. (B) Neutrophil immunostaining: the presence of parenchymal neutrophils was assessed using a specific antineutrophil antibody. There were occasional neutrophils in the lungs of WT and iNOS−/− control mice breathing spontaneously (arrows, e and f). Exposure to HVT produced an increase in interstitial and alveolar neutrophils in WT mice (g) but not in iNOS−/− mice (h). (C) Accumulation of lung nitrotyrosine product in response to MV: representative paraffin-embedded lungs from C57BL/6J (WT) and iNOS−/− mice were incubated with monoclonal antinitrotyrosine antibody for immunohistochemistry. There was no evidence of nitrotyrosine product in the lungs of WT and iNOS−/− control mice breathing spontaneously (i and j). Exposure to HVT produced accumulation of nitrotyrosine product, which was more pronounced in WT mice (k) as compared with iNOS−/− mice (l). Additional representative panels for nitrotyrosine immunostains for WT and iNOS−/− mice are shown in the online supplement. m represents a negative control (lung from LPS-treated animal stained with IgG alone), and n represents a positive control (lung from LPS-treated animal stained with antinitrotyrosine antibody and IgG).
Figure 8.

Assessment of neutrophil infiltration in lung parenchyma of C57BL/6J (WT) and iNOS−/− mice exposed to HVT. There was a significant increase in lung neutrophils in C57BL/6J (WT), but not in iNOS−/− mice, after exposure to HVT. Data represent means ± SEM of an average of 10 high-power fields for each exposure group. *p < 0.05 versus sham.
Figure 9.

Assessment of nitrotyrosine immunostaining in lung parenchyma of C57BL/6J (WT) and iNOS−/− mice exposed to HVT. Average was obtained through digital imaging of at least 20 fields/group (Nikon Eclipse E800 microscope with Pro+4.51 software; Nikon Instech Co., Kanagawa, Japan). *p < 0.05 versus sham.
Figure 10.

Immunolocalization of nitrotyrosine in response to HVT. Colocalization of nitrotyrosine (red signal, a–f), the endothelial cell marker CD34 (green signal; a, c, and e), and type II epithelial cell marker surfactant protein C (SPC; green signal; b, d, and f) in C57BL/6J exposed to HVT (a and b), LPS (c and d), and in control, untreated mouse lungs (e and f). Note the preferential colocalization of nitrotyrosine and CD34 in lungs from mice exposed to HVT (yellow signal, a) as compared with expression of nitrotyrosine in type II, SPC-positive cells (b). There was more nitrotyrosine accumulation in lungs of mice exposed to HVT (a and b) as compared with room-air control animals (e–f). Lungs of LPS-treated mice displayed intense nitrotyrosine expression, mostly present in septal space and presumably septal neutrophils (c–d).
Figure 11.

Effect of HVT on bronchoalveolar lavage (BAL) protein in C57BL/6J (WT) and iNOS−/− mice. BAL fluid was obtained at the end of a 2-hour exposure to HVT, and protein was measured. There was no effect of HVT on BAL protein in C57BL/6J (WT) and iNOS−/− mice. n = 8–10 mice/group.
Figure 12.

Effect of MV on pulmonary vascular leakage. Pulmonary vascular leakage was assessed by the extravasation of Evans blue dye albumin (EBA) into lung parenchyma 2 hours after MV. MV with LVT caused no significant extravasation of EBD in C57BL/6J mice (WT). In contrast, HVT caused a 50% increase in EBA lung accumulation in WT but no change in iNOS−/− mice. n = 5–6 mice/experimental condition. *p < 0.05 versus control animals; †p < 0.05 versus HVT in WT mice.
To confirm the pathogenic role of iNOS in capillary permeability in this model, C57BL/6J and iNOS−/− mice were exposed to MV with HVt with or without pretreatment with AG, a specific chemical inhibitor of iNOS. AG treatment had no effect on EBA lung accumulation in control, nonventilated, wild-type or iNOS-deficient mice (results not shown); however, it completely abrogated increased EBA lung accumulation in wild-type C57BL/6J mice in response to MV without affecting EBA accumulation in iNOS−/− mice. The latter mice were again protected from increased capillary permeability (Figure 13), as previously demonstrated.
Figure 13.

Effect of aminoguanidine (AG) treatment on MV-induced pulmonary vascular leakage. C57BL/6J and iNOS−/− mice were pretreated with AG (15 mg/kg, given intraperitoneally), a selective iNOS inhibitor, 1 hour before exposure to MV with HVT. AG treatment significantly prevented EBA lung accumulation in C57BL/6J mice (WT) produced by MV. n = 5–6 mice/experimental condition. *p < 0.05 versus sham; †p < 0.05 versus HVT in WT mice.
DISCUSSION
The consequences of NO release after induction of iNOS, an enzyme generally expressed under pathologic conditions, have been implicated in the pathogenesis of several diseases, including asthma, ALI, and circulatory shock. Although iNOS is generally believed to be induced by cytokines and inflammatory products, there have been recent reports of regulation of this enzyme by mechanical forces. For example, iNOS expression is increased in systemic smooth muscle cells exposed to shear stress (24) and in osteoblasts in response to mechanical loading (25). Similarly, eNOS expression is increased in bovine aortic endothelial cells in response to oscillatory shear stress (26) and in pulmonary vascular cells in response to circumferential stretch (27). On the basis of these findings, we determined whether cyclic mechanical stress induced by MV could alter the expression of lung iNOS and eNOS, and whether iNOS expression contributed to the development of capillary permeability related to VILI.
Our study indicates that iNOS mRNA and protein expression is significantly increased with 2 hours of MV, correlates with the degree of alveolar distension (Vt), and is accompanied by a significant increase in iNOS activity in C57BL/6J mice. eNOS protein expression was decreased in wild-type animals and relatively unchanged in iNOS−/− mice. However, there was a significant increase in eNOS activity in iNOS−/− mice in response to HVt. We speculate that this upregulation may be related to a potential direct effect of mechanical stress on eNOS expression, the lack of iNOS-derived NO, and/or the relative absence of nitrosative stress in the iNOS−/− mice. Indeed, changes in cell redox status (e.g., oxidative or nitrosative stress) have been reported as a mechanism of downregulation of eNOS activity through denitrosylation of eNOS (28) and therefore may explain a lack of similar increase in eNOS activity in the C57BL/6J mice in response to HVt.
MV with a Vt of 20 ml/kg produced ALI in wild-type animals as assessed by histologic changes, increased total cell count in BAL, parenchymal infiltration of neutrophils, evidence of nitrosative stress (accumulation of nitrotyrosine product), and increased capillary leakage. However, there was no evidence of epithelial injury (as reflected by a lack of increase in BAL protein), which is not surprising considering that efflux of protein into the alveolar space is significantly delayed (i.e., several hours) in relationship to increased capillary permeability, as demonstrated by Quinn and coworkers (29) in a rat model of MV using Vts similar to those used in the present study. There was overall less pulmonary injury (as reflected by a more modest increase in BAL cells and extravasation of EBA dye) in the present model as compared with a model of endotoxin inhalation (16). However, all changes were significantly attenuated in iNOS−/− mice, and pulmonary capillary leakage was prevented in these mice and in wild-type mice pretreated with the iNOS inhibitor AG, strongly suggesting that the increase in iNOS expression in C57BL/6J mice contributes to lung inflammation, nitrosative stress, and capillary leakage in this model.
The role of NOS in VILI has been the subject of previous investigations. A recent report by Frank and colleagues (11) demonstrated an increase in lung iNOS protein expression and BAL nitrite and nitrate levels in rats ventilated with HVt (30 ml/kg) for 1 hour followed by LVt (7 ml/kg) for 2 hours. Furthermore, inhibition of iNOS activity prevented the decrease in airspace fluid clearance caused by MV. In contrast, Choi and colleagues (12) demonstrated an increase in eNOS protein expression in lungs and kidneys in a rat model of HVt (20 ml/kg for 2 hours), however, with no detectable iNOS expression. Treatment of the animals with L-NAME, a nonspecific inhibitor of NOS, significantly attenuated the microvascular leak of lung and kidney and the proteinuria produced by HVt ventilation, which led the investigators to incriminate eNOS as the mediator of injury. Hammerschmidt and coworkers (30) also reported an increase in BAL nitrite levels but failed to detect any change in iNOS and eNOS mRNA expression in response to HVt MV in isolated rabbit lungs. A study by Broccard and colleagues (31), also in isolated perfused rabbit lungs, demonstrated a positive correlation between NO metabolites in BAL and lung vascular permeability. Finally, a recent study by Lang and coworkers (32), in a rabbit model of LPS-induced lung injury, suggests that hypercapnia (produced by changes in ventilator settings) increases lung iNOS expression and amplifies LPS-induced pulmonary inflammatory responses. In none of the above studies was specific NOS activity ever measured. Despite some methodologic differences, the present study is consistent with that of Frank and colleagues (11). However, in addition to finding increased iNOS protein expression, we also correlated overall changes in iNOS gene expression and activity to nitrosative stress and capillary permeability. Furthermore, the lack of lung injury, nitrosative stress, or increased capillary permeability in the iNOS-deficient mice strongly suggests a direct role of this NOS isoform in VILI.
In relation to the mechanisms of injury, it is unlikely that NO itself is directly responsible for the development of nitrosative damage. NO diffuses quite freely across membranes and its fate is only limited when it encounters hemoglobin, a target enzyme, such as smooth muscle guanylate cyclase (3), or less likely, thiol groups to form nitrosothiols (which results in spatial confinement of NO as suggested by Lancaster and Gaston [33]) through a tightly regulated cellular process. Different redox forms of NO, such as nitrosonium ion and nitroxyl anion, rather than NO itself, have been considered potential effectors of NOS (34). In addition, the fate of the NO molecule will also be limited in the presence of superoxide production (because the reaction of superoxide with NO is much greater than that of superoxide with its scavenger superoxide dismutase), and peroxynitrite will be formed (3). We have recently demonstrated that the superoxide-producing enzyme xanthine oxidoreductase is activated by mechanical stress in endothelial cells in vitro (cells subjected to cyclic stretch) and in vivo after exposure of mice to MV (35). Therefore, we speculate that lung damage and the resulting capillary permeability demonstrated in the present model are related to iNOS-derived NO reacting with superoxide (from activation of xanthine oxidoreductase or other lung oxidases) to form peroxynitrite in components of the alveolar–capillary membrane. The presence of lung nitrotyrosine (a footprint of peroxynitrite damage) at the site of iNOS increased expression in response to MV supports our contention that iNOS-derived NO is spatially confined, presumably by the local production of superoxide in response to mechanical stress.
Regarding potential lung toxicity mediated by NO and its end-products, it is noteworthy that inhaled NO has been used safely and with little evidence of overall toxicity in patients with acute respiratory distress syndrome (ARDS) and patients with pulmonary hypertension. However, there is evidence that inhaled NO causes some nitrotyrosine formation in animal models (32, 36) as well as in humans (37). Lamb and coworkers (37) demonstrated that patients with ARDS receiving inhaled NO had increased levels of 3-nitrotyrosine and 3-chlorotyrosine compared with similar patients not receiving inhaled NO. Formation of 3-nitrotyrosine and 3-chlorotyrosine are presumably mediated by peroxynitrite and possibly interaction between nitrite and hypochlorous acid, respectively. On the basis of these studies, we speculate that the beneficial effect or effects (e.g., improvement in oxygenation) of inhaled NO in patients with ARDS may be mitigated by potential toxicity of this agent, such as formation of nitrotyrosine residues in lung. We further surmise that the net effect of NO (whether from inhaled therapy or cellular source, such as shown in the present study) in the lung depends on a critical balance between NO and superoxide formation, the site(s) and sources of NO release, and associated conditions (e.g., concomitant localized production of superoxide by activated oxidases).
The mechanisms of iNOS upregulation in response to MV were not investigated and are beyond the scope of this study. However, several possibilities can be invoked. Type 2 NOS is generally induced by proinflammatory factors, such as interleukins (IL-1, IL-6, and IL-8), TNF-α, and γ-IFN. Animal (8) and human (6) studies have now shown that some of these cytokines are released in BAL and plasma in response to MV, and that a protective lung strategy (i.e., LVt) can prevent this inflammatory response (5, 8). Chemokine and cytokine release may be secondary to activation of nuclear factor–κB (38), a transcriptional factor also known to induce iNOS (39). To identify mechanical stress–induced candidate genes using schematic representation of cross-species (rat, murine, canine, and human) ortholog database and gene ontology processes, we recently identified inflammatory response ontologies in experimental MV and cell-stretch models. Ontology patterns were heavily represented by several cytokines (e.g., IL-1β and IL-6), cytokine receptors (IL-1 and IL-8 receptors), and chemokines (chemoattractant protein [MCP]-1 and macrophage inflammatory protein [MIP]-2α) in these models (40). Using microarray analysis to search for differentially expressed genes in lungs of C57BL/6J mice exposed to 2 hours of MV, we confirmed a dramatic increase in certain cytokines (twofold increase in IL-1β, TNF-α, and γ-IFN, and > 50-fold increase in IL-6) and a twofold increase in iNOS in the present MV model (results not shown). However, we were unable in the present study to detect IL-6, TNF-α, and γ-IFN, even after concentration of the BAL fluid (results not shown), which does not rule out a local release of these cytokines as a mechanism of iNOS induction. Taken together, these results suggest an inflammatory response as the underlying process in iNOS gene activation in response to MV.
Although an inflammatory response is potentially responsible for the induction of iNOS, a direct effect of mechanical stress cannot be excluded. There is overwhelming evidence linking cytoskeletal integrity to gene regulation, and a growing interest in iNOS regulation by the cytoskeleton (41). Disruption of the actin cytoskeleton upregulates iNOS expression in vascular smooth muscle cells (42). Inhibition of Rho proteins (important components of the organization of the cytoskeleton) also upregulates iNOS in epithelial cells through transcriptional and post-transcriptional events (43). Therefore, because mechanical stress involves endothelial cytoskeletal reorganization and activation of specific signaling pathways (44), we postulate that one potential mechanism for iNOS upregulation in our model is a direct effect of mechanical stress on the cytoskeleton, a possibility currently being tested in our laboratory using cultured pulmonary microvascular endothelial cells subjected to cyclic stretch.
Nitric oxide end-products (i.e., nitrite, nitrate, and nitrotyrosine) have been found in BAL of patients with trauma or suspected sepsis, either at risk for or with ARDS, and have been related to increased mortality (45). However, although all the patients in this study were on MV, they also had trauma or suspected sepsis. Our study would suggest that MV alone (without any additional trigger of the inflammatory cascade) can upregulate iNOS, thus contributing to formation of NO end-products and nitrosative stress. These changes correlate with increased Vt. However, we cannot rule out the possibility of pressure as a stimulus for iNOS induction in view of the significant increase in peak airway pressure in response to HVt ventilation. Nevertheless, it is more likely that volutrauma rather than barotrauma was responsible for the injury itself, as suggested by work by Dreyfuss and colleagues (46), using high-pressure/LVt (as provided by thoracoabdominal banding of ventilated rats) and low-pressure/HVt MV, in which lung injury was solely related to changes in lung volume (volutrauma) and not airway pressure (barotrauma). HVt (i.e., 20 ml/kg) was higher in this study compared with our previous study (i.e., 17 ml/kg) demonstrating lung injury in a similar model (10), but significantly lower compared with volumes (i.e., 30 ml/kg) reported in the study by Frank and colleagues (11) in rats.
Although we did not measure neuronal NOS, it is clear that the source of increased NO output in the present study was iNOS and not eNOS (at least for the C57BL/6J mice), as indicated by the changes in mRNA, protein, and respective NOS activities. We were able to identify several potential sources of iNOS by immunohistochemical staining. There was an increase in iNOS immunostaining involving predominantly endothelial but also epithelial cells. We have previously demonstrated that iNOS can be induced by cytokines and hypoxia in cultured pulmonary microvascular endothelial cells, and have postulated a role for this cellular source in endothelial damage (and potential capillary permeability) in inflammatory conditions such as sepsis and ARDS (14). We now postulate that these cells can be a significant source of iNOS-derived NO in response to mechanical stress. In that respect, the cellular source of iNOS in our model differs from that of a sepsis-induced ALI after cecal ligation and perforation described by Wang and coworkers (47) in which the authors studied iNOS+/+, iNOS−/−, and two reciprocally bone marrow–transplanted iNOS chimeric mice groups (iNOS+/+ donor bone marrow transplanted into iNOS−/− recipient mice and vice-versa) to demonstrate that inflammatory cells accounted for increased iNOS activity and injury. However, previous work from the same group using a similar approach of bone marrow–transplanted chimeric mice incriminated parenchymal cells (similar to the present study) as the predominant source of iNOS activity in LPS-induced lung injury (48). Taken together, these studies suggest that the specific source (inflammatory vs. lung parenchymal cells) of iNOS/nitrosative stress in ALI may vary according to the stimulus and mechanism of injury.
Mechanical stress–induced capillary permeability has been related to endothelial and epithelial plasma membrane breaks (46, 49). However, it is increasingly evident that, aside from mechanical disruption of the alveolar–capillary barrier, certain signaling pathways play a predominant role in the initiation of events leading to formation of intercellular gaps. For instance, inhibitors of myosin light-chain kinase and phosphodiesterase reduce the increased capillary coefficient filtration in animal models of MV-induced injury (50). Other signaling pathways altered by MV include tyrosine phosphorylation, focal adhesion formation (51), and activation of mitogen-activated protein kinase (7). Central to all these pathways is alteration in the cytoskeleton. In that respect, it is worth noting that peroxynitrite causes endothelial barrier dysfunction (52). The exact mechanisms involved in endothelial barrier disruption by NO end-products remain to be deciphered and will be best addressed with a combination of in vitro and in vivo studies.
In summary, we have shown that mechanical ventilation upregulates lung iNOS gene expression and activity in a mouse model of VILI. In addition, iNOS most likely contributes to ventilator-induced pulmonary capillary leakage through lung nitrosative stress.
Supplementary Material
Acknowledgments
None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
Supported by awards from the National Heart, Lung, and Blood Institute (NIH R01 HL049441 and P50 HL 73994).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
References
- 1.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 1993;329:2002–2012. [DOI] [PubMed] [Google Scholar]
- 2.Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Ding A, Troso T, Nathan C. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science 1992;256:225–228. [DOI] [PubMed] [Google Scholar]
- 3.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 1996;271:C1424–C1437. [DOI] [PubMed] [Google Scholar]
- 4.Kristof AS, Goldberg P, Laubach V, Hussain SN. Role of inducible nitric oxide synthase in endotoxin-induced acute lung injury. Am J Respir Crit Care Med 1998;158:1883–1889. [DOI] [PubMed] [Google Scholar]
- 5.Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000;342:1301–1308. [DOI] [PubMed] [Google Scholar]
- 6.Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 1999;282:54–61. [DOI] [PubMed] [Google Scholar]
- 7.Li LF, Yu L, Quinn DA. Ventilation-induced neutrophil infiltration depends on c-Jun N-terminal kinase. Am J Respir Crit Care Med 2004;169:518–524. [DOI] [PubMed] [Google Scholar]
- 8.Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest 1997;99:944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med 1999;160:109–116. [DOI] [PubMed] [Google Scholar]
- 10.McVerry BJ, Peng X, Hassoun PM, Sammani S, Simon BA, Garcia JG. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med 2004;170:987–993. [DOI] [PubMed] [Google Scholar]
- 11.Frank JA, Pittet JF, Lee H, Godzich M, Matthay MA. High tidal volume ventilation induces NOS2 and impairs cAMP-dependent air space fluid clearance. Am J Physiol Lung Cell Mol Physiol 2003;284:L791–L798. [DOI] [PubMed] [Google Scholar]
- 12.Choi WI, Quinn DA, Park KM, Moufarrej RK, Jafari B, Syrkina O, Bonventre JV, Hales CA. Systemic microvascular leak in an in vivo rat model of ventilator-induced lung injury. Am J Respir Crit Care Med 2003;167:1627–1632. [DOI] [PubMed] [Google Scholar]
- 13.Peng X, Birukov KG, Lavoie T, Ma SF, Garcia JG, Hassoun PM. Cyclic stretch upregulates inducible nitric oxide synthase in pulmonary endothelial cells [abstract]. Am J Respir Crit Care Med 2003;167:A117. [Google Scholar]
- 14.Zulueta JJ, Sawhney R, Kayyali U, Fogel M, Donaldson C, Huang H, Lanzillo JJ, Hassoun PM. Modulation of inducible nitric oxide synthase by hypoxia in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol 2002;26:22–30. [DOI] [PubMed] [Google Scholar]
- 15.Liao JK, Zulueta JJ, Yu FS, Peng HB, Cote CG, Hassoun PM. Regulation of bovine endothelial constitutive nitric oxide synthase by oxygen. J Clin Invest 1995;96:2661–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med 2004;169:1245–1251. [DOI] [PubMed] [Google Scholar]
- 17.Patterson CE, Rhoades RA, Garcia JG. Evans blue dye as a marker of albumin clearance in cultured endothelial monolayer and isolated lung. J Appl Physiol 1992;72:865–873. [DOI] [PubMed] [Google Scholar]
- 18.Tuder RM, Zhen L, Cho CY, Taraseviciene-Stewart L, Kasahara Y, Salvemini D, Voelkel NF, Flores SC. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol 2003;29:88–97. [DOI] [PubMed] [Google Scholar]
- 19.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 2000;106:1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA 1990;87:682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ungureanu-Longrois D, Balligand JL, Okada I, Simmons WW, Kobzik L, Lowenstein CJ, Kunkel SL, Michel T, Kelly RA, Smith TW. Contractile responsiveness of ventricular myocytes to isoproterenol is regulated by induction of nitric oxide synthase activity in cardiac microvascular endothelial cells in heterotypic primary culture. Circ Res 1995;77:486–493. [DOI] [PubMed] [Google Scholar]
- 22.Sade K, Schwartz I, Schwartz D, Wolman Y, Chernichovski T, Fireman E, Iaina A, Kivity S. Effect of montelukast pretreatment on inducible nitric oxide synthase mRNA expression in the lungs of antigen-challenged allergic mice. Clin Exp Allergy 2003;33:1741–1746. [DOI] [PubMed] [Google Scholar]
- 23.Okawa T, Asano K, Takahashi H, Hashimoto S, Anbe H, Sato A, Gafield RE. Expression of iNOS mRNA and inhibitory effect of NO on uterine contractile activity in rats are determined by local rather than systemic factors of pregnancy. J Pharmacol Sci 2004;95:349–354. [DOI] [PubMed] [Google Scholar]
- 24.Gosgnach W, Messika-Zeitoun D, Gonzalez W, Philipe M, Michel JB. Shear stress induces iNOS expression in cultured smooth muscle cells: role of oxidative stress. Am J Physiol Cell Physiol 2000;279:C1880–C1888. [DOI] [PubMed] [Google Scholar]
- 25.Watanuki M, Sakai A, Sakata T, Tsurukami H, Miwa M, Uchida Y, Watanabe K, Ikeda K, Nakamura T. Role of inducible nitric oxide synthase in skeletal adaptation to acute increases in mechanical loading. J Bone Miner Res 2002;17:1015–1025. [DOI] [PubMed] [Google Scholar]
- 26.Ziegler T, Silacci P, Harrison VJ, Hayoz D. Nitric oxide synthase expression in endothelial cells exposed to mechanical forces. Hypertension 1998;32:351–355. [DOI] [PubMed] [Google Scholar]
- 27.Kuebler WM, Uhlig U, Goldmann T, Schael G, Kerem A, Exner K, Martin C, Vollmer E, Uhlig S. Stretch activates nitric oxide production in pulmonary vascular endothelial cells in situ. Am J Respir Crit Care Med 2003;168:1391–1398. [DOI] [PubMed] [Google Scholar]
- 28.Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric oxide synthase in vascular endothelial cells. J Biol Chem 2005;280:19884–19894. [DOI] [PubMed] [Google Scholar]
- 29.Quinn DA, Moufarrej RK, Volokhov A, Hales CA. Interactions of lung stretch, hyperoxia, and MIP-2 production in ventilator-induced lung injury. J Appl Physiol 2002;93:517–525. [DOI] [PubMed] [Google Scholar]
- 30.Hammerschmidt S, Schiller J, Kuhn H, Meybaum M, Gessner C, Sandvoss T, Arnold K, Wirtz H. Influence of tidal volume on pulmonary NO release, tissue lipid peroxidation and surfactant phospholipids. Biochim Biophys Acta 2003;1639:17–26. [DOI] [PubMed] [Google Scholar]
- 31.Broccard AF, Feihl F, Vannay C, Markert M, Hotchkiss J, Schaller MD. Effects of L-NAME and inhaled nitric oxide on ventilator-induced lung injury in isolated, perfused rabbit lungs. Crit Care Med 2004;32:1872–1878. [DOI] [PubMed] [Google Scholar]
- 32.Lang JD, Figueroa M, Sanders KD, Aslan M, Liu Y, Chumley P, Freeman BA. Hypercapnia via reduced rate and tidal volume contributes to lipopolysaccharide-induced lung injury. Am J Respir Crit Care Med 2005;171:147–157. [DOI] [PubMed] [Google Scholar]
- 33.Lancaster JR Jr, Gaston B. NO and nitrosothiols: spatial confinement and free diffusion. Am J Physiol Lung Cell Mol Physiol 2004;287:L465–L466. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt HH, Hofmann H, Schindler U, Shutenko ZS, Cunningham DD, Feelisch M. No .NO from NO synthase. Proc Natl Acad Sci USA 1996;93:14492–14497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdulnour RE, Peng X, Han EJ, Hasan EJ, Kayyali U, Garcia JG, Hassoun PM. Mechanical stress upregulates xanthine oxidoreductase through a p38 MAP kinase-dependent pathway [abstract]. Am J Respir Cell Mol Biol 2005;2:A717. [Google Scholar]
- 36.Weinberger B, Fakhrzadeh L, Heck DE, Laskin JD, Gardner CR, Laskin DL. Inhaled nitric oxide primes lung macrophages to produce reactive oxygen and nitrogen intermediates. Am J Respir Crit Care Med 1998;158:931–938. [DOI] [PubMed] [Google Scholar]
- 37.Lamb NJ, Quinlan GJ, Westerman ST, Gutteridge JM, Evans TW. Nitration of proteins in bronchoalveolar lavage fluid from patients with acute respiratory distress syndrome receiving inhaled nitric oxide. Am J Respir Crit Care Med 1999;160:1031–1034. [DOI] [PubMed] [Google Scholar]
- 38.Held HD, Boettcher S, Hamann L, Uhlig S. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med 2001;163:711–716. [DOI] [PubMed] [Google Scholar]
- 39.Marks-Konczalik J, Chu SC, Moss J. Cytokine-mediated transcriptional induction of the human inducible nitric oxide synthase gene requires both activator protein 1 and nuclear factor kappaB-binding sites. J Biol Chem 1998;273:22201–22208. [DOI] [PubMed] [Google Scholar]
- 40.Grigoryev DN, Finigan JH, Hassoun P, Garcia JG. Science review: searching for gene candidates in acute lung injury. Crit Care 2004;8:440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marczin N, Jilling T, Papapetropoulos A, Go C, Catravas JD. Cytoskeleton-dependent activation of the inducible nitric oxide synthase in cultured aortic smooth muscle cells. Br J Pharmacol 1996;118:1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hattori Y, Kasai K. Disruption of the actin cytoskeleton up-regulates iNOS expression in vascular smooth muscle cells. J Cardiovasc Pharmacol 2004;43:209–213. [DOI] [PubMed] [Google Scholar]
- 43.Witteck A, Yao Y, Fechir M, Forstermann U, Kleinert H. Rho protein-mediated changes in the structure of the actin cytoskeleton regulate human inducible NO synthase gene expression. Exp Cell Res 2003;287:106–115. [DOI] [PubMed] [Google Scholar]
- 44.Birukov KG, Jacobson JR, Flores AA, Ye SQ, Birukova AA, Verin AD, Garcia JG. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am J Physiol Lung Cell Mol Physiol 2003;285:L785–L797. [DOI] [PubMed] [Google Scholar]
- 45.Sittipunt C, Steinberg KP, Ruzinski JT, Myles C, Zhu S, Goodman RB, Hudson LD, Matalon S, Martin TR. Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;163:503–510. [DOI] [PubMed] [Google Scholar]
- 46.Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema: respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 1988;137:1159–1164. [DOI] [PubMed] [Google Scholar]
- 47.Wang LF, Patel M, Razavi HM, Weicker S, Joseph MG, McCormack DG, Mehta S. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am J Respir Crit Care Med 2002;165(12):1634–1639. [DOI] [PubMed] [Google Scholar]
- 48.Wang LF, Mehta S, Weicker S, Scott JA, Joseph M, Razavi HM, McCormack DG. Relative contribution of hemopoietic and pulmonary parenchymal cells to lung inducible nitric oxide synthase (iNOS) activity in murine endotoxemia. Biochem Biophys Res Commun 2001;283:694–699. [DOI] [PubMed] [Google Scholar]
- 49.West JB, Tsukimoto K, Mathieu-Costello O, Prediletto R. Stress failure in pulmonary capillaries. J Appl Physiol 1991;70:1731–1742. [DOI] [PubMed] [Google Scholar]
- 50.Parker JC. Inhibitors of myosin light chain kinase and phosphodiesterase reduce ventilator-induced lung injury. J Appl Physiol 2000;89:2241–2248. [DOI] [PubMed] [Google Scholar]
- 51.Bhattacharya S, Sen N, Yiming MT, Patel R, Parthasarathi K, Quadri S, Issekutz AC, Bhattacharya J. High tidal volume ventilation induces proinflammatory signaling in rat lung endothelium. Am J Respir Cell Mol Biol 2003;28:218–224. [DOI] [PubMed] [Google Scholar]
- 52.Knepler JL Jr, Taher LN, Gupta MP, Patterson C, Pavalko F, Ober MD, Hart CM. Peroxynitrite causes endothelial cell monolayer barrier dysfunction. Am J Physiol Cell Physiol 2001;281:C1064–C1075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.