

Abstract
Motivation: The need for accurate and efficient tools for computational RNA structure analysis has become increasingly apparent over the last several years: RNA folding algorithms underlie numerous applications in bioinformatics, ranging from microarray probe selection to de novo non-coding RNA gene prediction.
In this work, we present RAF (RNA Alignment and Folding), an efficient algorithm for simultaneous alignment and consensus folding of unaligned RNA sequences. Algorithmically, RAF exploits sparsity in the set of likely pairing and alignment candidates for each nucleotide (as identified by the CONTRAfold or CONTRAlign programs) to achieve an effectively quadratic running time for simultaneous pairwise alignment and folding. RAF's fast sparse dynamic programming, in turn, serves as the inference engine within a discriminative machine learning algorithm for parameter estimation.
Results: In cross-validated benchmark tests, RAF achieves accuracies equaling or surpassing the current best approaches for RNA multiple sequence secondary structure prediction. However, RAF requires nearly an order of magnitude less time than other simultaneous folding and alignment methods, thus making it especially appropriate for high-throughput studies.
Availability: Source code for RAF is available at:http://contra.stanford.edu/contrafold/
Contact: chuongdo@cs.stanford.edu
1 INTRODUCTION
The secondary structure adopted by an RNA molecule in vivo is a vital consideration in many bioinformatics analyses. In PCR primer design, stable secondary structures can obstruct proper binding of the primer to DNA (Dieffenbach et al., 1993); in RNA folding pathway studies, secondary structure forms the basic scaffold on which more complicated 3D structures organize (Brion and Westhof, 1997); and in computational non-coding RNA gene prediction, RNA secondary structural stability provides the characteristic signal for distinguishing real RNA sequence from non-functional transcripts (Eddy, 2002).
To date, the most powerful non-experimental methods for determining RNA secondary structure rely primarily on position-specific patterns of nucleotide covariation in multiple homologous RNA sequences. Specifically, enrichment for complementarity in pairs of columns from an RNA multiple alignment, especially when primary sequence is not conserved, provides strong evidence for potential base-pairings in the RNA's in vivo structure. A primary limitation of covariation analysis, however, is the difficulty of obtaining reliable sequence alignments for divergent RNA families. This shortcoming is especially relevant in the detection of ncRNA genes, as secondary structural constraints often exist even when primary sequence conservation is lacking (Torarinsson et al., 2006).
In this article, we describe RNA alignment and folding (RAF), a new algorithm for predicting RNA secondary structure from a collection of unaligned homologous RNA sequences. Algorithmically, RAF belongs to a category of RNA secondary structure prediction methods which simultaneously align and fold RNA sequences. By optimizing a pair of unaligned RNA sequences for both sequence homology and structural conservation concurrently, simultaneous alignment and folding approaches sidestep the usual problem of needing accurate sequence alignments before the folding is done. By exploiting sparsity in the set of likely base pairings and aligned nucleotides, RAF achieves O(L2) running time for sequences of length L, improving significantly upon the O(L4) running times of typical simultaneous folding and alignment approaches.
The main contribution of RAF, however, is its application of discriminative machine learning techniques for parameter estimation to the problem of simultaneous alignment and folding. Unlike previous methods, RAF's scoring model does not rely on ad hoc combinations of thermodynamic free energies for structural features (Mathews et al., 1999) with arbitrary alignment match and gap penalties (Hofacker et al., 2002), nor does RAF attempt the ambitious task of simultaneously modeling the evolutionary history of both sequences and structure (Knudsen and Hein, 2003). Instead, RAF defines a fixed set of basis features describing aspects of the alignment, RNA secondary structure, or both. RAF then poses the task of learning weights for these features as a convex optimization problem, giving rise to efficient algorithms with guaranteed convergence to optimality.
The concept of using discriminative methods for parameter estimation rather than relying solely on parameters compiled from experimental measurements originated with the CONTRAfold (Do et al., 2006b) program, and later also became the basis of the CG (Andronescu et al., 2007) method. In a manner analogous to these two previous methods for single sequence secondary structure prediction, RAF demonstrates that automatic learning of parameters can also confer benefits to multiple sequence structure prediction accuracy.
2 METHODS
The RAF algorithm consists of four components: (1) a simple yet flexible objective function for pairwise alignment and folding of unaligned RNA sequences; (2) a fast Sankoff-style inference engine for maximizing this objective function via sparse dynamic programming; (3) a simple progressive strategy for extending the pairwise algorithm to handle multiple unaligned sequence inputs; and (4) a max-margin framework for automatically learning model parameters from training data. We describe each of these in turn.
2.1 The RAF scoring model
We begin our description of the algorithm by describing a scoring scheme for alignments and consensus foldings of two sequences. Let a and b be a pair of unaligned input RNA sequences. We refer to a candidate alignment and consensus secondary structure of a and b collectively as a parse. Formally, a parse y for a pair of sequences a and b is a set whose elements consist of base pairings (ai, aj) belonging to sequence a, base pairings (bk, bl) belonging to sequence b, and aligned positions (ai, bk) between a and b.
For a given parse y from the space of all valid1 parses 𝒴, RAF uses a simple scoring scheme which takes into account aligned positions and conserved base pairings. Specifically, RAF defines the score, Score(y;w), of such a parse y to be
where 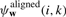 and
and 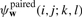 are scoring terms for aligned positions and conserved base pairs, respectively, and where ℬ(y) is the set of all conserved base pairings. In turn, RAF models each scoring term as a linear combination of arbitrary basis features (Appendix A.1):
are scoring terms for aligned positions and conserved base pairs, respectively, and where ℬ(y) is the set of all conserved base pairings. In turn, RAF models each scoring term as a linear combination of arbitrary basis features (Appendix A.1):
 |
where w∈ℝnaligned+nPaired=ℝn is a vector of scoring parameters.
2.2 Fast pairwise alignment and folding
Given the scoring scheme described in the previous section, the problem of simultaneous alignment and folding reduces to the optimization problem,
| (1) |
In principle, the solution to (1) follows immediately from the original dynamic programming algorithm for simultaneous alignment and folding presented by Sankoff (1985). Sankoff's algorithm, however, has an O(L3K) time complexity and O(L2K) space complexity for K sequences of length L, rendering it impractical for all but the smallest multiple folding problems. Therefore, most programs for RNA simultaneous alignment and folding use heuristics to reduce time and memory requirements while minimally compromising alignment and structure-prediction quality. Some heuristics used in previous programs have included incorporating structural information into a single alignment scoring matrix (Dalli et al., 2006), disallowing multi-branch loops (Gorodkin et al., 1997), and precomputing potential conserved helices prior to alignment (Tabei et al., 2006; Touzet and Perriquet, 2004).
The most popular heuristics, however, involve reduction of the portion of the dynamic programming matrices (which we call the DP region) that must be computed. For example, some methods restrict the DP region to a strip of fixed width about the diagonal (Hofacker et al., 2004; Mathews and Turner, 2002) or about an initial alignment path (Kiryu et al., 2007). Other methods rely on external single-sequence folding and probabilistic alignment programs to generate base pairing probability matrices (Torarinsson et al., 2007; Will et al., 2007) or alignment match posterior probability matrices (Kiryu et al., 2007), and then exploit the sparsity of these matrices in order to reduce the amount of computation required.
The RAF algorithm adopts the last of these strategies. Namely, RAF uses a single-sequence RNA secondary structure prediction program CONTRAfold; Do et al., 2006b) and a pairwise RNA sequence alignment program (CONTRAlign; Do et al., 2006a),2 respectively, to construct a constraint set 𝒞 of allowed base pairs and aligned positions in a and b. Given a constraint set 𝒞, RAF then replaces (1) with the reduced inference problem,
| (2) |
where 𝒴𝒞={y∈𝒴:y⊆𝒞} is the space of valid parses, restricted to those which contain only base pairings and alignment matches from the constraint set 𝒞 (Fig. 1).
Fig. 1.

Sparsity patterns in posterior probability matrices. Panels (a) and (b) illustrate the pairwise pairing posterior probabilities for two different sequences (such as generated by a single-sequence probabilistic or partition function–based RNA folding program). Panel (c) shows the alignment match probabilities for these sequences (such as generated by a probabilistic HMM). In each panel, the darkness of each square represents the posterior confidence in the corresponding base pairing or alignment match. While the single sequence folder or the pairwise sequence aligner may not be able to identify the single correct folding or alignment, respectively, the set of likely candidate base pairings and matched positions, nonetheless, is extremely sparse.
To obtain the set of allowed base pairings, RAF uses the implementation of McCaskill's algorithm (McCaskill, 1990) from CONTRAfold in order to compute the posterior probability of each possible base pairing in sequence a, and similarly for sequence b. All base pairs with posterior probability at least ɛpaired are then retained. Similarly, to determine the set of allowed aligned positions, RAF retains those matches whose posterior probability, according to a version of the CONTRAlign program adapted for RNAs, is at least ɛaligned. If these cutoffs ɛaligned and ɛpaired are chosen to be too low, then the reduction of the dynamic programming space achieved for 𝒴𝒞 will not be significant. Conversely, a higher cutoff could also degrade performance by excluding portions of the DP matrix which actually correspond to the true parse of the input sequences. A similar approach for pruning the space of candidate alignments and folds via fold and alignment envelopes was implemented in the Stemloc (Holmes, 2005) program. A number of other programs exploit either base-pairing sparsity (Torarinsson et al., 2007; Will et al., 2007) or alignment sparsity (Dowell and Eddy, 2006; Harmanci et al., 2007; Kiryu et al., 2007) separately.
Assuming O(c) and O(d) bounds on the number of candidate base pairing and alignment partners, respectively, per position of both sequences, we show that the time complexity of the RAF algorithm scales quadratically in the length of the sequences, while the space complexity scales linearly (Appendix B.1). A comparison table of asymptotic time and space complexity of a number of modern RNA simultaneous folding and alignment approaches is shown in Table 1. In practice, we find that RAF's scaling reflects the theoretical bounds, achieving running times often an order of magnitude faster than current simultaneous alignment and folding methods.3
Table 1.
Comparison of computational complexity of RNA simultaneous folding and alignment algorithms
| Algorithm | Time complexity | Space complexity |
|---|---|---|
| Sankoff | O(L6) | O(L4) |
| FOLDALIGN | O(L4) | O(L4) |
| LocARNA | O(c2L4) | O(c2L2) |
| Murlet | O(d2L2+d3L3/κ6) | O(d2L2) |
| RAF | O(min(c,d)·cd2L2) | O(min(c,d)·cdL) |
Here, L denotes the sequence length, c is the number of candidate base pairs per position, d is the number of candidate alignment matches per position and κ is the minimum allowed distance between adjacent helices.
2.3 Extension to multiple alignment
Using the RAF pairwise alignment subroutine, we can also address the problem of aligning two alignments. Let S and T be two sets of sequences that we wish to align; furthermore, we denote their corresponding alignments as A and B.
To align a pair of alignments, we first define new basis features 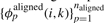 and
and 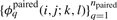 to simply be the average over all pairs of sequences s∈S and t∈T of the basis features for aligning s and t, remapped to the coordinates of the alignments A and B. Second, we define the new constraint set 𝒞 for aligning the two alignments to be the union over all pairs of sequences s∈S and t∈T of the constraint sets for each pair, again remapped to the alignment coordinates. Finally, using these new features and our new constraint set, we simply call the existing RAF subroutine for fast-pairwise alignment and folding.
to simply be the average over all pairs of sequences s∈S and t∈T of the basis features for aligning s and t, remapped to the coordinates of the alignments A and B. Second, we define the new constraint set 𝒞 for aligning the two alignments to be the union over all pairs of sequences s∈S and t∈T of the constraint sets for each pair, again remapped to the alignment coordinates. Finally, using these new features and our new constraint set, we simply call the existing RAF subroutine for fast-pairwise alignment and folding.
Using this new subroutine for aligning alignments, we can then perform multiple alignment in RAF using a standard progressive strategy (Feng and Doolittle, 1987). Specifically, we cluster the sequences with a UPGMA (Sneath and Sokal, 1962) tree-building procedure, using the expected accuracy similarity measure (Do et al., 2005). Finally, we perform progressive alignment by aligning subgroups of sequences according to the tree.
2.4 A max-margin framework
Given a set of training examples, 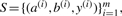 , the parameter estimation problem is the task of identifying a vector of weights w=(w1,w2,…,wn)∈ℝn for which the RAF inference algorithm, as described in the previous section, will yield accurate alignments and consensus structures. In this section, we present a max-margin framework for parameter estimation in RAF.
, the parameter estimation problem is the task of identifying a vector of weights w=(w1,w2,…,wn)∈ℝn for which the RAF inference algorithm, as described in the previous section, will yield accurate alignments and consensus structures. In this section, we present a max-margin framework for parameter estimation in RAF.
2.4.1 Formulation
In the max-margin framework, our goal is to obtain a parameter vector w for which running the RAF inference algorithm will generate accurate alignments and consensus structures. Clearly, this goal is met if for each training example (a(i),b(i),y(i)) from our training set S,4
| (3) |
In such a case, we would be guaranteed that the maximum of (2) is attained for y*=y(i) (provided the true parse y(i) belongs to 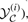 ), and hence our inference procedure would necessarily return the correct alignment and consensus folding. This intuition is captured in the following convex optimization problem:
), and hence our inference procedure would necessarily return the correct alignment and consensus folding. This intuition is captured in the following convex optimization problem:
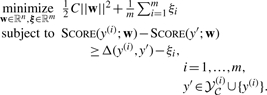 |
(4) |
Here, C is a regularization constant, and Δ(y(i),y′) is a non-negative distance measure between pair of parses, conventionally referred to as the loss function, which takes value 0 if and only if its two arguments are equal (Section 2.4.2).
The inequality constraints play the role of (3)—they try to ensure that the training output y(i) scores higher than any alternative incorrect parse y′ by some positive amount Δ(y(i),y′). In cases where this condition is not achieved, the objective function incurs a penalty of ξi. Finally, the regularization term (½)C‖w‖2 is a penalty used to prevent overfitting.5
2.4.2 The loss function
The loss function Δ(y(i),y′) in (4) plays two significant roles. Technically, the loss function establishes an appropriate scale for the parameters of the problem and prevents the trivial solution, w=0. Intuitively, however, the loss function also helps to make the max-margin optimization robust. By choosing a loss function that takes large positive values for incorrect candidate outputs y′ that differ from the true output y(i) in a very critical way, but that takes small positive values for incorrect candidate outputs y′ whose errors are more forgivable, the loss function allows the user to implement a notion of ‘cost’ for different types of mistakes in the max-margin model.
For RAF, we defined the loss function by restricting our attention to four types of parsing errors: (1) false positive base-pairings ((ai, aj)∈y′ ∖ y(i), or similarly in sequence b), (2) false negative base-pairings ((ai, aj)∈yi ∖ y′, or similarly in sequence b), (3) false positive aligned matches ((ai, bk)∈y’ ∖ y(i)) and (4) false negative aligned matches ((ai, bk)∈y(i) ∖ y′). Then, we set
 |
The numbers γFN paired, γFP paired, γFN aligned and γFP aligned are hyperparameters, chosen by the user prior to training the RAF algorithm, which allow the user to express her preference for models with either high sensitivity or high specificity for base-pairing positions and aligned nucleotides.6
2.4.3 Optimization algorithm
At first glance, the constrained optimization problem stated in (4) appears to be a standard convex quadratic program and hence solvable using off-the-shelf packages for convex programming. In reality, for each training example, the optimization problem has an exponential number of inequalities, one corresponding to each possible candidate parse y′ of the input sequences! Despite our use of constraints sets to reduce the set of allowed candidate outputs, in most cases, this space is still too large to enumerate.
One approach to deal with this problem is an iterative algorithm known as constraint generation (or column generation), as used in the program CG (Andronescu et al., 2007). In this approach, the parameter vector wt at each time t is the solution to a reduced version of (4) in which only a small subset of the constraints are retained. Next, one checks if wt violates any of the constraints of the original full optimization problem by more than an prescribed tolerance of ɛ. If so, the worst violated constraint is added to the current set of constraints to form a new reduced optimization problem, whose solution, in turn, gives the next iterate wt+1. If not, the optimization algorithm terminates. Each of the optimization problems in the sequence requires a quadratic programming solver.
Here, we take a simpler approach based on the recent SVM training algorithm of (Shalev-Shwartz and Singer, 2007) and Shalev-Shwartz et al., (2007). Omitting details, we begin by converting (4) into an equivalent unconstrained problem: namely, minimize (with respect to w∈ℝn),
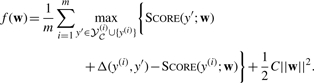 |
(5) |
Next, we use strong duality from optimization theory in order to derive an upper bound B on the norm of the optimal solution of our unconstrained problem (Appendix C.1). Finally, we actually run the optimization procedure by applying the simple update rule,
| (6) |
starting from w1=0. Here, gt∈∂f(wt) is any subgradient of the objective function f(w) evaluated at w=wt, and the operator ΠB[·] projects a vector onto an origin-centered ball of radius B (i.e. ΠB[v]=(B/‖v‖)v if ‖v‖>B and ΠB[v]=v otherwise). Intuitively, the algorithm works much like a standard gradient descent procedure adapted for non-differentiable objective functions, but with the added twist that the projection operation ensures that the weight vector iterates stay with a region of the parameter space where the optimum is known to exist.
Given an existing routine for computing subgradients of the unconstrained objective, this algorithm can be implemented in a few lines of code with no complicated numerical optimization software. As shown by Singer and Shalev-Shwartz, the algorithm is also quite efficient, requiring only Õ(m/Cɛ) iterations to achieve ɛ accuracy on a training set of m examples. An online variant of the algorithm, in which the subgradients gt in each step are computed based only on a randomly sampled subset of the training data (e.g. a single example), achieves an Õ(1/Cɛ) expected running time, independent of m, the size of the training set.
2.4.4 Subgradient computation
Finally, we show how to compute a subgradient gt∈∂f(wt). In order to simplify notation, define an n-dimensional vector Φ(y) whose pth component is
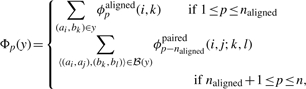 |
from which it follows that Score(y;w)=wTΦ(y). We can apply the usual rules for computing subgradients see, e.g. Bertsekas et al., 2003) to obtain
| (7) |
where 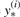 is simply any y′ which attains the maximum in the ith term of the summation in (5), for w=wt. Each ‘loss-augmented’ maximization, in turn, is easily performed by modifying the original RAF inference procedure to incorporate an appropriately defined additional scoring matrix, φ0(i, j; k, l), with fixed weight w0=1.
is simply any y′ which attains the maximum in the ith term of the summation in (5), for w=wt. Each ‘loss-augmented’ maximization, in turn, is easily performed by modifying the original RAF inference procedure to incorporate an appropriately defined additional scoring matrix, φ0(i, j; k, l), with fixed weight w0=1.
3 RESULTS
To evaluate the performance of RAF on real data, we collected training and testing data from a variety of sources. In particular, for training, we obtained Rfam 8.1 (Griffiths-Jones et al., 2005), a database of alignments and covariance models for RNA families along with annotated secondary structures where available. For testing, we obtained BRAliBASE II (Gardner et al., 2005), a benchmark set for RNA alignment programs. We also obtained a testing set of RNA families used by the authors of the recent program, MASTR (Lindgreen et al., 2007).
An important concern in the validation of RNA alignment programs is the confounding factor that unless cross-validation is properly performed, the performance that one sees on any given validation set is not likely to be a reliable judge of the program's performance on future data. Even in cases where the training and evaluation tests are disjoint but still contain sequences from the same RNA family, evaluation can still give misleading results, because the weights learned for loop lengths and composition will be biased toward specific properties of that RNA family.
To be absolutely sure of no contamination between training and testing data, we preprocessed our Rfam training set of alignments and consensus structures (October 2007 version, 607 families) by excluding all families for which either of the two testing databases contained an example from that family. We then also removed all families for which only automatically predicted consensus structures were known, leaving a total of 154 families. Finally, we generated a training set 𝒯1 of up to 10 randomly sampled pairwise alignments with consensus structures from each remaining family (1361 pairwise alignments in total), a training set 𝒯2, of up to 10 randomly sampled sequences with structures from each family (1179 sequences in total), and a training set 𝒯3, containing one randomly sampled five-way multiple alignment from each family (118 multiple alignments in total).
RAF uses two external programs, CONTRAlign (Do et al., 2006a) and CONTRAfold (Do et al., 2006b), to compute alignment match and base-pairing posterior probabilities, respectively. To ensure proper cross-validation, CONTRAlign was retrained from scratch using 𝒯1, and CONTRAfold was retrained using 𝒯2. Finally, the RAF algorithm itself was trained using all pairwise projections of each multiple alignment of 𝒯3. Our strict cross-validation procedure significantly reduces both the size and coverage of the training sets used for CONTRAlign and CONTRAfold, and thus places RAF at a significant disadvantage in the comparisons shown here. Nonetheless, as shown in the following sections, RAF performs well, indicating its ability to generalize for sequences not present in the training set.
3.1 Alignment and base-pairing constraints
To observe the effects of different cutoffs ɛaligned and ɛpaired, we computed the proportions of reference base pairings and reference aligned matches recovered for varying cutoff constraints. In addition, we also computed the sparsity ratio (i.e. the maximum number of pairing partners or matching partners for any nucleotide, averaged over the entire training set) for each cutoff. A plot of these two values for training set 𝒯3 is shown in Figure 2. As seen in the figure, nearly complete coverage of base pairings and alignment matches can be retained when each sparsity factor is roughly 10.7
Fig. 2.

Trade-off between sparsity factor and proportion of reference base-pairings or aligned matches covered when varying the cutoffs ɛpaired and ɛaligned. This graph was made using training set 𝒯3.
3.2 Evaluation metrics
To evaluate the quality of the resulting alignments, we used five different scoring measures:
(1) the standard sum-of-pairs (SP) score (Thompson et al., 1999), which computes the proportion of matches in a reference alignment which are present in the predicted alignment,
(2) sensitivity (Sens), the proportion of base pairings in a reference parse which are recovered in the predicted parse,
(3) specificity or positive-predictive value (PPV), the proportion of base pairings in a predicted parse which are also present in the reference parse, and
(4) the Matthews correlation coefficient (MCC) (Matthews, 1975), which we approximate as
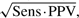 , following Gorodkin et al., (2001).
, following Gorodkin et al., (2001).
3.3 Comparison of accuracy
In our first accuracy assessment, we evaluated RAF as well as a number of other current RNA secondary structure prediction programs using the BRAliBASE II dataset. In particular, the first dataset from BRAliBASE II contains collections of 100 five-sequence subalignments, sampled from five specific Rfam families (5S rRNA, group II intron, SRP, tRNA and U5). For each of these alignments, we ran a number of current multiple-sequence RNA secondary structure prediction programs, including Murlet v0.1.1 (Kiryu et al., 2007), LocARNA v1.2.2a (Will et al., 2007), and RNA Sampler v1.3 (Xu et al., 2007). Wherever any of these programs required access to external pairing-posterior probabilities, we used ViennaRNA v1.7 (Hofacker et al., 1994). The results of the comparison are shown in Table 2.
Table 2.
Performance comparison on BRAliBASE II datasets. The best number in each column is marked in bold
| Dataset | Program | Time (s) | SP | Sens | PPV | MCC |
|---|---|---|---|---|---|---|
| 5S rRNA | Murlet | 687 | 0.94 | 0.70 | 0.70 | 0.70 |
| LocARNA | 812 | 0.93 | 0.55 | 0.60 | 0.57 | |
| RNA Sampler | 2361 | 0.90 | 0.55 | 0.64 | 0.59 | |
| RAF | 87 | 0.95 | 0.66 | 0.66 | 0.66 | |
| group II intron | Murlet | 962 | 0.78 | 0.75 | 0.76 | 0.75 |
| LocARNA | 250 | 0.74 | 0.79 | 0.65 | 0.72 | |
| RNA Sampler | 1626 | 0.72 | 0.77 | 0.65 | 0.71 | |
| RAF | 48 | 0.78 | 0.83 | 0.65 | 0.73 | |
| SRP | Murlet | 20548 | 0.88 | 0.75 | 0.78 | 0.76 |
| LocARNA | 22467 | 0.85 | 0.66 | 0.70 | 0.68 | |
| RAF | 1290 | 0.87 | 0.72 | 0.71 | 0.70 | |
| tRNA | Murlet | 525 | 0.93 | 0.86 | 0.90 | 0.88 |
| LocARNA | 246 | 0.95 | 0.86 | 0.90 | 0.88 | |
| RNA Sampler | 763 | 0.92 | 0.93 | 0.91 | 0.92 | |
| RAF | 52 | 0.94 | 0.81 | 0.85 | 0.83 | |
| U5 | Murlet | 1772 | 0.84 | 0.69 | 0.75 | 0.72 |
| LocARNA | 549 | 0.80 | 0.56 | 0.61 | 0.58 | |
| RNA Sampler | 4084 | 0.77 | 0.75 | 0.70 | 0.72 | |
| RAF | 99 | 0.82 | 0.83 | 0.79 | 0.81 |
As seen from the table, on the BRAliBASE II benchmark, RAF attains comparable accuracy to the other methods, achieving either the best or second-best overall accuracy according to MCC on four out of the five datasets. The running time of the method, however, is dramatically faster than the other algorithms, often taking an order of magnitude less time than many of the other programs.
We also obtained the dataset used in the benchmarking of the MASTR RNA secondary structure prediction program. For a number of different programs, pre-generated predictions for each input file are available for download on the MASTR website. In addition to scoring these pre-generated predictions, we also generated and scored predictions using Murlet and RAF. The results are shown in Table 3. In this benchmark set, RAF obtains the highest overall MCC.
Table 3.
Performance comparison on MASTR benchmarking sets. The best number in each column is marked in bold.
| Program | SP | Sens | PPV | MCC |
|---|---|---|---|---|
| CLUSTAL W+Alifold | 0.81 | 0.57 | 0.73 | 0.65 |
| FoldalignM | 0.78 | 0.38 | 0.81 | 0.55 |
| LocARNA | 0.75 | 0.41 | 0.77 | 0.56 |
| MASTR | 0.84 | 0.64 | 0.73 | 0.68 |
| Murlet | 0.89 | 0.62 | 0.78 | 0.70 |
| RNAforester | 0.53 | 0.55 | 0.55 | 0.55 |
| RNA Sampler | 0.82 | 0.65 | 0.70 | 0.67 |
| RAF | 0.88 | 0.68 | 0.77 | 0.72 |
We emphasize, however, that benchmarking results such as these should be taken with a grain of salt; both the BRAliBASE II and MASTR benchmarking sets are extremely restricted in their coverage of the space of RNA families, choosing to focus on a few individual RNA families only. As a result, methods carefully tuned to the benchmarks may perform less well on diverse RNA families not found in either of these benchmarks. By using cross-validation, we improve the chances that RAF's validation results really do indicate reliable out-of-sample performance.
We also note that the performance of RAF on particular RNA families is often closely related to the accuracy of the underlying alignment and single-sequence models used to derive folding and alignment constraints. Because the tools involved in the RAF pipeline all rely on automatic parameter learning, RAF allows the possibility of learning custom parameter sets well-suited for predictions on particular RNA families.
4 DISCUSSION
We presented RAF, a new tool for simultaneous folding and alignment of RNA sequences which exploits sparsity in base pairing and alignment probability matrices and max-margin training in order to achieve faster running times and higher accuracy than previous tools.
Besides its speed, one principal advantage of the RAF meth-odology is its use of a flexible scoring function for combining an arbitrary set of functions into a coherent objective function for alignment scoring. The ability to introduce new basis scoring functions into the RAF scoring model means that there remains a rich space of possible features to explore.
In addition, the use of the max-margin framework to identify relevant linear combinations of scoring functions has other promising potential applications. For example, Wallace et al. (2006) recently introduced M-Coffee, a meta-algorithm for protein sequence alignment, which combines the results of several different protein sequence alignment programs using the T-Coffee framework. The difficulty of identifying appropriate weights for the various programs used in the M-Coffee scoring scheme (i.e. some heuristically derived tree-based weights the authors tried did not give a significant improvement in accuracy over flat weights), led the authors to rely on a uniform weight model, treating programs known to be more accurate on equal footing with less accurate aligners. The max-margin framework developed in this paper obviates the need for heuristically-derived weights altogether.
ACKNOWLEDGEMENTS
C.B.D. was supported by an NSF Graduate Research Fellowship. C.S.F. was supported by an A*STAR National Science Scholarship. This material is based in part upon work supported by the NSF under grant number EF-0312459.
Conflict of Interest: none declared.
APPENDIX
A.1 RAF features
The features used by the RAF program, as evaluated in this article, consist of alignment features, 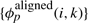 and pairing features,
and pairing features,  . Specifically, the alignment features, φaligned(i, k)∈ℝ4 for a candidate alignment match (ai, bk) are
. Specifically, the alignment features, φaligned(i, k)∈ℝ4 for a candidate alignment match (ai, bk) are
 |
(A1) |
The pairing features, φpaired(i, j; k, l)∈ℝ4 for a conserved base pairing 〈(ai, aj), (bk, bl)〉 are given by φpaired(i, j; k, l)=φpaired(ai, aj)+φpaired(bk, bl). In turn, φpaired(ai, aj)∈ℝ4 is given by
 |
(A2) |
and similarly for φpaired(bk, bl). Thus, the model contains a total of eight features whose weights must be learned. Here, the posterior probabilities for aligned positions and base-pairing positions are computed using the CONTRAlign (Do et al., 2006a) and CONTRAfold (Do et al., 2006b) programs, respectively.
B.1 The RAF inference engine
In the section, we describe the RAF inference engine for fast approximate simultaneous alignment and consensus folding for pairs of sequences. In particular, we first present some exact recurrences for alignment and folding, and then use restrictions on the set of allowed base pairings and aligned positions to achieve an improvement in computational complexity.
B.1.1 Recurrences
First, we describe a straightforward O(L6) dynamic programming recurrence for computing the optimal simultaneous alignment and consensus fold for a pair of sequences a and b.
To compute the optimal parse of a and b, we construct 2 four-dimensional matrices, S and D. Here, Si, j; k, l denotes the optimal score for aligning and folding ai+1ai+2…aj with bk+1bk+2…bl. Furthermore, Di, j; k, l denotes the optimal score for aligning and folding these same substrings, subject to the additional constraint that the outermost positions (ai+1,aj) and (bk+1,bl) form conserved base pairs.
For 0≤i≤j≤|a| and 0≤k≤l≤|b|, we have
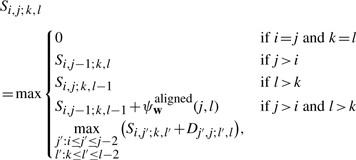 |
(B1) |
and for 0≤i<i+2≤j≤|a| and 0≤k<k+2≤l≤|b|,
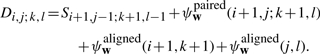 |
(B2) |
Here, recurrence (B1) takes the form of a standard Needleman-Wunsch procedure for aligning the substring ai+1ai+2… aj with bk+1bk+2…bl, with an extra case to handle bifurcations in the base-pairing structure of the RNAs. At the end of the recurrence, S0,|a|;0,|b| gives the score of the optimal alignment and consensus fold of the input sequences a and b. By using traceback pointers in the standard way, the optimal parse can be recovered easily once the recurrence has been evaluated.
In the next section, we explore how these recurrences may be sped up considerably if a constraint set 𝒞 of allowed base pairings and aligned positions is known ahead of time. For complexity analysis, we assume O(c) and O(d) bounds on the number of candidate base pairing and alignment partners per sequence position, respectively.
B.1.2 Exploiting base-pairing sparsity
LocARNA (Will et al., 2007) was the first program for simultaneous alignment and folding of RNA to take advantage of base pairing sparsity in a manner that significantly improved in both running time and memory usage. In this section, we recount the innovations of LocARNA as they are applied in RAF. In the next section, we extend these ideas to also account for alignment sparsity.
First, observe that since all parses in 𝒴𝒞 contain only conserved base pairings, the evaluation of (B2) may be restricted to only those Di, j; k, l cells for which both (ai+1,aj)∈𝒞 and (bk+1,bl)∈𝒞. Similarly, the inner loop for considering bifurcations in (B1) may also be restricted to only those j′ and l′ for which both (aj′+1,aj)∈𝒞 and (bl′+1,bl)∈𝒞. Since the bottleneck in the dynamic programming complexity is the number of executions of the innermost loop in (B1), it follows that restricting the considered bifurcations in the manner described above yields an O(c2L4) running time; in particular, for each i and k, computing all values of Si,•;k,• takes O(c2L2) time as each entry of the D matrix is touched at most once. This optimization was originally implemented as part of the LocARNA (Will et al., 2007) and FoldAlignM (Torarinsson et al., 2007) algorithms.
Second, consider the task of computing all entries in the D matrix. From (B2), we see that the values Di,•;k,• depend only on Si+1,•;k+1,•. Similarly, from (B1), the values Si+1,•;k+1,• depend only on Dj′,j;l′,l for j′≥i+1 and l′≥k+1. Thus, ordering computations in the following way allows the recurrences to be evaluated in a single pass:
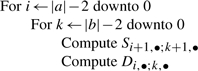 |
Furthermore, since Si+1,•;k+1,• is only needed while computing Di,•;k,• (but not for any later values of i and k), we need only to retain one Si+1,•;k+1,• matrix in memory at any given time while computing the D matrix. This observation was originally incorporated in the LocARNA program of Will et al., (2007).
Finally, observe that once the D matrix has been computed, the score S0,|a|;0,|b| of the optimal parse is easily obtainable in O(c2L2) time by recomputing S0,•;0,•. Likewise, computing the full traceback requires at most O(c2L3) time, negligible relative to the cost of computing the D matrix itself. Thus, we obtain an overall O(c2L4) time complexity with O(c2L2) space complexity (for storing the D matrix).
B.1.3 Exploiting alignment sparsity
To exploit sparsity in the set of allowed aligned positions in 𝒞, we again use the strategy of limiting the DP region. We accomplish this by first considering the simpler problem of computing the reduced DP region 𝒜 (known as the alignment envelope) for pairwise sequence alignment without folding scores. Using 𝒜, we then define a reduced DP region for our original alignment and folding task.
For the first step, consider the following restatement of recurrence (B1) using the notation 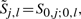 , where we have omitted the case involving bifurcations/base pairing:
, where we have omitted the case involving bifurcations/base pairing:
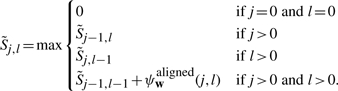 |
As before, 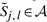 represents the optimal score of aligning a1a2…aj to b1 b2 … bl. Here, our goal is to find 𝒜, the minimal set of cells containing no holes,8 such that for every parse y∈𝒴𝒞, there exists some DP path through 𝒜 corresponding to an alignment with the same set of aligned positions. Under the assumption that 𝒜 contains no holes, we can represent 𝒜 by keeping track of its boundaries: for each j∈{0,1,…,|a|}, let 〈{𝒜}.First[j], 𝒜.Last[j]〉 denote the first and last positions l∈{0,1,…,|b|} such that
represents the optimal score of aligning a1a2…aj to b1 b2 … bl. Here, our goal is to find 𝒜, the minimal set of cells containing no holes,8 such that for every parse y∈𝒴𝒞, there exists some DP path through 𝒜 corresponding to an alignment with the same set of aligned positions. Under the assumption that 𝒜 contains no holes, we can represent 𝒜 by keeping track of its boundaries: for each j∈{0,1,…,|a|}, let 〈{𝒜}.First[j], 𝒜.Last[j]〉 denote the first and last positions l∈{0,1,…,|b|} such that 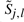 .
.
We compute these boundaries in linear time using the following procedure. First, we adjust the boundaries to include 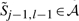 and
and 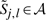 for each candidate aligning pair (aj,bl)∈𝒞. In addition, we also include the corners
for each candidate aligning pair (aj,bl)∈𝒞. In addition, we also include the corners 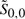 and
and 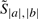 in 𝒜. Finally, we force the boundaries of 𝒜 to satisfy the monotonicity conditions
in 𝒜. Finally, we force the boundaries of 𝒜 to satisfy the monotonicity conditions
in such a way that guarantees all DP cells 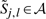 are accessible via some DP path from
are accessible via some DP path from 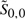 to
to 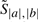 .
.
For the second step, we define the reduced DP region for our original simultaneous alignment and folding recurrences as the set ℛ of all positions Si, j; k, l such that 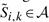 and
and 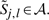 . To use this reduced DP region ℛ, then, we simply force Si, j; k, l=−∞ for all Si, j; k, l ∉ ℛ. Under this restriction, we can reduce the amount of computation performed in the recurrence (B1) by iterating only over cells Si, j; k, l∈ℛ, and similarly, restricting the evaluation of the D matrix in (B2) to only those cells Di, j; k, l for which Si+1,j−1;k+1,l−1∈ℛ. To ensure that each allowed parse belongs to 𝒴𝒞, we could penalize any base pairing or aligned position not in 𝒞 by −∞. In practice, we instead augment 𝒞 to include all aligned matches allowed by ℛ, since this can be done at no increase in computational complexity.
. To use this reduced DP region ℛ, then, we simply force Si, j; k, l=−∞ for all Si, j; k, l ∉ ℛ. Under this restriction, we can reduce the amount of computation performed in the recurrence (B1) by iterating only over cells Si, j; k, l∈ℛ, and similarly, restricting the evaluation of the D matrix in (B2) to only those cells Di, j; k, l for which Si+1,j−1;k+1,l−1∈ℛ. To ensure that each allowed parse belongs to 𝒴𝒞, we could penalize any base pairing or aligned position not in 𝒞 by −∞. In practice, we instead augment 𝒞 to include all aligned matches allowed by ℛ, since this can be done at no increase in computational complexity.
To analyze the new computational complexity of the algorithm, we begin by bounding the size of D matrix in two different ways. First, for each of the O(cL) base pairs (ai, aj)∈𝒞, there are O(d) aligning partners for ai and O(d) aligning partners for aj, giving a total size of O(cd2L). Alternatively, for each of the O(dL) aligning pairs (ai, bk)∈𝒞, there are O(c) base-pairing partners for ai and O(c) base-pairing partners for bk, giving a total size of O(c2dL). Thus, the size of the D matrix is O(min(c,d)·cdL).
As in Section B.1.2, the space complexity of the algorithm is dominated by cost of storing the D matrix, and hence, is O(min(c,d)·cdL). Similarly, the time complexity can be estimated as the number of evaluations of the innermost loop in the bifurcation case of (B1). Since the innermost loop touches each entry of the D matrix at most once for each i and k, and since there are O(dL) choices of (ai, bk)∈𝒜, it follows that the time complexity of the algorithm is O(min(c,d)·cd2L2).9
C.1 Norm bound
In this section, we derive a bound on the maximum norm of the optimal parameter vector w* for (4). From standard arguments (see, e.g. Taskar et al., 2003), the dual optimization problem is
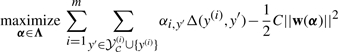 |
where
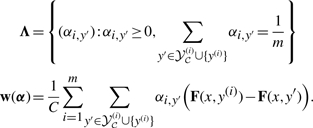 |
By strong duality, for any solutions (w*,ξ*) and α* of the primal and dual optimization problems, respectively, the values of the primal and dual objectives must be equal, i.e.,
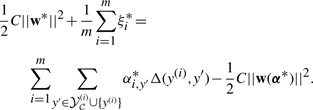 |
(C1) |
Now, suppose that Di∈ℝ for i=1,…,m satisfy
| (C2) |
In the case of the RAF loss function, for example, we can use
Then the KKT optimality condition w*=w(α*), the primal constraint that 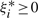 for i=1,…,m, and (C1) imply that
for i=1,…,m, and (C1) imply that
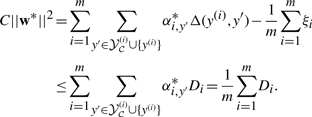 |
Therefore, 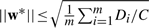 . ▪
. ▪
Footnotes
1We say that a parse y of inputs a and b is valid provided that (1) each nucleotide of a and b base pairs with at most one other nucleotide in the same sequence; (2) each nucleotide aligns with at most one nucleotide in the opposite sequence; (3) neither sequence contains pseudo-knotted base pairings; (4) the alignment of the two sequences does not contain rearrangements or repeats; and (5) all base pairings are conserved.
2The original CONTRAlign program was designed for protein sequences. We adapted this for RNAs by removing all protein-specific features (e.g. hydrophobicity), modifying the underlying alphabet (A, C, G and U) and simply retraining on the appropriate training set.
3We note that the method described here bears some relation to the ‘candidate list’ algorithm of Wexler et al. (2007), which maintains sparse lists of potential bifurcation points for single sequence folding. By showing that the number of relevant bifurcation points has a negligible dependence on sequence length, the authors provide an effectively quadratic time algorithm for single-sequence folding. Here, our algorithm also relies on sparsity of bifurcation point candidates when dealing with pairwise alignment and folding, but unlike in the previous algorithm, the candidates are provided explicitly via the constraint set 𝒞.
4Note that our notation hides the dependencies of the Score function on each of the input sequences a(i) and b(i), and similarly for the unconstrained and constrained space of parses, 𝒴(i) and 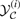 .
.
5By default, we used C=1. We found that when running the online Pegasos optimization algorithm (Section 2.4.3) for a fixed number of iterations, the resulting generalization performance for RAF is relatively insensitive to the value of C used, provided that C is not too large.
6By default, we used γFN paired=10, and γFP paired=γFN aligned=γFP aligned=1 in order to emphasize prediction of correct base pairings.
7In practice, we found that using cutoffs of ɛaligned∼0.01 and ɛpaired ∼ 0.002 gave a good trade-off between speed and accuracy of our algorithm when using CONTRAlign and CONTRAfold; these cutoffs correspond roughly to average sparsity factors of ∼10 each, respectively.
8That is, 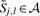 whenever
whenever 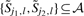 for some j1<j<j2, or
for some j1<j<j2, or 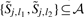 for some l1<l<l2.
for some l1<l<l2.
9Note that in these bounds, we assume an O(c) bound on the number of base-pairing partners per position, and an O(d) bound on the number of aligning partners per position. A weaker condition would be to assume an O(cL) bound on the total number of candidate base-pairing partners for sequences a and b and similarly, an O(dL) bound on the total number of candidate aligned positions; under these conditions, we obtain a worst-case space complexity of O(min(c,d)2 L2) and a worst case time complexity of O(min(c,d)2dL3).
REFERENCES
- Andronescu M, et al. Efficient parameter estimation for RNA secondary structure prediction. Bioinformatics. 2007;23:19–28. doi: 10.1093/bioinformatics/btm223. [DOI] [PubMed] [Google Scholar]
- Bertsekas DP, et al. Athena Scientific; 2003. Convex analysis and optimization. [Google Scholar]
- Brion p, Westhof E. Hierarchy and dynamics of RNA folding. Annu. Rev. Biophys. Biomol. Struct. 1997;26:113–137. doi: 10.1146/annurev.biophys.26.1.113. [DOI] [PubMed] [Google Scholar]
- Dalli D, et al. STRAL: progressive alignment of non-coding RNA using base pairing probability vectors in quadratic time. Bioinformatics. 2006;22:1593–1599. doi: 10.1093/bioinformatics/btl142. [DOI] [PubMed] [Google Scholar]
- Dieffenbach CW, et al. General concepts for PCR primer design. PCR Methods Appl. 1993;3:30–37. doi: 10.1101/gr.3.3.s30. [DOI] [PubMed] [Google Scholar]
- Do CB, et al. PROBCONS: probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–340. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do CB, et al. RECOMB. 2006a. CONTRAlign: discriminative training for protein sequence alignment; pp. 160–174. [Google Scholar]
- Do CB, et al. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics. 2006b;22:e90–e98. doi: 10.1093/bioinformatics/btl246. [DOI] [PubMed] [Google Scholar]
- Dowell RD, Eddy SR, et al. Efficient pairwise RNA structure prediction and alignment using sequence alignment constraints. BMC Bioinformatics. 2006;7:400. doi: 10.1186/1471-2105-7-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Computational genomics of noncoding RNA genes. Cell. 2002;109:137–140. doi: 10.1016/s0092-8674(02)00727-4. [DOI] [PubMed] [Google Scholar]
- Feng DF, Doolittle RF. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol. 1987;25:351–360. doi: 10.1007/BF02603120. [DOI] [PubMed] [Google Scholar]
- Gardner PP, et al. A benchmark of multiple sequence alignment programs upon structural RNAs. Nucleic Acids Res. 2005;33:2433–2439. doi: 10.1093/nar/gki541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodkin J, et al. Finding the most significant common sequence and structure motifs in a set of RNA sequences. Nucleic Acids Res. 1997;25:3724–3732. doi: 10.1093/nar/25.18.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodkin J, et al. Discovering common stem-loop motifs in unaligned RNA sequences. Nucleic Acids Res. 2001;29:2135–2144. doi: 10.1093/nar/29.10.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmanci AO, et al. Efficient pairwise RNA structure prediction using probabilistic alignment constraints in Dynalign. BMC Bioinformatics. 2007;8 doi: 10.1186/1471-2105-8-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL, et al. Fast folding and comparison of RNA secondary structures (The Vienna RNA Package) Monatsh. Chem. 1994;125:167–188. [Google Scholar]
- Hofacker IL, et al. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- Hofacker IL, et al. Alignment of RNA base pairing probability matrices. Bioinformatics. 2004;20:2222–2227. doi: 10.1093/bioinformatics/bth229. [DOI] [PubMed] [Google Scholar]
- Holmes I. Accelerated probabilistic inference of RNA structure evolution. BMC Bioinformatics. 2005;6 doi: 10.1186/1471-2105-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu H, et al. Murlet: a practical multiple alignment tool for structural RNA sequences. Bioinformatics. 2007;23:1588–1598. doi: 10.1093/bioinformatics/btm146. [DOI] [PubMed] [Google Scholar]
- Knudsen B, Hein J, et al. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 2003;31:3423–3428. doi: 10.1093/nar/gkg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgreen S, et al. MASTR: multiple alignment and structure prediction of non-coding RNAs using simulated annealing. Bioinformatics. 2007;23:3304–3311. doi: 10.1093/bioinformatics/btm525. [DOI] [PubMed] [Google Scholar]
- Mathews DH, Turner DH. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J. Mol. Biol. 2002;317:191–203. doi: 10.1006/jmbi.2001.5351. [DOI] [PubMed] [Google Scholar]
- Mathews DH, et al. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Matthews BW. Comparison of predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- Sankoff D. Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J. Appl. Math. 1985;45:810–825. [Google Scholar]
- Shalev-Shwartz S, Singer Y. 2007. Logarithmic regret algorithms for strongly convex repeated games, 2007. [Google Scholar]
- Shalev-Shwartz S, et al. ICML. 2007. Pegasos: Primal estimated sub-gradient solver for svm; pp. 807–814. [Google Scholar]
- Sneath PH, Sokal RR. Numerical taxonomy. Nature. 1962;193:855–860. doi: 10.1038/193855a0. [DOI] [PubMed] [Google Scholar]
- Tabei Y, et al. SCARNA: fast and accurate structural alignment of RNA sequences by matching fixed-length stem fragments. Bioinformatics. 2006;22:1723–1729. doi: 10.1093/bioinformatics/btl177. [DOI] [PubMed] [Google Scholar]
- Taskar B, et al. NIPS 16. 2003. Max-margin markov networks. [Google Scholar]
- Thompson JD, et al. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 1999;27:2682–2690. doi: 10.1093/nar/27.13.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torarinsson E, et al. Thousands of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA structure. Genome Res. 2006;16:885–889. doi: 10.1101/gr.5226606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torarinsson E, et al. Multiple structural alignment and clustering of RNA sequences. Bioinformatics. 2007;23:926–932. doi: 10.1093/bioinformatics/btm049. [DOI] [PubMed] [Google Scholar]
- Touzet H, Perriquet O. Nucleic Acids Res., 32 (Web Server) 2004. CARNAC: folding families of related RNAs; pp. W142–W145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace IM, et al. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 2006;34:1692–1699. doi: 10.1093/nar/gkl091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler Y, et al. A study of accessible motifs and RNA folding complexity. J. Comput. Biol. 2007;14:856–872. doi: 10.1089/cmb.2007.R020. [DOI] [PubMed] [Google Scholar]
- Will S, et al. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007;3 doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, et al. RNA Sampler: a new sampling based algorithm for common RNA secondary structure prediction and structural alignment. Bioinformatics. 2007;23:1883–1891. doi: 10.1093/bioinformatics/btm272. [DOI] [PubMed] [Google Scholar]