

Abstract
Despite a recent surge of interest in database-independent peptide identifications, accurate de novo peptide sequencing remains an elusive goal. While the recently introduced spectral network approach resulted in accurate peptide sequencing in low-complexity samples, its success depends on the chance of presence of spectra from overlapping peptides. On the other hand, while multistage mass spectrometry (collecting multiple MS 3 spectra from each MS 2 spectrum) can be applied to all spectra in a complex sample, there are currently no software tools for de novo peptide sequencing by multistage mass spectrometry. We describe a rigorous probabilistic framework for analyzing spectra of overlapping peptides and show how to apply it for multistage mass spectrometry. Our software results in both accurate de novo peptide sequencing from multistage mass spectra (despite the inferior quality of MS 3 spectra) and improved interpretation of spectral networks. We further study the problem of de novo peptide sequencing with accurate parent mass (but inaccurate fragment masses), the protocol that may soon become the dominant mode of spectral acquisition. Most existing peptide sequencing algorithms (based on the spectrum graph approach) do not track the accurate parent mass and are thus not equipped for solving this problem. We describe a de novo peptide sequencing algorithm aimed at this experimental protocol and show that it improves the sequencing accuracy on both tandem and multistage mass spectrometry.
Availability: The open-source implementation of our software is available at http://proteomics.bioprojects.org.
Contact: bandeira@ucsd.edu
Supplementary information:: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The coupling of tandem mass spectrometry (MS2) with database search tools (Perkins et al., 1999; Tanner et al., 2005; Yates et al., 1995) is the enabling core behind high-throughput protein identification (Aebersold and Mann, 2003). Unfortunately, this successful strategy is not applicable to unknown protein sequences. Particularly important examples of value derived from initially unknown proteins include antibody drugs such as Herceptin™ or Avastin™ (Haurum, 2006) and drugs derived from venom proteins (Pimenta and De Lima, 2005). Antibodies illustrate the scenario where the universe of possible protein sequences is very large and constantly altered by recombination and somatic hypermutation (Maizels, 2005). Additionally, drugs derived from venom proteins exemplify benefits of exploring the viable proteins already probed by natural biodiversity. Another recently emerged application of de novo peptide sequencing is proteogenomics, using mass spectra to predict new genes and alternatively spliced variants (Edwards, 2007; Gupta et al., 2007; Tanner et al., 2007). Proteogenomics studies would greatly benefit from searching 6-frame translations of entire genomes that amounts to roughly 6 billion amino acids for the mammalian genomes. The database search approaches are impractical for such large searches while de novo-based approaches to protein identifications (e.g. Alves and Yu, 2005) are not seriously affected by the database size.
MS-based studies of unknown proteins rely on de novo peptide sequencing techniques that attempt to recover the peptides directly from the spectra. Unfortunately, despite decades of research, accurate de novo peptide sequencing remains an elusive goal with only 30–40% of all peptides (depending on the sample and peptide size) reconstructed correctly (Pevtsov et al., 2006). While algorithms have been developed to find a best-‘scoring’ peptide for a given spectrum (Bafna and Edwards, 2003; Chen et al., 2001; Dancík et al., 1999; Fischer et al., 2005; Frank and Pevzner, 2005; Ma et al., 2003), their sequencing accuracy is still affected by incomplete fragmentation, noise and ambiguity in ion-type assignments. In abundant, low-complexity samples, these difficulties may be attenuated by generating overlapping peptides and combining the resulting MS2 spectra to yield higher accuracy de novo sequences—examples include 16O/18O labeling (Shevchenko et al., 1997) or Shotgun Protein Sequencing (Bandeira et al., 2004, 2007a). While Shotgun Protein Sequencing was shown to work well for low-complexity samples it remains unclear how it will scale up for complex samples. Multistage MS (e.g. generating multiple MS3 spectra for each MS2 spectrum) could be an ideal method to address the potential limitations of Shotgun Protein Sequencing in a controlled and even experiment-driven fashion. However, while various groups recently demonstrated the advantages of multistage MS for database search (Kalkum et al., 2003; Olsen and Mann, 2004; Ulintz et al., 2008), there are currently no software tools for de novo interpretation of multistage MS data. As a result, the multistage MS data are still manually interpreted, not unlike tandem MS in mid-1990s, before the first de novo peptide sequencing tools were developed.
Given a spectrum s, de novo peptide sequencing algorithms attempt to find a peptide π that best ‘explains’ the spectrum (e.g. maximizes the probability of generating the spectrum s). In multi-spectra peptide sequencing, one attempts to find a peptide π that best explains multiple spectra s1, … ,sk at once. Multistage MS is an example of the multi-spectra peptide sequencing problem. In certain aspects, interpretation of multistage MS data is more challenging than interpretation of multiple tandem mass spectra (as in Bandeira et al., 2007a) since MS3 spectra typically have lower quality than MS2 spectra. The manual usage of MS3 spectra as an aid to de novo sequencing dates back to a decade ago (Lin and Glish, 1998). To the best of our knowledge, the only automated approach to multistage MS is a heuristic approach proposed by Zhang and McElvain (2000). However, the sequencing accuracy of this approach remains unknown since Zhang and McElvain were limited to only 42 sets of multistage spectra and the accuracy gains from the proposed heuristics were described qualitatively rather than quantitatively. Moreover, this approach has not enthused further MS2/MS3 sequencing efforts because no implementation is publicly available. Our multi-spectra peptide sequencing algorithm was benchmarked on a set of 602 MS2 spectra with varying numbers of dependent MS3 spectra (yeast dataset). To further evaluate our algorithm we also tested it on 10 517 MS2 spectra for which spectra from overlapping prefix/suffix peptides were available.
Sequencing an MS2 spectrum in conjunction with k dependent MS3 spectra entails searching for the best-scoring peptide while considering every possible combination of fragment types for the MS3 spectra (i.e. was the MS3 generated from a b- or a y-ion). Our approach explores this search space by using dynamic programming to find the best peptide for each of 2k possible MS3 fragment-type assignments. We build on a probabilistic model (Frank and Pevzner, 2005) to score peptide-spectrum matches and extend it to include the particularities of MS2/MS3 sequencing. By incorporating these readily available experimental capabilities into a rigorous algorithmic framework, we show that overlapping spectra can significantly increase de novo sequencing accuracy and even make it almost error free when enough ‘usable’ MS3 spectra are available. As an addition (or alternative) to overlapping spectra, we also propose an efficient dynamic programming algorithm for de novo peptide sequencing with accurate peptide masses and evaluate the resulting contribution to de novo sequencing accuracy.
2 METHODS
Datasets The yeast dataset was acquired from a trypsin digestion of a yeast whole-cell lysate on a Thermo LTQ-FT instrument configured for the acquisition of up to five MS3 spectra from each MS2 spectrum with a doubly charged precursor; precursor masses for the MS3 spectra were restricted to masses higher than the MS2 precursor mass. As a result, we obtained a total of 3184 MS2 spectra with 15 770 dependent MS3 spectra. These spectra were searched against a yeast database using InsPecT (details are provided in Supplementary Materials) and resulted in the identification of 890 out of all 3148 MS2 spectra (28%), having a total of 4447 dependent MS3 spectra.1. In general, we observed that MS3 spectra tend to have less explained intensity and less b/y-ion peaks than MS2 spectra (details shown in Table 1). The shewanella dataset was acquired on Thermo LTQ instruments over many LC/MS/MS runs with different multi-dimensional separation techniques (Gupta et al., 2007). For the purpose of this study we arbitrarily selected 34 326 MS2 spectra from different peptides, resulting in 10 517 MS2 spectra with between one and five other MS2 spectra from prefix/suffix peptides. From a computational perspective, this shewanella MS2 dataset mimics the yeast MS2/MS3 dataset with unrealistically high quality of MS3 spectra and thus allows one to establish an upper bound on the performance of our multistage MS approach.
Table 1.
Spectrum ion statistics for the yeast dataset
| Type of spectra | Number of spectra | Intensity (%) |
Number of peaks | p (b) | p (y) | ||||
|---|---|---|---|---|---|---|---|---|---|
| b | y | Satellite | Unexplained | ||||||
| a) | MS/MS (MS2) | 890 | 13 | 30 | 20 | 37 | 542 | 0.81 | 0.84 |
| MS/MS/MS (MS3) | 4447 | ||||||||
| from y-ions | 2592 | 11 | 17 | 23 | 49 | 62 | 0.41 | 0.51 | |
| from b-ions | 1039 | 11 | 1 | 25 | 64 | 47 | 0.27 | 0.09 | |
| b) | Scored MS2 | 890 | 28 | 28 | 23 | 31 | 80 | 0.81 | 0.81 |
| Scored MS3 | 4447 | ||||||||
| from y-ions | 2592 | 20 | 20 | 16 | 44 | 41 | 0.53 | 0.53 | |
| from b-ions | 1039 | 14 | 3 | 22 | 61 | 36 | 0.30 | 0.12 | |
| c) | Merged (MS2 + MS3) | 602 | 66 | 8 | 7 | 19 | 83 | 0.89 | 0.77 |
| d) | Merged + sequenced | 602 | 92 | 1 | 1 | 6 | 11 | 0.82 | 0.01 |
a) Ion statistics for RAW spectra. b) Ion statistics after replacing each peak's raw intensity with a likelihood score (Frank and Pevzner, 2005). c) Ion statistics of the consensus spectra obtained by merging each MS2 spectrum with its dependent MS3 spectra (after scoring). This category has less spectra (602) than the others (890) mainly because not all MS2 spectra had at least one ‘usable’ MS3 spectrum (details in main text). d) Ion statistics for the peaks selected by de novo sequencing on the merged spectra.
Spectrum identification Similar to the protocol used for the identification of spectra in the shewanella dataset (Gupta et al., 2007), InsPecT (Tanner et al., 2005) was used to search a database containing 7517 Saccharomyces cerevisiae protein sequences (SwissPROT, October 8, 2006) and sequences of common contaminant proteins. The database additionally contained 7517 decoy protein sequences used to enforce the selected 5% false-discovery rate. InsPecT was configured to allow for 0.5 Da fragment mass tolerance and 2.5 Da precursor mass tolerance; the high accuracy of the experimental precursor masses was used to confirm identifications rather than to restrict the space of possible peptides. The set of allowed modifications was oxidation (M), phosphorylation (S, T and Y), acetylation (N-term), deamidation (Q) and 13C615N2 SILAC label (K). Since our approach does not address sequencing with post-translational modifications, we discarded all modified peptides with a single exception of the abundant SILAC-K whose mass was added as a 21st amino acid.
The search identified 890 out of all 3148 MS2 spectra (28%). However, only 1282 MS3 spectra out of 15 770 (8%) resulted in a significant match to the database. To further increase the number of annotated MS3 spectra, we matched each MS3 precursor mass to the theoretical fragment masses from the peptide assigned to the parent MS2 spectrum. Note that this procedure does not increase the number of identified peptides, but rather allows us to better characterize the data and evaluate the algorithms described below. The selection of unambiguous MS3 precursor mass matches resulted in 1039 MS3 spectra annotated as prefix fragments (b-ions), 2592 MS3 spectra annotated as suffix fragments (y-ions) and 320 spectra annotated as prefix/suffix fragments after loss of H2O or NH3. All dependent MS3 spectra from all identified MS2 spectra were used for de novo sequencing.
The ion statistics for all identified yeast spectra (shown in Table 1) allow us to quantify the differences between MS2 and MS3 spectra. In general, the latter tend to have less explained intensity and less b/y-ion peaks. Note that these observations are not entirely surprising, because we only consider the MS2 spectra that had a strong match to the database while most MS3 spectra were identified only by their precursor mass.
We note that the different number of spectra in Table 1(b, c) stems from the fact that 202 MS2 spectra did not generate any usable MS3 spectrum and also because 86 MS2 spectra contained modified residues not considered for our current de novo sequencing purposes. The only exception to the latter was SILAC-labeled Lysine because of the high number of peptides containing this modification.
De novo-peptide sequencing problem The best de novo sequencing algorithms use probabilistic models that capture multiple features such as peak intensities and expected propensities of the different ion types (Dancík et al., 1999; Fischer et al., 2005; Frank and Pevzner, 2005; Ma et al., 2003). We start by introducing a model that seemingly has nothing to do with de novo peptide sequencing, but rather describes a very general probabilistic process that transforms one Boolean string into another. We will show later that this process not only generalizes the probabilistic model from (Dancík et al., 1999) but also allows one to study de novo peptide sequencing from multiple spectra.
Let s=s1, … ,sn be a Boolean string called a spectrum and π=π1, …,πn be a Boolean string called a peptide. The probability of peptide π generating spectrum s is defined as P(s∣π)=Πi=1n P(si∣πi), where P(x∣y) is a 2 × 2 matrix.
Given a spectrum s and a set of strings Π, we are interested in solving the optimization problem of finding maxπ∈Π P(s∣π). Below we focus on the sets Π that are relevant in the context of tandem MS. Let V={1, …, n} and G(V,E) be a topological ordering of a Directed Acyclic Graph (DAG) such that i<j for every directed edge (i,j) in E. Every path from 1 to n in graph G corresponds to a G-peptide π=π1, …,πn such that πi=1 iff vertex i belongs to the path. We are interested in the following peptide sequencing (PS) problem.
PS problem Given a spectrum s and a DAG G, find a G-peptide Π maximizing P(s∣ π) over all G-peptides.
We impose no restrictions on the graph G(V,E), but in practical applications it is usually assumed that (i,j)∈E iff (j−i) equals the integer mass of an amino acid. Such graphs are referred to as spectrum graphs (Bartels, 1990; in MS, they usually encode all peptides of a given parent mass n).
The relation between this abstract Boolean strings model and de novo peptide sequencing is straightforward. In reality, an MS/MS spectrum can be represented as a string of ones (peak present) and zeros (no peak present), with a 0/1 for every consecutive 1 Da interval. Similarly, sequences of amino acid masses (peptides) can also be represented as strings of zeros and ones. Every amino acid can be represented as a string of α−1 zeros followed by a single one, where α is the integer amino acid mass. Then, a peptide is simply a concatenation of Boolean strings corresponding to its amino acids. In this context, θ≈0.05 (probability of observing a noise peak) and ρ≈0.7 (probability of observing a b-ion) represent typical values of θ and ρ for ion-trap MS/MS spectra (see table in Fig. 1A). This somewhat simplistic Boolean string model can be modified for any mass resolution, peptide-fragmentation rules and peak intensities (Bafna and Edwards, 2003; Chen et al., 2001; Frank and Pevzner, 2005). Moreover making this model more realistic typically does not affect the algorithmic solution. In particular, (Bandeira et al., 2007b) recently showed how spectral alignment can be used to separate between b/y-ions and thus generate strings as in this model.
Fig. 1.

(A) Probability P(x∣y) of a peptide symbol y generating a spectrum symbol x. Thus, the probability of a spectrum s=s1, … ,sn being generated by a peptide π=π1, …,πn is defined as P(s∣π)=Πi=1nP(si∣πi). (B) Construction of a HMM 𝒢 for a graph G. The bold (hidden) path corresponds to the 𝒢-peptide 10101001.
The PS Problem has an easy solution first described by (Dancík et al., 1999). We will find it convenient to cast the approach in (Dancík et al., 1999) as an application of the Viterbi algorithm (Durbin et al., 1999; Viterbi, 1967) in an appropriately constructed hidden Markov model (HMM) 𝒢. Figure 1 shows a graph G with every edge (i,j) substituted by a path with new j−i−1 traversal vertices that starts at i and ends at j. The resulting graph with vertex set V∪T (where V={1, …, n} and T is the set of all traversal vertices) represents the hidden states of the HMM 𝒢 and all possible transitions between the states. The emission probabilities of the HMM are defined by the matrix P(x∣y) with P(x∣y=0) for T states and P(x∣y=1) for all other states (see Fig. 1A). The transition probabilities of all edges in this HMM are defined to be 1.2 Finding an optimal path in this HMM is a straightforward application of the Viterbi algorithm.
The model above does not capture the fact that MS/MS spectra represent both prefix ions (b-ions series) and suffix ions (y-ions series). To reflect this we represent peptides as strings in 3-letter alphabet: 1 (theoretical b-cut), −1 (theoretical y-cut) and 0 (no cut). Given a peptide π=π1, …,πn, we define its reverse as the peptide π*=−πn, …,−π1, i.e. 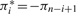 . We now redefine the probability of peptide π generating spectrum as s as
. We now redefine the probability of peptide π generating spectrum as s as 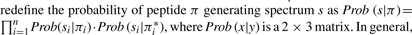 , where Prob(x∣y) is a 2 × 3 matrix. In general, k ions can be modeled by k+1 parameter columns in this matrix; note that using additional ion-types only changes the emission probabilities but not the construction of the HMM.
, where Prob(x∣y) is a 2 × 3 matrix. In general, k ions can be modeled by k+1 parameter columns in this matrix; note that using additional ion-types only changes the emission probabilities but not the construction of the HMM.
However, this and other formulations of the peptide sequencing problem encode a particular bias toward peptides that have b-ions bi, bj such that bi and bj add up to the peptide mass. In these cases, both ions use the same masses in the spectrum to artificially increase the score of the peptide but do so with conflicting ion type assignments (because bi=yj and bj=yi). Peptides that do not have such pairs of b-ions are referred to as anti-symmetric peptides and efficient algorithms are available to find the maximum scoring anti-symmetric peptide for any given spectrum (Bafna and Edwards, 2003; Chen et al., 2001).3 Nevertheless, ambiguous b/y-ion assignments remain one of the main sources of de novo sequencing errors. We show below how MS3 spectra can help resolve these ambiguities.
Although this model still essentially amounts to maximizing the weighted number of matched masses between a spectrum and a peptide, it already captures enough detail to allow us to describe the proposed extensions for combining MS2/MS3 spectra. In practice, this same framework is used to model more elaborate events to take into consideration the intensity of the peaks in the spectrum and to account for the presence/absence of other ion types (e.g. b−H2O ions). As shown in Table 1, using more elaborate scoring from (Frank and Pevzner, 2005), one can replace raw intensities with peak scores to significantly increase the signal-to-noise ratio over all collected spectra. The resulting scored spectra have more intensity assigned to true fragment masses and feature a much smaller number of noise peaks while simultaneously retaining almost all b/y-ions.
Multi-spectra peptide sequencing problem The simultaneous sequencing of spectra from multiple peptides (e.g. PEPTIDE, PEPTID and EPTIDE) requires solving two problems: (i) finding the correct multiple alignment between all spectra (described in the next subsection) and (ii) reconstructing a maximal-scoring peptide from the aligned spectra. In the following we assume that the spectra are already aligned (as shown in Fig. 2) and describe the problem of peptide sequencing from multiple aligned spectra. We do not limit ourselves to MS2/MS3 (when all MS3 spectra correspond to prefixes or suffixes of the peptide corresponding to MS2 spectrum), but rather consider arbitrary sets of overlapping peptides.
Fig. 2.

Conceptual illustration of peak scoring on an MS2 spectrum (e.g. from PEPTIDE) aligned with its dependent MS3 spectra (e.g. S1 from PEPTID and S2 from EPTIDE). Since the correct alignment between MS2/MS3 spectra is not known a priori, our algorithm finds the highest scoring peptide over all possible alignments. M indicates the parent mass of the MS2 spectrum and complementary masses (e.g. b/y-ions whose masses add up to the corresponding parent mass) are connected by arcs under each spectrum. Colors indicate the sets of masses accounted for when scoring different peaks; the colored squares in the equations represent the summed scores of all peaks of the same color; violet peaks indicate the b-ion masses dictated by the parent masses of the MS3 spectra. Intuitively, the score of a mass m summarizes all corroborating evidence for it being generated from a prefix fragment. The red and blue peaks illustrate the contribution of the MS3 spectra to the separation of b/y-ions in the MS2 spectrum—intuitively, if m is a b-ion and M−m is not then the red peaks should be more prominent than the blue peaks.
MS3 spectra contain additional information in the form of corroborating fragmentation peaks at the expected fragment masses in the MS2 and all MS3 spectra. When the MS3 spectra are aligned with the MS2 spectrum, the corroborating b-ions match ‘vertically’ while the matching y-ions are found at different positions depending on the parent mass of each MS3 spectrum (Fig. 2). As such, we now consider the problem of finding the most probable peptide π=π1, …,πn for a set of multiple (possibly overlapping) spectra s1, …,sk. Thus, the alignment between the MS2/MS3 spectra defines a substring mapping where 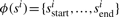 is the sequence of consecutive numbers from
is the sequence of consecutive numbers from 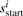 to
to 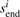 (
(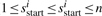 ) such that the substring
) such that the substring 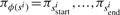 generates the spectrum si.
generates the spectrum si.
We now consider the following Multi-spectra peptide sequencing (MSPS) problem.
MSPS problem Given spectra s1, …,sk and a substring mapping φ, find a G−peptide Π maximizing 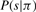 .
.
It is easy to see that the HMM constructed for the PS Problem (described above) can also solve the MSPS problem. The only difference between the two HMMs resides in the types of symbols emitted by the hidden states. Let S(i) be the set of all spectrum values generated from πi (defined by spectra s1, … ,sk and the mapping φ). Then, each hidden state now emits a set of independent values S(i)={vj}, with P(vj∣πi) given by the table in Figure 1 as before: P(x∣y=0) for T states and P(x∣y=1) for all other states. Assuming all vj are independent observations, the probability of observing any given set S(i) is ∏vj∈S(i) P(vj,πi). Once again, the MSPS problem can be solved by an anti-symmetric sequencing algorithm on 𝒢 (Bafna and Edwards, 2003; Chen et al., 2001). We note that although multiple ion-type assignments can be avoided in the MS2 spectrum, the resulting peptide may be somewhat biased by conflicting ion-type assignments in the MS3 spectra. While these conflicts could be readily avoided when only one overlapping spectrum is available (as described in Bandeira et al., 2007b), extending the algorithm to more overlapping spectra would lead to a significant computational burden unlikely to result in relevant gains in terms of sequencing accuracy.
A possible change to the MSPS problem would be to require the start/end positions of each spectrum as mandatory 1s in the returned peptide Π. This modified problem can also be solved using the strategy described above if we modify the HMM 𝒢 by removing all T states at the start/end positions of every spectrum. As such, every path through this modified HMM 𝒢 will be forced to use the states corresponding to these start/end positions and thus results only in peptides that are consistent with the alignment of MS3 spectra.
Aligning MS2/MS3 spectra Under common experimental conditions, the highest intensity peaks in MS2 spectra typically correspond to y- and, to a lesser extent, b-ions. Another common source of high-intensity peaks are doubly charged ions. However, on MS2 from doubly charged peptides one can avoid doubly charged fragment ions by considering only peaks with a mass higher than that of the precursor ion. By restricting the selection of MS3 precursor ions to this high-mass region our experimental setup (i) implicitly selected singly charged MS3 precursors from doubly charged MS2 precursors and (ii) biased toward MS3 spectra generated from b/y-ions.
As such, determining the correct alignment between an MS3 spectrum and its parent MS2 spectrum essentially reduces to determining the b/y-ion type of the MS3 precursor ion.
Given an MS2 spectrum from a particular peptide (e.g. PEPTIDE), the generation of an MS3 spectrum from one of its b-ions yields additional information about the corresponding prefix peptide (e.g. PEPTID). The converse reasoning applies to MS3 spectra from y-ions and suffix peptides (e.g. PTIDE). As such, the assignment of an MS3 spectrum to the correct ion type allows one to match the corroborating fragmentation from the same peptide regions and thus reinforce the confidence in co-occurring fragment masses. Since the correct ion-type assignments are not known in advance, one needs to explore 2k possible combinations of assignments—an easy task since k is usually small. Using the set of peaks with matching masses between the MS2 and MS3 spectrum (in either ion-type assignment), we define an MS3 spectrum as usable if the summed scores of the matched peaks include at least 25% of the total summed peak scores (in each spectrum).
Using the peptide sequencing framework described here, our approach explores all 2k possible ion-type assignments for an MS2 spectrum with k usable dependent MS3 spectra and selects the combination of assignments resulting in the highest scoring peptide. The same algorithm was used for the case of overlapping MS2 spectra, with the peptide dependencies (but not the b/y-ion assignments) determined from InsPecT identifications (see Bandeira et al., 2007b; for a blind approach to the detection of spectra from overlapping peptides).
De novo-peptide sequencing with accurate parent masses Nearly all de novo peptide sequencing algorithms use spectrum graphs (Bartels, 1990; Chen et al., 2001; Dancík et al., 1999; Fernández-de Cossío et al., 1995; Frank and Pevzner, 2005) for interpreting the spectra. The spectrum graph approach, while very useful, has an important (and often overlooked) shortcoming when it comes to handling the parent-mass tolerance. Indeed, due to errors in mass measurements, the lengths of edges in the spectrum graph may deviate from the exact masses of amino acids by as much as 0.5 Da for ion-trap instruments. In the case when all errors are +0.5 Da (unlikely but possible case), this can result in 0.5 × n error in the parent mass for a peptide of length n. Such large deviations may result in optimal but inadequate solutions thus reducing the accuracy of de novo peptide sequencing. While such inconsistent solutions represent a minor nuisance in the low-accuracy setting, they turn into a major problem in the case of highly accurate parent mass measurements. Indeed, in this case it becomes necessary to find the best solution with a given parent mass rather than the best solutions among all peptides encoded by the spectrum graph.
The accurate handling of accurate parent masses (with tolerances of 0.05 Da or lower) can be achieved by adding a third dimension to the standard anti-symmetric recursion (Chen et al., 2001; Dancík et al., 1999) to keep track of the cumulative mass errors (Fig. 3).
Fig. 3.

Dynamic programming recursion for de novo peptide sequencing of a spectrum S with accurate parent mass M. The variable S[u,v,k] represents the maximum gapped-peptide score over all anti-symmetric gapped-peptides with a prefix ending at peak u, a suffix starting at peak v and with cumulative mass error k; a gapped-peptide at position S[u,v,k] is defined as a (possibly incomplete) peptide consisting of a prefix peptide ending at peak u, a gap of mass m(v)−m(u) and a suffix peptide starting at peak v. Used notation: m(x)—mass of peak x; s(x)—score of peak x; 𝒜—set of amino acid masses; δ is the mass error incurred by modeling the peak mass difference m(x)−m(x′) with the amino acid mass a. In reality, user-specified fragment mass tolerances are allowed when appropriate. Also, the set of amino acid masses 𝒜 may include summed masses of pairs of amino acids (to allow for missing ions in the spectrum).
In practical terms, the memory requirements for the third dimension spanned by k can be controlled by binning the cumulative mass errors at a chosen resolution r. For typical values of t=0.5 Da, r=0.01 Da and T≪t this results in a need for 100× more memory than would have otherwise been needed. The memory requirements are on the order of O(m2/r), where m is the number of peaks in the spectrum. Since m is usually ≤100, these requirements can be easily met on any current desktop computer.5 We further note that the mass-error tolerances can easily be adjusted for each individual peak by varying the allowed range for k. In particular, peaks derived from the parent masses of overlapping peptides may be set to much lower mass tolerances than peaks from fragment masses.
3 RESULTS
When analyzed in isolation, individual MS3 spectra are less useful than individual MS2 spectra. Lower amounts of substrate, low charge and a bias toward shorter peptides result in spectra with generally inferior-ion statistics (Table 1) and consequently a much smaller percentage of identified spectra—8% of all MS3 spectra versus 28% of all MS2 spectra in the yeast dataset (using traditional database search). However, the combination of dependent MS3 spectra with the parent MS2 spectrum promptly reveals matching peaks from true peptide fragments and non-matching peaks from unexplained noise masses. Capitalizing on this corroborating fragmentation leads to a significant increase in signal-to-noise ratio with 19% intensity in non-explained peaks versus 31%/49% in MS2/MS3 spectra. Also, the distinct locations of b/y-ions in the aligned MS2/MS3 spectra (Fig. 2) allow one to separate between these ion types. Separating b/y-ions alleviates the uncertainties in peak ion-type assignments and thus reduces the probability of high-scoring incorrect peptide identifications. Furthermore, the parent masses of the MS3 spectra create a strong bias toward the corresponding b-ion masses. Since the overwhelming majority of all usable MS3 spectra come from true fragment masses, the resulting set of essentially mandatory b-ions significantly reinforces the score of the correct peptide, while severely reducing the probability of an incorrect high-scoring peptide match (see Fig. S1 Supplementary Materials). This feature is very important for de novo-based approaches to peptide identifications (like RAId-DbS, Alves and Yu, 2005). The main bottleneck of such approaches is a large set of full-length suboptimal peptides that need to be generated to ensure that the correct peptide is searched against a database. As shown in Figure S1, multistage mass spectrometry has a potential to reduce the number of such peptides by several orders of magnitude. The gains obtained from merging MS3 spectra with the corresponding parent MS2 spectra are summarized in Table 1c.
Our analysis revealed that scored MS3 spectra sometimes have b/y-ions absent in MS2 spectra. Such additional fragmentation is especially important in a de novo sequencing context because one implicity searches for the best peptide match over the space of all possible peptides of the observed parent mass. The combined contributions of the factors described above results in a strong bias toward the correct peptide sequence reflected in a significant increase in the average percentage of correctly predicted amino acids (from 85.7 to 90.7%, Fig. 4). Moreover, we note that this increase in sequencing accuracy is not achieved at the cost of making less predictions—sequencing MS2/MS3 spectra resulted in 4922 amino acid predictions versus 4772 for MS2 spectra only. As expected, having more usable MS3 spectra results in increasing de novo sequencing accuracy, generating almost-error-free sequences as soon as four usable MS3 spectra are available.
Fig. 4.

Improvements in de novo peptide sequencing from overlapping spectra and accurate parent masses. (A) Changes in amino acid prediction accuracy, defined as percentage of correct amino acids out of all amino acid predictions; (B) Changes in number of spectra yielding a correct tag of six amino acids or longer (see Bandeira et al., 2007b, for a discussion of how these long tags could suffice for peptide identification). Four different scenarios were found to result in significantly different sequencing results (shown in four different colors), with the yeast dataset mostly resembling the blue subset of the shewanella dataset (charge two precursors with average peptide length ≈11.5). Values for single spectra are marked with triangle and the corresponding de novo sequencing gains (from additional overlapping spectra) are marked with a diamond of the same color and in the same column; values from the same peptide subset are connected by either dashed (single spectra) or full (multiple spectra) lines. The plots for the single peptides are not perfectly horizontal since there are small variations in the spectral quality of subsets of MS2 spectra with i dependent MS3 spectra (for 1≤i≤5).
As illustrated by the blue lines in Figure 4, the gain in de novo sequencing accuracy is considerably higher when combining multiple MS2 spectra from overlapping peptides (shewanella dataset). The larger gains seem to be due to two distinct factors—(i) the lower average quality of MS2 spectra not yielding MS3 spectra (as compared to yeast MS2 spectra) and (ii) the better ion statistics of the MS2 spectra from overlapping peptides (as compared to yeast MS3 spectra). These two factors are clearly illustrated in Figure 4 by the differences between the blue/green dashed (case i) and full (case ii) lines. Nevertheless, increasing numbers of overlapping spectra also resulted in the the same trend of better sequencing accuracy.6 In addition to these blue/green cases where the peptides from the yeast and shewanella datasets were most similar, we also observed significant improvements in the traditionally difficult de novo sequencing of long peptides (13+ amino acids). Since these peptides tend to generate less singly charged b/y-ion fragments in the MS2 spectrum (especially for triply charged precursors), these cases gain the most from the additional fragmentation in the additional spectra from overlapping peptides. Thus, the sequencing accuracy of long peptides becomes essentially indistinguishable from that of short peptides and doubly charged precursors. Also, while the sequencing accuracy on MS2 spectra from triply charged precursors still remains slightly inferior, it roughly approaches (or exceeds, when accurate parent masses are available) the sequencing accuracy previously achievable only for spectra from doubly charged precursors.
The higher sequencing accuracy in combined MS2/MS3 spectra increases the number of spectra for which one can recover long subsequences (i.e. tags) of at least six consecutive amino acids. The availability of MS3 ameliorates but does not completely eliminate the difficulties in de novo sequencing of complete peptides caused by missing fragment masses and high-intensity noise peaks. Nevertheless, one is often able to confidently recover tags that may uniquely identify the peptide using a simple (and very efficient) text-based database search. Even when these long tags happen to match multiple locations in larger databases, the number of possibilities tends to be very small and can be quickly resolved by matching the N/C-terminal masses (tags are usually recovered from the middle of the spectrum) and additional peaks not included in the tag. These factors are especially relevant when de novo sequencing is followed by homology-tolerant searches such as those enabled by MS-Alignment (Tsur et al., 2005). In these cases, the tag-based efficiency gains (Bandeira et al., 2007b; Tanner et al., 2005) should combine with the improved ion statistics shown in Table 1d to simultaneously deliver faster and more accurate results.
4 CONCLUSION
High-throughput studies of unknown proteins such as recombinant (Pham et al., 2006) or venom (Birrell et al., 2007) proteins remain burdened by low-throughput experimental procedures such as Edman sequencing and the limited accuracy of de novo sequencing of individual tandem mass spectra. While related approaches increase the sequencing accuracy by combining multiple MS2 spectra from overlapping peptides (Bandeira et al., 2004; Bandeira et al., 2007a; Shevchenko et al., 1997), the experimental setup described here requires fewer sample handling steps (no need to digest with multiple enzymes) and should thus be applicable to smaller amounts of substrate.
We have demonstrated the utility of MS3 spectra for de novo peptide sequencing and compared the resulting gains with those obtainable from overlapping MS2 spectra. Although the ion statistics in individual MS3 spectra are usually too weak to allow reliable identification via database searching, the combination of multiple MS3 spectra with the parent MS2 spectrum results in higher signal-to-noise ratios and much improved separability of b/y-ions. By combining the corroborating fragmentation in multiple spectra our approach leads to increasing accuracy with higher numbers of usable MS3 spectra and even achieves almost error-free sequencing as soon as four usable MS3 spectra are available. The algorithmic incorporation of accurate parent masses was also shown to contribute to higher sequencing accuracy.
When applied to sets of overlapping MS2 spectra, the approach described here also resulted in sequencing accuracy improvements and practically erased the distinction between de novo sequencing of short and long peptides. These gains were even more pronounced for spectra of long peptides, including the traditionally difficult case of spectra from triply charged precursors.
The major reason why database search is more accurate than de novo sequencing is that the former only matches each spectrum to a relatively small number of peptides in the database, while the latter searches the space of all possible peptides. This distinction is especially relevant because peptide fragmentation is usually incomplete and generally confounded by noise peaks. Rather than requiring an a priori guess of the set of possible peptides, the simultaneous analysis of sets of spectra from distinct fragments of the same peptide provides an independent experimental bias toward the correct peptide and largely reduces the set of possible high-scoring alternatives. Since more spectra lead to increasingly restricted sets of high-scoring peptides, it follows that increasing the number of usable MS3 spectra should result in yet higher sequencing accuracy. Experimentally, higher fragment mass accuracy would significantly reduce the chances of spurious peak matches and should seamlessly increase the percentage of usable MS3 spectra. Alternatively, more elaborate statistical models could be used to predict the ion-types of MS3 precursor ions and eventually allow the utilization of MS3 spectra from doubly charged precursor ions. We further note that the approach described here is applicable to peptides of any length and could, in principle, be used in the context of top-down or middle-down (via limited proteolysis) proteomics experiments. However, algorithmic extensions may be necessary to account for MS3 spectra of internal peptide fragments.
ACKNOWLEDGEMENTS
The authors would like to thank Ari Frank and Sangtae Kim for insightful discussions.
Funding: This project was supported by NIH grant NIGMS 1-R01-RR16522.
Conflict of Interest: none declared.
Footnotes
1A total of 1039 MS3 spectra were annotated as prefix fragments (b-ions), 2592 MS3 spectra were annotated as suffix fragments (y-ions) and 320 spectra were annotated as prefix/suffix fragments after loss of H2O or NH3.
2Although these transition probabilities do not add up to 1, we prefer not to normalize them. This keeps the resulting probabilities of hidden paths consistent with the Dančik model and does not affect the Viterbi algorithm for finding an optimal path.
3An alternative approach used to enforce strict parent-mass tolerances is the mass array data structure (Mo et al., 2007), which defines mass bins with resolution r=0.01 Da over the whole spectrum. However, we note that extending the mass-array approach to enforce the anti-symmetric condition would result in prohibitive-memory requirements on the order of O(M2/r), where M could be anywhere between 400 and 6000 (for triple-charged precursor ions).
4In practice, peptides that are not anti-symmetric are not excluded from consideration, but special care is taken to avoid multiple ion-type assignments to the same spectrum peaks in the scoring function.
5The only notable exceptions (≈7% of all identified MS3 spectra) were the selection of MS3 precursors from neutral-loss ions (e.g. b−H2O, y−NH3).
6We note that the lower average sequencing accuracy with 5 overlapping MS2 spectra (in the blue line, shewanella dataset) was due to inaccurate parent masses and was thus surpassed when accurate parent masses were available. Also, even with perfect sequencing accuracy, these spectra sometimes lacked tags of length six because of missing peaks, even though tags of length five were readily available.
REFERENCES
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Alves G, Yu YK. Robust accurate identification of peptides (raid): deciphering ms2 data using a structured library search with de novo based statistics. Bioinformatics. 2005;21:3726–3732. doi: 10.1093/bioinformatics/bti620. [DOI] [PubMed] [Google Scholar]
- Bafna V, Edwards N. On de-novo interpretation of tandem mass spectra for peptide identification. In Proceedings of the seventh annual international conference on Computational molecular biology; 2003. pp. 9–18. [Google Scholar]
- Bandeira N, et al. Shotgun protein sequencing by tandem mass spectra assembly. Anal. Chem. 2004;76:7221–7233. doi: 10.1021/ac0489162. [DOI] [PubMed] [Google Scholar]
- Bandeira N, et al. Shotgun protein sequencing: assembly of tandem mass spectra from mixtures of modified proteins. Mol. Cell Proteomics. 2007a;6:1123–1134. doi: 10.1074/mcp.M700001-MCP200. [DOI] [PubMed] [Google Scholar]
- Bandeira N, et al. Protein identification via spectral networks analysis. Proc. Natl Acad. Sci. USA. 2007b;104:6140–6145. doi: 10.1073/pnas.0701130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels C. Fast algorithm for peptide sequencing by mass spectroscopy. Biomedical and Environmental Mass Spectrometry. 1990;19:363–368. doi: 10.1002/bms.1200190607. [DOI] [PubMed] [Google Scholar]
- Birrell GW, et al. Mol. Cell Proteomics. 2007. The diversity of bioactive proteins in australian snake venoms. (in print). [DOI] [PubMed] [Google Scholar]
- Chen T, et al. A dynamic programming approach to de novo peptide sequencing via tandem mass spectrometry. J. Comput. Biol. 2001;8:325–337. doi: 10.1089/10665270152530872. [DOI] [PubMed] [Google Scholar]
- Dancík V, et al. De novo peptide sequencing via tandem mass spectrometry. J. Comput. Biol. 1999;6:327–342. doi: 10.1089/106652799318300. [DOI] [PubMed] [Google Scholar]
- Durbin R, et al. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press; 1999. [Google Scholar]
- Edwards NJ. Novel peptide identification from tandem mass spectra using ests and sequence database compression. Mol. Syst. Biol. 2007;3:102–102. doi: 10.1038/msb4100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-de Cossío J, et al. A computer program to aid the sequencing of peptides in collision-activated decomposition experiments. Comput. Appl. Biosci. 1995;11:427–434. doi: 10.1093/bioinformatics/11.4.427. [DOI] [PubMed] [Google Scholar]
- Fischer B, et al. Novohmm: a hidden markov model for de novo peptide sequencing. Anal. Chem. 2005;77:7265–7273. doi: 10.1021/ac0508853. [DOI] [PubMed] [Google Scholar]
- Frank AM, Pevzner PA. PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 2005;77:964–973. doi: 10.1021/ac048788h. [DOI] [PubMed] [Google Scholar]
- Gupta N, et al. Whole proteome analysis of post-translational modifications: applications of mass-spectrometry for proteogenomic annotation. Genome Res. 2007;17:1362–1377. doi: 10.1101/gr.6427907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurum JS. Recombinant polyclonal antibodies: the next generation of antibody therapeutics? Drug. Discov. Today. 2006;11:655–660. doi: 10.1016/j.drudis.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Kalkum M, et al. Detection of secreted peptides by using hypothesis-driven multistage mass spectrometry. Proc. Natl Acad. Sci. USA. 2003;100:2795–2800. doi: 10.1073/pnas.0436605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Glish GL. C-terminal peptide sequencing via multistage mass spectrometry. Anal. Chem. 1998;70:5162–5165. doi: 10.1021/ac980823v. [DOI] [PubMed] [Google Scholar]
- Ma B, et al. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003;17:2337–2342. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- Maizels N. Immunoglobulin gene diversification. Annu. Rev. Genet. 2005;39:23–46. doi: 10.1146/annurev.genet.39.073003.110544. [DOI] [PubMed] [Google Scholar]
- Mo L, et al. Msnovo: a dynamic programming algorithm for de novo peptide sequencing via tandem mass spectrometry. Anal. Chem. 2007;79:4870–4878. doi: 10.1021/ac070039n. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Mann M. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc. Natl Acad. Sci. USA. 2004;101:13417–13422. doi: 10.1073/pnas.0405549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Pevtsov S, et al. Performance evaluation of existing de novo sequencing algorithms. J. Proteome. Res. 2006;5:3018–3028. doi: 10.1021/pr060222h. [DOI] [PubMed] [Google Scholar]
- Pham V, et al. De novo proteomic sequencing of a monoclonal antibody raised against ox40 ligand. Anal. Biochem. 2006;352:77–86. doi: 10.1016/j.ab.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Pimenta AM, De Lima ME. Small peptides, big world: biotechnological potential in neglected bioactive peptides from arthropod venoms. J. Pept. Sci. 2005;11:670–676. doi: 10.1002/psc.701. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, et al. Rapid ‘de novo’ peptide sequencing by a combination of nanoelectrospray, isotopic labeling and a quadrupole/time-of-flight mass spectrometer. Rapid. Commun. Mass. Spectrom. 1997;11:1015–1024. doi: 10.1002/(SICI)1097-0231(19970615)11:9<1015::AID-RCM958>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Tanner S, et al. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 2005;77:4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- Tanner S, et al. Improving gene annotation using peptide mass spectrometry. Genome Res. 2007;17:231–239. doi: 10.1101/gr.5646507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsur D, et al. Identification of post-translational modifications by blind search of mass spectra. Nat. Biotechnol. 2005;23:1562–1567. doi: 10.1038/nbt1168. [DOI] [PubMed] [Google Scholar]
- Ulintz PJ, et al. Investigating ms2-ms3 matching statistics: A model for coupling consecutive stage mass spectrometry data for increased peptide identification confidence. Mol. Cell Proteomics. 2008;7:71–87. doi: 10.1074/mcp.M700128-MCP200. [DOI] [PubMed] [Google Scholar]
- Viterbi A. Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. IEEE Transactions on Information Theory. 1967;13:260–269. [Google Scholar]
- Yates JR, et al. Mining genomes: correlating tandem mass spectra of modified and unmodified peptides to sequences in nucleotide databases. Anal. Chem. 1995;67:3202–3210. doi: 10.1021/ac00114a016. [DOI] [PubMed] [Google Scholar]
- Zhang Z, McElvain JS. De novo peptide sequencing by two-dimensional fragment correlation mass spectrometry. Anal. Chem. 2000;72:2337–2350. doi: 10.1021/ac000226k. [DOI] [PubMed] [Google Scholar]