

Summary
The SV40 outer shell is composed of 72 pentamers of VP1. The core of the VP1 monomer is a β-barrel with jelly-roll topology, with extending N and C-terminal arms. A pentapeptide hinge, KNPYP, tethers the C-arm to the VP1 β-barrel core. The five C-arms that extend from each pentamer insert into the neighbouring pentamers, tying them together through different types of interactions. In the mature virion, this element adopts either of 6 conformations, according to their location in the capsid. We found that the hinge is conserved among 16 members of the polyomaviridae, attesting to its importance in capsid assembly and/or structure. We have used site-directed mutagenesis in order to gain an understanding into the structural requirements of this element: Y299 was changed to A, F and T and P300 to A and G. The mutants showed reduction in viability to varying degrees. Unexpectedly, assembly was only reduced to a small extent. Interestingly the data showed that the mutants were highly unstable. The largest effect was observed for mutations of P300, indicating a role of the proline in the virion structure. P300G was more unstable than P300A, indicating requirement for rigidity of the pentapeptide hinge. Y299T and Y299A were more defective in viability than Y299F, highlighting the importance of an aromatic ring at this position. Structural inspection showed that this aromatic ring contacts C-arms of neighbouring pentamers. Computational modelling predicted loss of stability of the Y mutants in concordance with the experimental results. This study provides insights into the structural details of the pentapeptide hinge that are responsible for capsid stability.
Keywords: SV40, major capsid protein VP1, site-directed mutagenesis, computational binding prediction, protein structure-function
Introduction
Viral capsids have been designed by long evolutionary processes to have a robust structure capable of protecting the viral genome in harsh external environment. In addition, viral capsids must recognize surface receptors of potential hosts and facilitate cellular entry and disassembly. Moreover, the capsid proteins contain the information for self-assembly, for encapsidation of the viral genome and for cell recognition during infection. Selection pressure for a minimal viral genome dictates that the capsid is built of small identical subunits.1 Thus changes in the amino acid sequence of the subunit will be multiplied many fold, and may affect any of the capsid properties.
SV40 is a member of polyomaviridae family. The SV40 structure has been solved at 3.1 Å resolution.2 The viral capsid, surrounding the viral minichromosome, is a T=7d icosahedral lattice, ~45 nm in diameter. It is composed of three viral-encoded proteins, VP1, VP2, and VP3. VP1 forms the outer shell while VP2 and VP3 bridge between the VP1 shell and the chromatin core. The VP1 monomers are tightly bound in pentamers through interdigitating β-strands.2 The pentamers are readily formed in the cytoplasm following mRNA translation via transitory S-S interactions.3 A single molecule of VP2 or VP3 is tightly attached to each pentamer at its inward facing cavity, through a region close to the C-terminus of VP2/3.4; 5 These and other findings indicate that VP15VP2/3 is the building block for SV40 capsid assembly. VP1 has a jelly-roll β-barrel structure,2; 6 with extending N- and C-terminal arm. The N-terminal arm carries the DNA-binding domain.7
The icosahedral capsid of members of the polyomaviridae family is built of 72 identical VP1 pentamers.8 12 of those are surrounded by 5 pentamers each (‘pentavalent pentamers’) and the other 60 by 6 pentamers each (‘hexavalent pentamers’). The puzzle how the variability in contacts between the identical building blocks is achieved has been solved by the elegant X-ray crystal structure study, which has shown that the pentamers are tied together via the long flexible C-arms.6 The C-arm, 60 amino acids long, extends from the β-barrel core and inserts into a monomer in a neighbouring pentamer forming three distinct kinds of interpentameric interactions. The C-arms assume 6 different conformations according to their position within the capsid. The respective monomers have been designated α, α′, α″, β, β′ and γ, where α are the monomers of the pentavalent pentamer (see Fig. 1 Top). Monomers forming interactions with the pentavalent pentamer are α′ and α″. β, β′ and γ are monomers that form interactions between hexavalent pentamers.2; 6
Fig. 1.
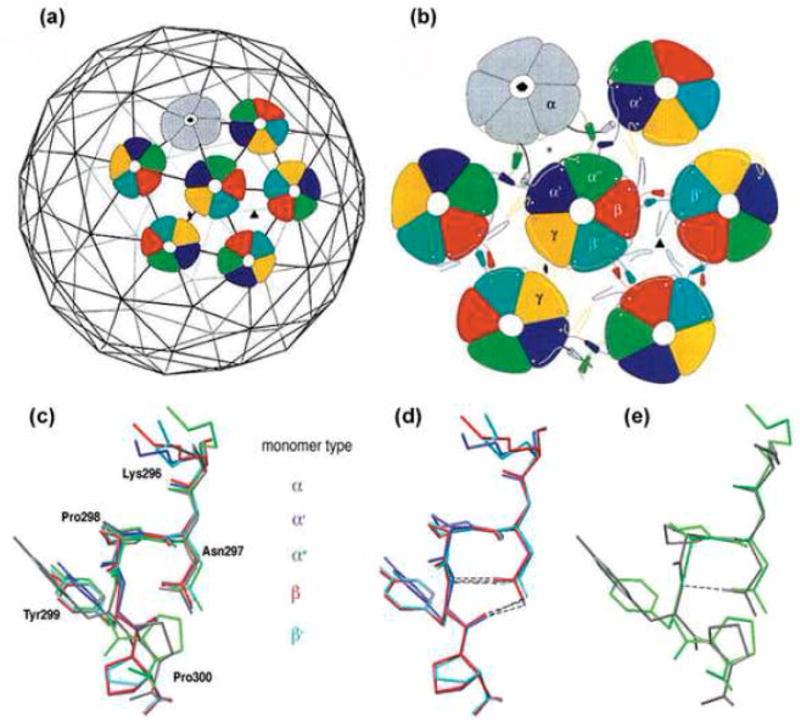
Top: Architecture of the virion shell (reprinted from2 with permission from Elsevier). (a) Arrangement of the pentavalent (grey) and hexavalent (coloured) pentamers on the T=7d icosahedral lattice. (b) Three distinct types of interactions between pentamers. α monomers (grey) of pentavalent pentamers and monomers, α′ and α″ of hexavalent pentamers (coloured) form a three-helix contact. The hexavalent pentamers interact through two-helix contact of monomers β- β′ and γ-γ. Bottom: The different conformations of the pentapeptide hinge. (c) Superimposition of the pentapeptide hinge of monomers α (grey), α′ (blue), α″ (green), β (red) and β′ (turquoise). Y299 and P300 have distinct orientations in the different monomers. The pentapeptide of γ monomers is not included, as its high B-factor indicates that it is less ordered.2 (d,e) The two well-ordered conformation groups. Superimposition of monomers β, α′ and β′ shows stabilization by two hydrogen bonds (d), while monomers α and α″ contain only one or no hydrogen bond, respectively (e). Superimpositions were prepared using Swiss-PDB-Viewer.23
The C-arms contain an α- helix (amino acids 301-312), except for the γ monomers. The α- helices of monomers α, α′ and α″ form an asymmetric three fold bundle that occupies the space between 3 pentamers by hydrophobic interactions. The helices of β, β′ and the corresponding region in γ- γ form two-fold tight hydrophobic contacts.2; 6
A pentapeptide hinge, KNPYP (amino acids 296-300), connects between the final β-strand of the jelly-roll core and the α-helix of the C-arm. This pentapeptide was termed ‘connector’ by Stehle.2 Since this term is used in viruses and bacteriophages for the element that participates in DNA packaging into pre-formed shells, we will refer to it here as a ‘hinge’. X-ray crystallography show that in the mature virion these residues adopt either of 6 conformations,2 which appear to dictate the conformations of the C-arms. Structural examination reveals that the hinge conformations diverge in particular in Y299 and P300 (Fig. 1 Bottom and9). To investigate the role of the hinge, we mutated amino acids Y299 and P300. Viability of the mutants was significantly reduced. We developed specific assays for assembly and stability, which, together with computational modelling and binding prediction allowed us to understand the role of these amino acids in the virus life cycle.
Results and Discussion
Conservation of the pentapeptide hinge
Comparison of 16 members of the polyomavirus family demonstrated that the 5 amino acids KNPYP of the pentapeptide hinge are almost fully conserved10 attesting to their importance in capsid structure and/or assembly. Only 3 of the 16 polyomaviruses show deviation from the KNPYP sequence. In 2 of the 3 only the lysine is changed: in BFDV and Wu viruses to arginine, conserving the positive charge, and in the POVMK virus to alanine. In addition, in the Wu virus, a novel polyomavirus lately discovered in respiratory secretions from human patients,11 P300 is changed to threonine.
As described in the introduction, the pentapeptide hinge assumes different conformations in the capsid. Superimposition of these conformers (Fig. 1 Bottom and9) demonstrates that amino acids Y299 and P300 are the most divergent. We therefore focused in the present study on these two residues. The pentapeptide of type γ monomers was found to be less ordered2 and has not been included in the present analyses.
Construction and expression of mutant VP1
We have mutated amino acids Y299 to F, T and A, and P300 to A and G. The plasmid pUC-SV40 was used for viral DNA propagation in E. coli and as a template for site directed mutagenesis. Mutagenesis was followed by back cloning into the original plasmid. The viral DNA was excised from the respective pUC-SV40 for studies in CV1 cells.
A critical question is whether the mutations affect protein expression or stability. Three days post-transfection, the level of the mutated VP1 proteins was determined by western analysis, in comparison to a WT control. Quantification of the results (Table 1) indicated that VP1 production and stability in the various mutants was reduced by less than 50% compared to the WT.
Table 1.
Analysis of the different mutants
| Virus | pfu/ml | Replication centres/ml | VP1 Productiona | Assemblya | Stability half lifeb(min) |
|---|---|---|---|---|---|
| SV40-WT | 1.2×109 (3) | 2.3×108 (9) | 100 | 100 (4) | 20.5 |
| SV40-P300A | 2.5×105 (2) | 7×106 (4) | 65 | 100 (2) | 5 |
| SV40-P300G | <102 (2) | 4.2×105 (4) | 63 | 67 (3) | 5 |
| SV40-Y299F | - | 2.8×107 (3) | 93 | 34 (1) | 14 |
| SV40-Y299T | - | 6.8×104 (3) | 60 | 22 (1) | 8.5 |
| SV40-Y299A | - | 4.5×102 (3) | 68 | 34 (1) | 6.5 |
| SV40-ΔC47 | <102 (2) | <102 | ND | 2 (1) | ND |
- calculated as % of WT
- at 37°C
The mutations reduce virus viability
Virions were prepared by transfection of CV1 cells with mutant virus DNA, excised from the respective pUC-SV40 plasmid and religated at low concentration (10μg/ml) to avoid multimer formation.
Virions were harvested by freeze-thaw 5 days later. Viability of the proline mutants was tested by the plaque assay. Table 1 shows that the titer of P300G was dramatically diminished (<102 pfu/ml, compared to 10 9 pfu/ml for the wild type). The P300A mutation had a milder effect, reducing the titer to 2.5× 105 pfu/ml (a reduction of 4 orders of magnitude compared to the wild type).
The plaque assay depends on multiple infection cycles, thus amplifying any effect on viability several fold. To obtain reliable estimates of the relative defect of the various mutants, we evaluated viability by scoring replication centres in the first infection cycle of the mutant virions, two days post infection. The replication-centres data for all the mutants (see Table 1) show that mutating P300 to A reduced viability by only 1–2 orders of magnitude, while viability of P300G was reduced by almost 3 orders of magnitude. Replication centres scores of Y299 mutants were spread over a wider range, in the order of F>T>A, suggesting that the aromatic ring is more important for Y299 function than the hydroxyl group.
Effects of the mutations on capsid assembly
Loss in viability might result from a defect in the assembly process, in capsid stability or in cell recognition and entry during infection. We developed an assay that tests for assembly. Mutant virions harvested by freeze-thaw were subjected to DNase I digestion, to remove non-assembled DNA. The amount of viral DNA that remained protected from DNAse I was quantified by RQ-PCR. We considered the possibility that DNA may be protected by VP1 molecules that are not assembled in capsid, due to its strong DNA-binding activity. To validate the method we have used a mutant VP1, SV40-ΔC47, with a 47 aa deletion of the C-terminal arm (R315 - Q361 at the end of the peptide chain) that cannot assemble. As the DNA binding domain is at the amino terminus of VP1,7 the deletion mutant in the C-arm retains its ability to bind DNA.12 The SV40-ΔC47 mutant scored as negative for assembly in our assay (Table 1). Similar results were obtained with and without DNAse I treatment before the RQ-PCR, indicating that the DNA molecules measured by this method were protected from DNase I digestion. The results (Table 1) show that capsid assembly was affected in all the mutants, except for P300A. However, assembly was reduced by less than an order of magnitude, not accounting for the dramatic effect in viability. These findings demonstrate that assembly per se is not greatly affected by the mutations, suggesting that the hinge assumes its specific conformation, and plays a critical role mostly after assembly.
Instability of the mutant virion particles
In an attempt to purify virion particles by equilibrium sedimentation in CsCl we observed that the mutants were highly unstable. As seen in Fig. 2, the WT virus peaked, as expected, at a density of 1.345 g/ml. On the other hand, P300G and Y299A showed only a small band at the expected density, with a large smear that extended towards the top of the gradient. Thus it appears that these mutants are falling apart in CsCl.
Fig. 2.
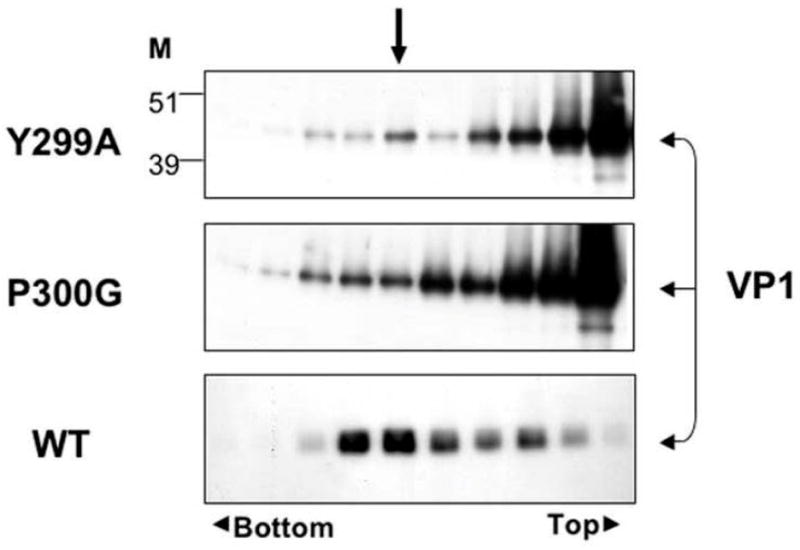
Instability of mutant capsids. Virions were harvested by di-detergent and centrifuged in CsCl density gradient. 250 microliter fractions were collected from the top. The refraction index of each fraction was measured and calculated for its density. Fractions were analyzed by western blot with anti-VP1 antibody. The arrow shows the fraction with density of 1.345 g/ml where full SV40 particles are expected.
To obtain a measure for mutant instability we have established a test based on the sensitivity of the packaged DNA to DNase I over time. Virion particles were prepared by freeze thaw method, and incubated with DNase I at 37°C and at 0°. The amount of SV40 DNA that remained protected at various time points was determined by RQ-PCR with primers spanning the polyA region. Experiments performed at 37°C (Fig. 3a) showed that the WT virus lost 80% of its DNA within 30 min (half life of 21 min), and stabilized at 20% of the original DNA amount. The unstable virus particles may represent immature virions that are sensitive to nucleases at 37°C as previously reported.13 The experiment further demonstrates that proline mutants were extremely unstable, with half-life of less than 5 min, and lost DNA protection almost completely within 10 min. The Y299 mutants showed intermediate DNase I protection, from Y299F > Y299T > Y299A, with a half-life of 14, 8.5 and 6.5 min, respectively (Table 1). Similar results were obtained when the RQ-PCR reaction was performed with another pair of primers, covering 470 bp within the VP1 encoding region (not shown). We concluded that the mutants were unstable either because of a distorted capsid structure or due to incomplete maturation (or both).
Fig. 3.
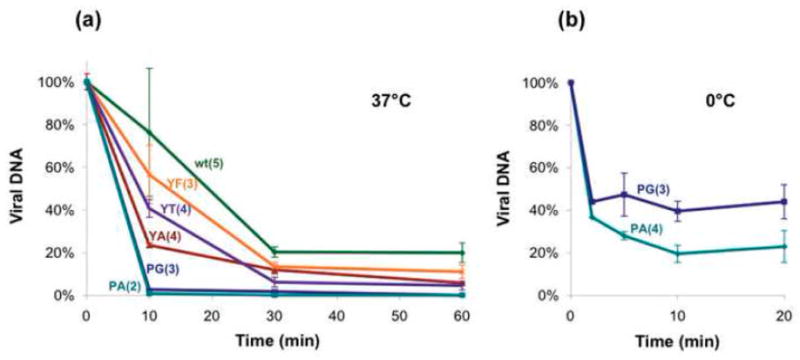
Stability analysis of the virions. Stability of mutants was measured as sensitivity to DNase I at 37°C (a) and at 0°C (b). Viral DNA amount was measured by RQ-PCR with primers covering the SV40 Poly A region. Results are presented as percentage of DNA at t=x relative to the DNA amount at the start point and are averages of several experimental repeats, as indicates in parentheses.
We next studied behaviour of the mutants at 0°. Remarkably, the P mutants showed sensitivity to DNase I even at 0° (Fig. 3b), with a half-life of about 2 min. Only 25–50% of the DNA of both P300A and P300G remained protected for longer time periods. At this temperature WT SV40 did not show any loss in DNA protection. Presumably, immature virion particles fully protect the DNA from nucleases at this temperature. The Y mutants were also fully DNA-protective at this temperature (data not shown).
The role of P300 in stabilizing the pentapeptide conformation
Proline restricts the conformational freedom of the protein backbone and by this induces a rigid bend into the pentapeptide hinge. Mutations, in particular to glycine, are expected to increase the flexibility of the hinge region.
Inspection of the crystal structure revealed two discrete hinge conformations, as previously described.2 This results in a difference of more than 60° between the emerging Cα helix orientations. This in turn affects the orientation of the tripartite helix formed by C-arms of monomers α, α′ and α″ (see below). Thus P300 appears to participate in determination of the angle of the emerging C-arm during capsid assembly. Consequently the C-arm is directed to form contacts only with other pentamers and prevented from contacting of the pentamer from which it emerges.2
The viability data of the P300 mutants (as measured by scoring replication centres), demonstrate that the glycine mutation has reduced viability by two-three orders of magnitude, compared to alanine. Glycine does not interfere with bending, on the contrary - it confers significant flexibility to the peptide chain. Thus this finding suggests that backbone flexibility is deleterious to the function of the hinge, presumably by relaxing the fixed relative orientations of the emerging C-arm. As previously suggested2 the constraint imposed by P300 (and P298) prevents back-insertion of the C-arm into the parental monomer. It is possible that in the mutant capsid some of the C-arm links are not formed, resulting in its instability. We further propose that once the capsid is assembled, rigidity of the proline and formation of hydrogen bonding within the hinge are critical to capsid stability.
Y299 stabilizes the hinge conformation and participates in interpentameric contacts
As previously reported,2 the hinge is stabilized by intramolecular hydrogen bonds between the backbone of Y299 and the side chain of N297: There are two hydrogen bonds in the hinge of monomers α′, β and β′, one hydrogen bond in α, and none in α? (Fig. 1d). These hydrogen bonds stabilize the different conformations of the hinge and lower the relative temperature B-factors.2 As these hydrogen bonds involve the backbone of Y299, they should not be affected by the mutations. However, as seen in Fig. 4a (see also14) for monomer α, the aromatic ring of Y299 stabilizes the hinge by interacting with the adjacent P298 and the nearby F303 of the same monomer (green). In concordance with statistically preferred orientations in other proteins,15 the side chains of P298, Y299 and F303 are positioned in an orientation that creates favourable stacking interactions between aromatic rings. Deletion of the ring in Y299T and Y299A disrupts these stacking interactions, as well as additional interaction with C-arms of monomers of other pentamers (see below).
Fig. 4.
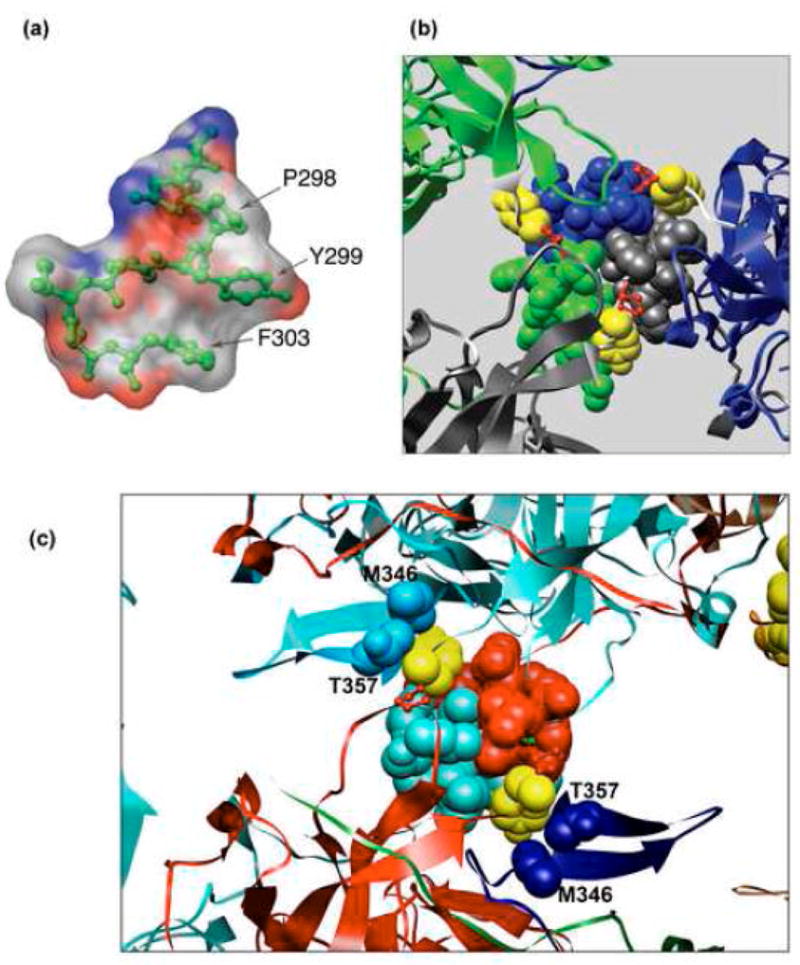
Interactions of Y299. (a). Stacking interactions within the α monomer involving Y299 stacked between P298 and F303. The surface is coloured according to CPK (Grey – carbon, blue – nitrogen, red – oxygen atoms). (b). Y299 covers the hydrophobic patch formed by the three C-terminal α helices. Representation of the hydrophobic interaction interface between VP1 monomer α (grey) α′ (blue) and α″ (green) tying the pentavalent pentamer (grey) with its neighbouring hexavalent pentamers. The three C-terminal α helices are represented as spheres. The monomer core is represented as ribbon. The hinge is shown in white, Y299 in yellow, and P300 in red. (c) Y299 interaction with distal part of the C-arm. The helices of monomers β (red) and β′ (turquoise) form two-fold, tight hydrophobic contacts between hexavalent pentamers. Y299 (yellow spheres) of these monomers interact with M346 and T357 (blue and light-blue spheres) which arrive from distant monomers in other pentamers, contributing to the interpentameric interaction. All Figures were created using CHIMERA.32
Computational analysis suggests that the Y299 stabilizes the capsid mainly through hydrophobic interactions with other pentamers.16 One of these interactions is shown in Fig. 4b, which illustrates a tri-partite hydrophobic helical bundle formed by the C-arm α-helices of monomers: α (grey), α′ (blue1) and α″ (green). The interactions between the three α-helices are mediated by a range of hydrophobic residues, including I301, L304, L305 and L308. Y299 (yellow) shields this hydrophobic patch from the solvent, by interacting with I301, L305, L308 and I309 (see Table16). These interactions appear to contribute to stability of the α - α′- α″ contact. The importance of this hydrophobic patch is highlighted by the high conservation of the hydrophobic character of positions 305, 308 and 309 in almost all polyomaviruses. Similar interactions are seen between Y299 of β with the α-helix of β′, and the reciprocal interaction between β′ and the α-helix of β (Fig 4c). Finally, Y299 of both α and β monomer-types also interacts with the distal part of C-terminal arm of distant monomer (see Table16). For example, Fig. 4c presents interactions of Y299 (in yellow) with positions M346 and T357 of β′ (light blue) and α′ (blue), thereby connecting three different pentamers. Interestingly, M346 too is almost fully conserved among 14/16 polyomaviruses. T357 in SV40 retains its hydrophobic character in 9 polyomaviruses. Since Y299 is highly conserved, the conservation of those distal amino acids suggests similar interpentameric interactions in other polyomaviridae family members. Y299 is at the centre of this hydrophobic patch and appears to anchor this structure in the mature virion (Fig. 4c). Thus, the aromatic ring of Y299 participates in both intramolecular and intermolecular interactions, as summarized in the Table.16
In order to estimate the effect on capsid stability, we modeled the structure of the mutations at position 299, and calculated the decrease in stability upon mutation using the Rosetta protocols for interface energy calculation and design.17; 18 The effect of a mutation was estimated by modeling the structure of the mutant and calculating the change in binding energy compared to the wild type structure. The structure of the capsid (pdb id 1SVA19) was used to model the three Y299 mutations, Y-F, Y-T and Y-A, in 5 of the 6 different chains (α, α′, α″ in the α interface; β and β′ in the β interface). The results are summarized in Fig. 5. Overall, a similar picture emerges from analysis of the different chains. In the wild type structure, Y299 interacts mostly through hydrophobic contacts and one hydrogen bond with its neighbors. Mutation to F is not predicted by Rosetta to significantly affect binding, since (i) the number of interactions changes only slightly, (ii) the solvation of the buried phenylalanine is improved compared to the less hydrophobic tyrosine, and (iii) this improved solvation most likely compensates for the loss of the hydrogen bond formed by the polar moiety of the Y299. In contrast, mutations to T or A are predicted to affect the binding more significantly, as they reduce the number of interactions mediated by position 299 (see Table16 and Fig. 5). The most dramatic effect is predicted for Y299A that results in the largest decrease in Van-der Waals interactions. In conclusion, our calculations reconfirm, and explain, the trend shown by the experiments (Table 1 and Fig. 3): While the mutation Y299F affects stability only slightly, mutations that truncate the side-chain (Y299T and Y299A) decrease stability significantly. Note that only side chain conformational changes were considered, and the effect of the mutations was assumed to be local. Since this simple model can reproduce the experimentally observed effects, it can be assumed that the mutants have a very local effect, and no major structural rearrangement is responsible for the observed impaired capsid viability.
Fig. 5.
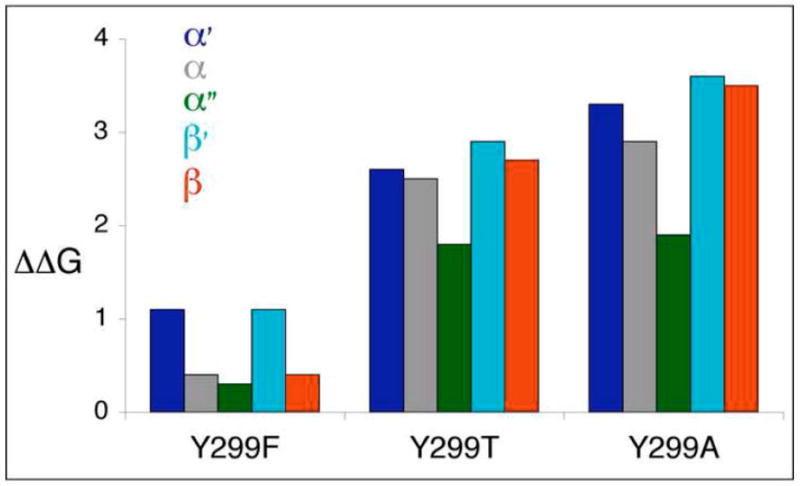
Calculated effect of mutations on binding. The predicted ΔΔG values of the mutations Y299F, Y299T, Y299A for each of the five tested chains, α (grey), α′ (blue), α″ (green), β (red) and β′ (turquoise) are shown (in Rosetta Units – an energy decrease of >1–1.5kcal/mol indicates significantly impaired binding). The effect of Y299F is negligible, while truncation of the sidechain to T, and even more to A, results in a significant loss of binding energy.
In conclusion, Y299 and P300 are critical to capsid structure. The P300 mutants lost the ability to protect the DNA, either due to distorted capsid structure or incomplete maturity. As previously proposed,2 the inflexibility introduced by prolines in the WT virus constrains the orientation of the emerging C-arm. During assembly, the hinge appears to function in orienting the C-helix away from its parent pentamer, facilitating contacts with C-helices of other pentamers. The different conformations of the hinge are most likely established after assembly and during virion maturation. In contrast to P300, Y299 functions in stabilizing the hinge via local interactions that include hydrogen bonding with N297, packing interactions with its hydrophobic neighbours, and stacking interactions via its aromatic ring. As long as the large, aromatic side-chain of position 299 is not removed, most of the interactions remain, and the effect on binding is negligible. Impaired binding is observed only upon removing of the interactions by mutation to smaller side chains. The present study sheds new light on the key role of the conserved pentapeptide hinge KNPYP in providing the capsid with the robustness needed to protect the genome outside the cell.
Materials and Methods
Cell lines
CV1 (ATCC # CCL70, African Green Monkey Kidney Fibroblasts) and CV1-PD were cultured in high glucose Dulbecco’s Modified Eagle’s Medium containing glutamine, penicillin, streptomycin, and 10% FBS. CV1-PD is a derivative of CV1, kindly obtained from J. Mertz.20
Vector construction and mutagenesis
To propagate viral DNA in E. coli and facilitate mutagenesis, we cloned the entire SV40 genome in pUC18, in which BamHI site was deleted by digestion with BamHI and filling in the sticky ends by Klenow. KpnI-linearized SV40 genome (GenBank accession No. J02400) was inserted in the KpnI site of the BamHI-deleted pUC18. The pUC-SV40 plasmid contains a single BamHI site in the SV40 late region.
All the point mutations were inserted by the QuickChange XL Site directed Mutagenesis Kit (Staratagene #200521). The primers for each mutation are shown in.21 Mutants were screened by colony hybridization 22 using their specific probes21 and presence of the mutations was verified by sequencing. 271 bp fragments containing the mutations, flanked by ApaI and BamHI sites, were back-cloned into the original pUC-SV40 plasmid, to remove potential undesired mutations introduced by the long PCR reaction. Following re-sequencing, DNA was prepared in E. coli and purified using Qiagene EndoFree Maxi Kit (#12362).
Deletion mutant SV40-Δ47, deleted in the distal 47 amino acids of VP1 (R315 to Q361), was constructed by recombineering.23; 24 This technology enables in vivo mutagenesis of plasmids (or the E. coli chromosome itself) by homologous recombination, using PCR products or synthetic oligonucleotides. This technology relies on a bacteriophage l - encoded recombination function, Red Beta, which efficiently recombines ssDNA sequences with homologies as short as 35 to 50 base pairs.23 An oligonucleotide (79 nucleotides) with flanking homology for the deletion was designed (Δ47 in21). The HME6 strain of E. coli,23 harbouring the λ Red system, was induced at 42°C for 15 min, to destroy the thermo-sensitive CI857 repressor and express λ Red functions. These cells were electroporated with both pUC-SV40 and Δ47 oligonucleotide, cultured at 32°C for 1 hr and plated on LB-Amp plates for selection. Mutants were selected by colony hybridization using specific primers.21
Propagation of mutant virions
The bacterial sequences were excised off pUC-SV40 by digestion with KpnI and religation at a low DNA concentration (10 μg/ml) as previously described.25 The ligated viral DNA was transfected into logarithmically growing cultures of CV1 or CV1-PD using the Polyfect reagent according to manufacturer protocol (Qiagen, #301105). Five days post-transfection virions were harvested by three freeze–thaws, or by the di-detergent method, and were stored at −20°C.
Equilibrium sedimentation in a CsCl gradient
Cells were harvested at the 5th day post-infection by the di-detergent method,26 by adding Triton X-100 and Deoxycholate to the culture medium to final concentrations of 1% and 0.5%, respectively. The cell suspension was centrifuged at 9,500 rpm (10,000×g) for 30 min at 4°C to precipitate debris. The virus was sedimented by centrifugation at 80,000×g for 4 hr at 4°C. Viral pellet was allowed to resuspend in PBS overnight at 4°C, sonicated and centrifuged to clarify the virus suspension. At this point viruses were purified by equilibrium sedimentation in a CsCl gradient. CsCl was added to the virus and the density was adjusted to 1.34 g/cm3, (refractive index of 1.366). The samples were centrifuged at 40,000 rpm for 38 hr at 4°C using an SW50.1 rotor (Beckman). Then, fractions (250 ml) were collected from the top. The fractions were dialyzed against Tris-NaCl buffer (10mM Tris-Hcl pH 7.5, 10mM NaCl) and stored at −20°C.
Virus titration
Titration was performed by the standard plaque assay27 or by scoring for replication centres in CV1-PD cells infected at different dilutions.25 Replication centres were scored two days post infection, at the peak of viral DNA replication, using in situ hybridization with SV40 DNA labelled with [α-32P]-dCTP.22
Assay for assembly
To evaluate assembly, the amount of DNA protected from DNase I by the assembled capsids was measured by real quantitative PCR (RQ-PCR). Virus stocks, 45 μl, harvested by freeze-thaw, were treated with 1U DNase I in DNase I buffer and incubated for 10 min on ice. The DNAse was inactivated and removed by chloroform treatment and centrifugation. RQ-PCR was performed with or without dissociation of the capsid by incubation with 25 mM EGTA, 25 mM EDTA and 25 mM DTT for 30 minutes at 37°C.28 Both methods gave similar results.
Assay for virion stability
Virion stability was evaluated by incubation with DNase I for different time periods. 40 μl of DNAse I buffer (Promega) were added to 10 μl of virus harvested by freeze-thaw, and kept on ice until the addition of 2 units of RQ1 DNase I (Promega). The mixture was incubated at 37°C or on ice, as indicated. Aliquots were withdrawn at different time points and the tubes were placed at 95°C for 15 min to stop the reaction. The DNA content was quantified by RQ-PCR.
Real time Quantitative PCR (RQ-PCR)
Viral DNA was quantified using 2 pairs of primers. The first pair flanks the SV40 PolyA region (157 bp fragment, PolyA-F and PolyA-R21). The second pair is from the VP1 structural gene (470 bp fragment, VP1-F and VP1-R21). The PCR reaction was performed in the LightCycler 2.1 Instrument (Roche), in 20 μl buffer, composed of: 1xFastStart PCR reaction buffer (Roche), 2 μg BSA, 0.5 mM dNTP’s, 5 μl DNA sample, 1xSYBR Green (Roche, 1988131), 1 unit of FastStart Taq DNA polymerase (Roche, #12 032 945 001), 5 mM Mg++ or 1.5 mM Mg++ and 10 ng or 10 pmol of each primer for the PolyA primer set or the VP1 primer set, respectively. As a standard we used SV40 DNA with PolyA primers or VP1 primers. The PCR reaction program was: 95°C 10′, followed by 35 cycles of 95°C 10″, 57°C 30″, 72°C 20″, 77°C 2″.
Protein analysis
Total protein was harvested 3 days post transfection by 5 min incubation of cells in lysis buffer (50 mM Tris-Hcl pH 7.4, 5% beta mercaptoethanol, 10% glycerol, 2% SDS). The samples were boiled for 15 min and spun to remove insoluble proteins. The supernatant was sonicated to shear cellular DNA. Total protein was measured by Bradford assay (Bio-rad #500-0006).
Proteins were separated on 4–12% SDS–PAGE (NuPAGE) and electroblotted onto a PVDF membrane (Immobilon-P, Millipore). Western analysis was performed using rabbit polyclonal anti-VP1 antibody,28 and HRP- anti rabbit as secondary antibody. Normalization of protein amounts was done by analysis of the same immunoblots with mouse monoclonal anti β-actin (Sigma- A 5441). Quantification was performed by densitometry, using the TINA program.
Computational modelling of mutations and their effects
Conservation of polyoma capsid sequences was evaluated by a multiple sequence alignment created with clustalW.29 The molecular graphics programs SPDBV,30 RasMol,31 and Chimera32 were used for inspection of the capsid structure, and for the creation of figures.
Rosetta was used to model the mutations at the interface, and to calculate the effect on binding.17; 18 The binding energies of the wild type and mutant structure were compared, as described before (see also.16) The structure of the mutant was created by choosing side chain conformations for the mutated residue, as well its neighboring residues, from an extended backbone-dependent rotamer library that included also the original side chain conformation as option.18 For more details and the exact command lines used, see.33 The main terms in the Rosetta energy function consist of a softened Lennard-Jones potential, hydrogen bonds and solvation.34 The effect of a mutation is expressed as DDG, which describes the change in binding energy of the mutant protein complex, relative to the wt complex (mutations with ΔΔG=1–1.5 kcal/mol are considered to significantly affect binding). We evaluated the effect of the different mutations by calculating the ΔΔG values for mutations at the different interfaces that include Y299, namely in chains α, α′, α″ for the α interface and β and β′ in the β interface). In each case, the binding energy of the chain with the Y to the other chains was calculated separately, thereby allowing us to estimate whether the effect would be on all different Y299 positions within the capsid, or only on a sub-population of structurally similar residues. The resolution of the crystal structure is not high (3.1Å), and therefore we applied a short minimization protocol to create a clash-free starting structure. We minimized all torsion angles of interface residues, as well as the rigid body orientation, while constraining the Cα positions to their original conformation, as detailed.33 Calculations were performed using the Rosetta release 2.3.0, obtainable at the Rosetta website.35
Acknowledgments
Prof. Amos Oppenheim contributed ideas and know-how through helpful discussions and guided and assisted with the recombineering procedure. We wish to thank Dr. Gali Prag for helpful discussions. Seth Salpeter assisted in construction and preparations of the pUC-SV40 and P300 mutants. We wish to thank the anonymous reviewer for most helpful comments. This work was supported in part by US Public Health Service Grant CA100479, and by the U.S.-Israel Binational Science Foundation (BSF) grant number 2005050. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crick FH, Watson JD. Structure of small viruses. Nature. 1956;177:473–5. doi: 10.1038/177473a0. [DOI] [PubMed] [Google Scholar]
- 2.Stehle T, Gamblin SJ, Yan Y, Harrison SC. The structure of simian virus 40 refined at 3.1 A resolution. Structure. 1996;4:165–82. doi: 10.1016/s0969-2126(96)00020-2. [DOI] [PubMed] [Google Scholar]
- 3.Li PP, Nakanishi A, Clark SW, Kasamatsu H. Formation of transitory intrachain and interchain disulfide bonds accompanies the folding and oligomerization of simian virus 40 Vp1 in the cytoplasm. Proc Natl Acad Sci U S A. 2002;99:1353–8. doi: 10.1073/pnas.032668699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen XS, Stehle T, Harrison SC. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. Embo J. 1998;17:3233–40. doi: 10.1093/emboj/17.12.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon-Shaag A, Ben-Nun-Shaul O, Roitman V, Yosef Y, Oppenheim A. Cellular Transcription Factor Sp1 Recruits the SV40 Capsid Proteins to the Viral Packaging Signal ses. J Virol. 2002;76:5915–5924. doi: 10.1128/JVI.76.12.5915-5924.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liddington R, Yan Y, Moulai J, Sahli R, Benjamin T, Harrison S. Structure of simian virus 40 at 3.8-A resolution. Nature. 1991;354:278–284. doi: 10.1038/354278a0. [DOI] [PubMed] [Google Scholar]
- 7.Li PP, Nakanishi A, Shum D, Sun PC, Salazar AM, Fernandez CF, Chan SW, Kasamatsu H. Simian virus 40 Vp1 DNA-binding domain is functionally separable from the overlapping nuclear localization signal and is required for effective virion formation and full viability. J Virol. 2001;75:7321–9. doi: 10.1128/JVI.75.16.7321-7329.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker TS, Drak J, Bina M. Reconstruction of the three-dimensional structure of simian virus 40 and visualization of the chromatin core. Proc Natl Acad Sci U S A. 1988;85:422–6. doi: 10.1073/pnas.85.2.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Supplemental video 1.
- 10.Supplemental figure 1.
- 11.Gaynor AM, Nissen MD, Whiley DM, Mackay IM, Lambert SB, Wu G, Brennan DC, Storch GA, Sloots TP, Wang D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoSPathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roitman-Shemer V, Stokrova J, Forstova J, Oppenheim A. Assemblages of simian virus 40 capsid proteins and viral DNA visualized by electron microscopy. Biochem Biophys Res Commun. 2007;353:424–430. doi: 10.1016/j.bbrc.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 13.Coca-Prados M, Yu HY, Hsu MT. Intracellular forms of simian virus 40 nucleoprotein complexes. IV. Micrococcal nuclease digestion. J Virol. 1982;44:603–9. doi: 10.1128/jvi.44.2.603-609.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Supplemental video 2.
- 15.Misura KM, Morozov AV, Baker D. Analysis of anisotropic side-chain packing in proteins and application to high-resolution structure prediction. J Mol Biol. 2004;342:651–64. doi: 10.1016/j.jmb.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 16.Supplemental Table 2.
- 17.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A. 2002;99:14116–21. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Kuhlman B. RosettaDesign server for protein design. Nucleic Acids Res. 2006;34:W235–8. doi: 10.1093/nar/gkl163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1sva.
- 20.Farrell ML, Mertz JE. Hormone response element in SV40 late promoter directly affects synthesis of early as well as late viral RNAs. Virology. 2002;297:307–18. doi: 10.1006/viro.2002.1478. [DOI] [PubMed] [Google Scholar]
- 21.Supplemental Table 1.
- 22.Sambrook J, Fritsh EF, Maniatis T. Molecular cloning, a laboratory manual. 2. 1. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1989. [Google Scholar]
- 23.Costantino N, Court DL. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc Natl Acad Sci U S A. 2003;100:15748–53. doi: 10.1073/pnas.2434959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu Rev Genet. 2002;36:361–88. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- 25.Dalyot-Herman N, Ben-nun-Shaul O, Gordon-Shaag A, Oppenheim A. The simian virus 40 packaging signal ses is composed of redundant DNA elements which are partly interchangeable. J Mol Biol. 1996;259:69–80. doi: 10.1006/jmbi.1996.0302. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg BH, Deutsch JF, Ungers GE. Growth and purification of SV40 virus for biochemical studies. J Virol Methods. 1981;3:167–76. doi: 10.1016/0166-0934(81)90051-3. [DOI] [PubMed] [Google Scholar]
- 27.McCutchan JH, Pagano JS. Enhancement of infectivity of simian virus 40 deoxyribonucleic acid with diethyl-aminoethyl-dextran. J N• at Cancer Inst. 1968;41:351–357. [PubMed] [Google Scholar]
- 28.Sandalon Z, Oppenheim A. Self assembly and protein-protein interactions between the SV40 capsid proteins produced in insect cells. Virol. 1997;237:414–421. doi: 10.1006/viro.1997.8796. [DOI] [PubMed] [Google Scholar]
- 29.http://www.ebi.ac.uk/Tools/clustalw2/.
- 30.http://expasy.org/spdbv/.
- 31.http://www.umass.edu/microbio/rasmol/index2.htm.
- 32.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Supplemental Command lines.
- 34.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 35.http://www.rosettacommons.org/software/index.html.