

Abstract
Compounds that stabilize p53 could suppress tumors providing a additional tool to fight cancer. Mdm2, and the human ortholog, Hdm2 serve as ubiquitin E3 ligases and target p53 for ubiquitylation and degradation. Inhibition of Hdm2 stabilizes p53, inhibits cell proliferation and induces apoptosis. Using HTS to discover inhibitors, we identified three new alkaloids, isolissoclinotoxin B, diplamine B, and lissoclinidine B from Lissoclinum cf. badium. Lissoclinidine B inhibited ubiquitylation and degradation of p53, and selectively killed transformed cells harboring wild-type p53, suggesting this compound could be used to develop new treatments.
1. Introduction
The tumor suppressor p53 plays a central role in protecting cells from tumor development by inducing cell cycle arrest, senescence, and apoptotic cell death in response to diverse stresses, such as DNA damage or oncogene activation.1,2,3,4 Within normal cells, p53 is maintained at low levels in normal cells by ongoing ubiquitylation and proteasomal degradation. Ubiquitylation occurs as a result of the sequential action of three enzymes, E1 (ubiquitin-activating enzyme), E2 (ubiquitin conjugating enzyme), and E3 (ubiquitin ligase). Recognition of ubiquitylated proteins by the proteasome requires the formation of polyubiquitin chains, which are linked through Lys 48 of ubiquitin (K48). E3s, which interact with E2, recognize and target specific protein substrates like p53 for ubiquitylation. The largest class of E3s identified contains a specific zinc binding protein fold known as a RING finger (RF) motif. These RF-E3s mediate the direct transfer of ubiquitin from the E2 to the protein substrate, and in some cases they have also been shown to regulate their own stability through auto-ubiquitylation.5
Mouse double minute 2 (mdm2), whose human ortholog is Hdm2, is one of the principal cellular regulators responsible for targeting the degradation of p53, and maintaining low p53 levels during normal growth and development.6,7,8,9 Hdm2 belongs to the RF-E3 family and targets both p53 and itself for proteasomal degradation through ubiquitylation. 1–4 The importance of Hdm2 in regulating p53 is illustrated by the observation that deletion of Mdm2 is embryonic lethal only in mice retaining expression of functional p53. Mice containing a simultaneous deletion of both Mdm2 and p53 develop normally, suggesting that Hdm2 is essential for negative regulation of the growth-inhibitory activities of p53 during development.10,11 Under normal conditions, p53 is tightly controlled by Hdm2 through an auto-regulatory feedback loop. First, cellular stress causes an increase in active p53, which in turn, activates the transcription of Hdm2 and thereby increases the level of Hdm2. Next Hdm2 diminishes p53’s ability to transactivate target genes by binding to p53 and inhibiting its translocation to the nucleus. There is evidence that a significant percentage of those tumors that retain wild type p53 exhibit Hdm2 amplification or disrupt the pathways that control the stabilization and activation of p53 through other means.12 Therefore, restoration or enhancement of p53 function through inhibiting Hdm2’s E3 activity represents a potentially efficacious approach for inducing apoptotic cell death in human tumors containing wild-type p53.
For this reason, we developed a high throughput screen to identify inhibitors of the Hdm2 E3 activity.13 Using this assay to screen the NCI natural product extract library, we identified an extract of the ascidian Lissoclinum cf. badium collected off the coast of Papua New Guinea that inhibited the E3 activity of Hdm2. Ascidians of the genus Lissoclinum have yielded many different classes of natural products, including cyclic peptides,14–16 diterpenoids,17 pyridoacridine alkaloids,18–20 polyethers,21,22 benzopentathiepins,23,24 and dimerized aromatic amines.20,25,26 In this study, we describe the isolation and characterization of five alkaloids (1–5). Three of these compounds are new and two, diplamine B (4) and lissoclinidine B (5) stabilize Hdm2 and p53 in cells. Moreover, 5 selectively kills transformed cells expressing wild-type p53, suggesting that 5 could form the basis for the development of therapeutically useful compounds.
2. Chemistry
Compound 1 was isolated as yellow amorphous solid. High resolution ESI-TOF MS analysis suggested a molecular formula of C11H15NO2S5. The 1H NMR spectrum (CD3OD) of 1 was uncomplicated, with an aromatic singlet at δH 7.00 (1H), methyl singlets at δH 3.96 (3H) and 2.98 (6H), and multiplets at δH 3.29 (2H), 3.30 (1H), and 3.32 (1H). The 13C NMR spectrum of 1 contained ten signals, six of which appeared to arise from aromatic carbons. Although our data did not exactly match that for any known compound, the NMR data for 1 corresponded well to that of the synthetic benzopentathiepin derivative isolissoclinotoxin A (6).27 The obvious differences were the appearances of the six-proton signal at δH 2.98 and a carbon signal at δC 43.58 found in 1 that were not seen in 6. The HMBC spectrum of 1 displayed correlations from the signal at δH 7.00 to the quaternary carbon signals at δC 133.94, 150.18, 151.58, and 136.52, which was consistent with the presence of a pentasubstituted benzene ring containing two oxygenated carbons. An HMBC correlation between the methoxyl proton signal at δH 3.96 and the signal at δC 151.58 proved that the methoxyl group was attached to the aromatic ring. Other HMBC correlations (shown in Figure 1) were consistent with the presence of a (dimethylamino)ethyl moiety in 1. Placement of substituents around the aromatic ring plus the presence of five sulfur atoms in the molecular formula suggested 1 belonged to the benzopentathiepin family previously isolated from Lissoclinum.27,28 The confirmation of the sulfur ring system in 1 was supported by the presence of the peak for [M−S2+H]+ in the high resolution ESI mass spectrum (m/z 290.0356, calcd 290.0343).27 To account for the remaining oxygen atom, acetylation of 1 using acetic anhydride and pyridine yielded the acetate ester 7, suggesting the presence of a hydroxyl group in 1. ROESY correlations (Figure 1) supported the assignment of the substitution pattern for 1. Thus, 1 was identified as a dimethylamino analogue of 6, and we have given it the name isolissoclinotoxin B. Compounds 2 and 3 were isolated from Hdm2-inhibitory fractions but were found to have very little activity in our assay. These compounds (2–3) were identified as varacin (2) and N,N-dimethyl-5-methylvaracin (3) by comparison of our NMR data with those found in the literature. 19,28,29
Figure 1.

Key 2D NMR correlations of 1, 4 and 5.
Compound 4 was isolated as an orange film. HRESI-TOFMS analysis indicated 4 had a molecular formula of C18H15N3OS. The 1H NMR spectrum of 4 in CD3OD consisted of nine signals, six of which corresponded to protons attached to aromatic carbons. The three-proton singlet at δH 2.68 suggested the presence of a thiomethyl group in 4. The remaining two signals corresponded to a pair of coupled methylenes at δH 3.37 and 3.89. The 13C NMR spectrum displayed eighteen signals, fifteen of which appeared to arise from aromatic carbons. Of the aromatic carbons, nine were quaternary, including one that was oxygenated (δC 181.05). COSY analysis of 4 indicated that the six downfield protons formed two isolated spin systems: a 1,4-butadienyl moiety, and a pair of vinylic protons. A single ROESY correlation indicated that two protons at δH 8.80 and 8.90 (H-4 and H-5), which did not show a COSY correlation, were in close proximity to each other. This information suggested that 4 was a member of the diplamine class of pyridoacridine alkaloids, which have previously been reported from other members of the genus Lissoclinum.
A comparison of 1H and 13C NMR data for 4 with that of diplamine (8)29 and lissoclin B (9)20 suggested all three were closely related. The chemical shifts for the aromatic carbons of 4 differed from the corresponding literature values for 8 and 9 by no more than 3%. HMBC correlations observed for 4 (Figure 1) were completely consistent with the proposed skeleton, although HMBC correlations to all quaternary carbons were not detected. The spectrum clearly lacked signals for the acetyl group reported in 8. Therefore, 4 was assigned the structure of a deacetylated analogue of diplamine, which we have named diplamine B.
Compound 5 was isolated as a purple film. HRESI-TOFMS analysis indicated that 5, like 4, had a molecular formula of C18H15N3OS. The 1H NMR spectrum of 5 contained eight signals (a ninth signal was observed by HSQC analysis), but the spectrum had several differences compared to that of 4. The most significant change was the disappearance of a SCH3 singlet observed in 4 that was replaced by a methylene singlet at δH 6.11 in 5. The 13C NMR spectrum of 5 consisted of eighteen signals, with fifteen apparent aromatic carbon signals. COSY and ROESY analysis elucidated two spin systems in close proximity, as was found in 4. From HMBC analysis of 5, the two-proton singlet at δH 6.11 showed correlations to two sp2 carbons (δC 124.67 and 143.95). This information and the HMBC analysis (Figure 1) suggested that 5 was deacetylated lissoclinidine (10), characterized by a five-member benzoxathiole ring.19,20 We have named this compound lissoclinidine B.
Given the ability of pyridoacridine alkaloids containing a quinone carbonyl at C-8 and a thiomethyl group at C-9 to rearrange to form lissoclinidine-type isomers,20 the possibility arose that 5 was a chromatographic artifact arising from the purification process. In our initial isolation of 5, 1H NMR signals characteristic of 5 were observed in fractions from the earliest stage of the isolation process, and we ultimately isolated more than 4 mg of material. In a repeated isolation procedure, additional care was taken to avoid prolonged exposure of fractions to acidic conditions and ambient light. The yield of 5 was lower (2 mg), but it was still detectable by LC-MS and NMR from the beginning of the isolation. We proposed that 5 is therefore, in our case, both a natural product as well as a product of the rearrangement of 4. This was consistent with the results of Appleton et al.,19 who provided strong evidence that 10 is a component of living samples of Lissoclinum notti.
3. Biological Characterization of Compounds
Compounds 1–5 were tested in the Hdm2 electrochemiluminescence assay, and these data are summarized in Table 2. Compound 1 was shown to have an IC50 of 58.6 µM, while 4 and 5 had IC50’s of 101.3 and 98.1 µM, respectively. Compound 3 was weakly active with an IC50 of 120.8 µM, while 2 was essentially inactive (IC50 >295 µM). The presence of a phenol at C-3 of 1 appears to improve the activity of 1 over that of 2–3. The dimethylamine moiety found in 1 and 3 also appears to increase Hdm2 inhibition of the benzopentathiepins. Since 4 and 5 are equally active, this suggests that the functionality required for their activity lies in the aromatic ring system and sidechain, and may not be affected by substituent differences at C-8, C-9, or N-11.
Table 2.
Hdm2 Inhibitory Activity of 1–5.
| Compound | IC50 (µM) |
|---|---|
| 1 | 58.6 ± 4 |
| 2 | >295 |
| 3 | 120.8 ± 9 |
| 4 | 101.3 ± 4 |
| 5 | 98.1 ± 6 |
To determine whether the activity observed in vitro is also found in cells, we tested compounds 1–5 for effects on cellular p53 and Hdm2. Tert-immortalized human retinal pigment epithelial (RPE) cells were incubated with compounds 1–5 and cellular p53 and Hdm2 levels were determined by immunoblotting.
Compounds 2 and 3 had no discernable cellular effects (data not shown). Compound 1 was toxic to RPE cells even at 10 µM while at 1 µM did not increase p53 levels (data not shown). On the other hand, compounds 4 and 5 increased p53 and Hdm2 in a dose-dependent manner and at 10 µM exhibited greatest increase in p53 and Hdm2 similar to 50 µM of the proteasome inhibitor N-acetyl-leucyl-leucyl-norleucinal (ALLN) (Figure 2). It is interesting to note that 4 and 5 displayed more potent activity in cells (10 µM) than they did in the cell free assay system (101.3 and 98.1 µM, resp.). This enhanced activity in cells was also seen in the subsequent cell-based studies that follow.
Figure 2.

Diplamine B (4) and lissoclinidine B (5) increased p53 and Hdm2 in cells in a dose-dependent manner. RPE cells were treated with 50 µM ALLN, or 1–10 µM 4 or 5 for 8 h. Total levels of p53, Hdm2 and β-actin, which serves as an equal loading control, were assessed by immunoblotting using antibodies directed against each of these proteins.
To ensure that the increase in Hdm2 was not the result of p53-dependent transcriptional activation, p53−/−mdm2−/− mouse embryo fibroblasts (MEFs) were transiently transfected with plasmid encoding Hdm2 under the control of a p53- independent CMV promoter. After treatment with 10 µM 4 or 5 for 8 h, cellular Hdm2 was determined. Both 4 and 5 increased Hdm2 in MEFs (Figure 3), demonstrating that 4 and 5 function at least in part by stabilizing Hdm2. In conducting these experiments it became apparent that compound 4 was fairly unstable and degraded over time. However, since the effects and structures of the two compounds are quite similar, 5 was selected for further evaluation. To directly investigate whether 5 blocks Hdm2-mediated p53 ubiquitylation, U2OS (a human osteosarcoma) cells were transiently co-transfected with plasmids encoding p53 and Hdm2 prior to incubation with ALLN or 5. Co-transfection with p53 and Hdm2 induced loss of p53 (Figure 4, compare lanes 2 and 3). Consistent with its activity as a general proteasome inhibitor, ALLN resulted in the accumulation of p53, including higher molecular weight forms of the protein indicative of ubiquitylation (Figure 4, lanes 3 and 4). Incubation with 5 similarly blocked p53 degradation in a dose-dependent manner. Strikingly, however, ubiquitylated forms of p53 were absent in 5-treated cells (Figure 4, lanes 5–8). This result was confirmed in an additional experiment with HCT116 colon carcinoma cells expressing wild-type p53, which also showed that 5 prevented the accumulation of ALLN-induced ubiquitylated p53 following treatment with both 5 and ALLN (data not shown). This result indicates that 5 inhibits Hdm2-mediated ubiquitylation and degradation of p53.
Figure 3.

Diplamine B (4) and lissoclinidine B (5) stabilized Hdm2 in MEFs. p53−/− mdm2−/− MEFs were transiently transfected with Hdm2 plasmid under the control of a CMV promoter. Forty-eight h after transfection, the cells were treated with 10 µM 4 or 5 for 8 h. Cellular Hdm2 and β-actin were determined by immunoblotting.
Figure 4.
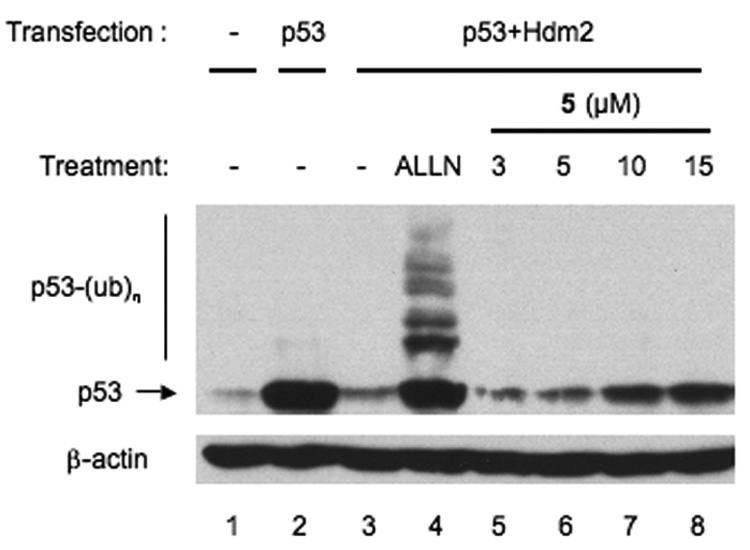
Lissoclinidine B (5) inhibits Hdm2-mediated p53 degradation. U2OS cells were transiently co-transfected with plasmids encoding p53 and Hdm2. Sixteen h after transfection, the cells were treated with 50 µM ALLN, or 3–15 µM 5 for 8 h. Cellular p53 and β-actin were determined by immunoblotting. p53-(Ub)n: ubiquitylated forms of p53.
The capacity of p53 to induce cell cycle arrest or apoptosis is dependent on its transcriptional activity. 2 To examine whether 5 activates p53-dependent transactivation, U2OS-pG13 cells that stably express endogenous wild-type p53 and a p53-responsive luciferase reporter (reporter pG13)30,31 were employed. Treatment with 5 led to a dose-dependent increase in luciferase activity (Figure 5). This data suggests that the stabilization of p53 by 5 results in activation of p53-dependent transcription.
Figure 5.
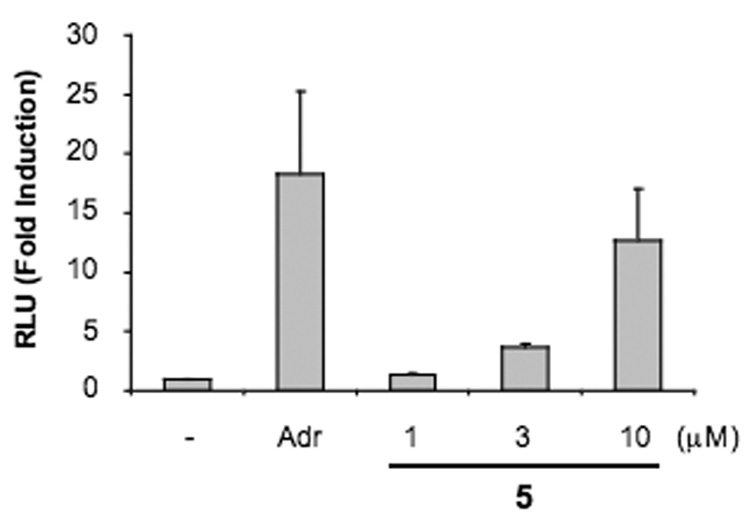
Lissoclinidine B (5) activates p53 transcription. U2OS-pG13 cells were incubated with 1 µg/ml adriamycin, or 1–10 µM 5 for 22 h. After the incubation, luciferase activity was assessed. Data represents average and standard deviation of three independent experiments. RLU: relative light unit. Adr: adriamycin.
Previous studies have shown that oncogenic transformation can sensitize cells to p53-induced apoptosis.32 To determine whether 5 selectively kills transformed cells, we compared RPE (retinal pigment epithelial) cells with RPE cells transformed with adenovirus E1A (RPE-E1A) which interacts with the retinoblastoma (Rb) tumor suppressor gene product but not p53.33 While 5 had no effect on parental RPE cells, it resulted in a clear dose-dependent increase in cell death in the RPE-E1A cells (Figure 6). Based on our previous work,13 we would predict that p53-expressing cells will be differentially susceptible to apoptosis in response to agents that inhibit the E3 activity of Hdm2. A well- established system for measuring p53-dependent cell death is to use MEFs (mouse embryo fibroblasts) transformed with adenovirus E1A and activated Ha-ras.33 To evaluate whether 5 induces cell apoptosis in a p53-dependent manner, we used p53-deficient (A9) and wild-type p53 MEFs (C8). After incubation with 5 for 20 h, a marked increase in cell death in C8 cells was noted while p53-deficient A9 cells were relatively resistant (Figure 7A). Consistent with apoptotic cell death, cleavage of the poly (ADP-ribose) polymerase (PARP) was observed in response to 5 specifically in the p53-expressing C8 cells but not A9 cells (Figure 7B). Similarly, p53-dependent killing was found when HCT116-p53+ cells were compared to HCT116-p53− cells (data not shown). Consistent with a primary role in inhibiting Hdm2 and thereby activating p53, these data indicate that 5 induces cell apoptosis in a p53-dependent manner.
Figure 6.

Lissoclindine B (5) selectively kills transformed cells. Parental RPE cells and RPE-E1A cells were incubated with 1 µg/ml adriamycin, or 3–25 µM 5 for 17 h and cell death was assessed by trypan blue exclusion. Data represents average and standard deviation of three independent experiments.
Figure 7.

A. Lissoclinidine B (5) selectively kills cells expressing wild-type p53. p53-deficient (A9) or wild-type p53 (C8) MEFs were treated with 5–20 µM 5 for 20 h. Cell death was measured by trypan blue exclusion. Data represents average and standard deviation of three independent experiments. B. Lissoclinidine B (5) induces apoptosis in C8 cells. A9 and C8 cells were incubated with 5–20 µM 5 for 14 h. Cellular PARP and β-actin were evaluated by immunoblotting using anti-PARP and anti-β-actin specific antibodies.
4. Discussion
We report here the isolation of five compounds and the biological characterization of one new alkaloid 5 as an inhibitor of Hdm2 E3 activity in a cell free system. In cells, 5 is more potent which suggests that it does more than inhibit the E3 function of Hdm2 and indeed, may act on other pathways. Within the Hdm2 pathway, this compound increases cellular p53 and Hdm2 levels, inhibits auto-ubiquitylation of Hdm2 in cells, and inhibits Hdm2-mediated ubiquitylation of p53. Most importantly, 5 selectively induces apoptotic cell death in transformed cells and cells harboring wild-type p53.
p53 is the most frequently inactivated tumor suppressor gene in human malignancies. The RF-E3 Hdm2 plays a major role in modulating p53 activity by targeting it for ubiquitin-mediated proteasomal degradation.34 Therefore restoring wild-type p53 activity through inhibiting Hdm2 E3 activity is an attractive approach to treating malignancies that retain wild-type p53. Several different approaches aimed at inhibiting the activity of Hdm2 have been attempted. One approach is to inhibit Hdm2 gene expression through the use of antisense oligonucleotides.35 These reagents specific for Hdm2 have shown anti-tumor ability both in vitro and in vivo.36 A second approach employed by several groups to inhibit the binding of Hdm2 to p53. Early experiments have shown that p53-derived peptides inhibit the Hdm2-p53 interaction.37 More recently, several chemical nonpeptide compounds have been identified, which include Nutlin,38 RITA,39 MI-63,40 and Syl-155.41 These compounds show promise in the treatment of tumors harboring a wild-type p53 and with amplified cellular Hdm2 levels. We have taken a different approach, in which we have sought to identify small molecule inhibitors of Hdm2’s catalytic activity in order to inhibit Hdm2-mediated p53 degradation.13,40,42 Diplamine B (4) and lissoclinidine B (5) are new inhibitors of Hdm2 auto-ubiquitylation and clearly stabilize both p53 and Hdm2 in cells at low micromolar concentrations.
More than half of the current clinically used chemotherapeutic agents are derived from natural products.43 In an analysis of the 79 new chemical entities approved for cancer therapy by the FDA between 1981 and 2002, sixty-five (82%) were identified as originating from small organic molecules and 48 of 65 (74%) were natural products or natural product derivatives.43 At least three classes of natural products, chlorofusin,44 polycyclic compounds,45 and chalcone derivatives46 have been reported to efficiently inhibit the formation of the Hdm2-p53 complex. More recently, our group identified natural product sempervirine inhibits auto-ubiquitylation of Hdm2 and stabilizes p53 and Hdm2 in cells.13 Here we report lissoclinidine B (5) clearly inhibits auto-ubiquitylation of Hdm2, stabilizes p53 and Hdm2, and selectively kills transformed cells and cells harboring wild-type p53. Further studies will be needed to understand the mechanistic details of this compound’s effect on Hdm2.
5. Experimental
5.1. General Experimental Procedures
UV spectra were recorded using a Beckman DU-600 spectrophotometer. IR spectra were recorded for neat samples with a Perkin-Elmer Spectrum 2000 FT-IR spectrophotometer. NMR spectra were obtained on a Varian Inova 500 spectrometer operating at 500.2 MHz for 1H and 125.8 MHz for 13C. High resolution mass spectra were recorded with a Applied Biosystems QSTAR hybrid triple-quad time-of-flight mass spectrometer with an electrospray ionization source. The calibration solution used was Renin substrate, calibrated on masses 110.0713 and 879.9723, both within 5 ppm. Low resolution mass spectra were recorded on a Hewlett-Packard Series 1100 MSD LC-MS with an electrospray ionization source. Electrochemiluminescence plates, cell lysates, and reagents for the Hdm2 assay were purchased from MesoScale Discovery.
5.2. Reagents and antibodies
The proteasome inhibitor N-acetyl-leucyl-leucyl-norleucinal (ALLN) was purchased from Calbiochem (La Jolla, CA). Adriamycin and the anti-β-actin antibody were purchased from Sigma (St. Louis, MO). Antibodies recognizing Hdm2 (Ab-1 (OP46) and Ab-2 (OP115)) were purchased from Oncogene (Boston, MA). Antibodies recognizing p53 (sc-126) and poly (ADP-ribose) polymerase (PARP) (sc-7150) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
5.3. Cell lines and plasmids
Human retinal pigment epithelial (RPE), RPE-E1A, p53−/−mdm2−/− mouse embryo fibroblasts (MEFs), U2OS, U2OS-pG13, C8, A9 cells were as described previously.30 Plasmids encoding Hdm2 and p53 genes have been described previously.7, 47
5.4. Marine Sample Extraction
The ascidian sample, used in this work, was collected for the NCI, by Pat Colin of the Coral Reef Research Foundation in the Coral Sea, 90 miles southwest of Port Moresby, Papua New Guinea, on October 22, 1993. The taxonomist was Françoise Monniot and a voucher is at the Smithsonian Division of Worms under the collector number 0CDN1811. The frozen sample was pulverized at the National Cancer Institute in dry ice by use of a worm-fed grinder (hamburger mill), the powder produced was allowed to stand at −30 °C until the CO2 sublimed, and the mass was then extracted at 4 °C with deionized water (1 L) by stirring (30 rpm) for 30 min. The mixture was centrifuged at room temperature and the supernatant was separated and then lyophilized to give the aqueous extract. The insoluble portion from the centrifugation was lyophilized and then statically extracted overnight at room temperature with 1 L of 1:1 MeOH-CH2Cl2. The organic phase was filtered, the pellet was washed with a 10% volume of fresh MeOH, and the combined organic phase was concentrated to dryness at <35 °C by rotary evaporation and then finally dried under vacuum at room temperature to give the organic extract as a gum.
5.5. Isolation of Compounds
The crude extract (2.02 g), which showed activity in our initial Hdm2 screening assay (IC50 of 71 µg/mL), was fractionated first using wide-pore C4-bonded silica gel with a step gradient of aqueous methanol to afford six fractions, one of which (33% MeOH(aq), 248 mg) had activity better than that of the crude extract. This fraction was fractionated further by LH-20 with 1:1 MeOH:MeCN as a mobile phase, which yielded seven fractions (fractions A–G), three of which had improved activity (fractions B–D).
Fraction B (72 mg) was subjected to flash chromatography using a step gradient (20%–100% MeOH(aq) with 0.05% TFA) to afford eight fractions, three of which had significant activity (fractions B-2, B-5, and B-6; 6.8, 29.1, and 17.0 mg, respectively). Fraction C (30 mg) was separated by this same flash chromatography method to afford eight fractions, five of which had good activity in the Hdm2 assay (fractions C-2, C-3, C-4, C-5, and C-6; 1.8, 2.7, 3.7, 9.9, and 12.0 mg, respectively). Fraction D (10 mg) was fractionated by use of C18 SPE using a step gradient (50–100% MeOH(aq) with 0.05% TFA) to afford three fractions, one of which (the 50% MeOH(aq) wash) was purified further by reversed-phase C18 HPLC (gradient, 2 mL/min, 10%–50% MeOH(aq) with 0.05% TFA over 20 min) to afford 5 (15.9 min, 2.2 mg).
Compounds 1 and 2 (4.8 mg and 2.3 mg, respectively) were purified by reversed phase C18 HPLC purification of 15.7 mg of fraction B-6 (gradient, 2 mL/min, 40%–80% MeOH(aq) with 0.05% TFA over 30 min; 1: 20.0 min; 2: 27.0 min). Compound 1 was also obtained from fraction B-5. Fraction B-2 consisted of a complicated mixture of several compounds related to 1, and these fractions were not purified further due to lack of material. Fraction B-7 (50% MeOH(aq) wash, 4 mg), which was 3 times less active than 1 in the Hdm2 assay, was shown to contain 3 along with several minor impurities, based on LC-MS and 1H NMR analysis. Compound 4 (5.4 mg) was isolated from fractions C-5 and C-6 by reversed phase C18 HPLC (gradient, 2 mL/min, 45%–62% MeOH(aq) with 0.05% TFA over 26 min, 14.5 min) followed by C4 HPLC (gradient, 2 mL/min, 45%–62% MeOH(aq) with 0.05% TFA over 30 min, 25.5 min). Fraction C-3 consisted mainly of 5, which appeared to be responsible for the activity of this fraction. Fractions C-2 and C-4 consisted of complicated mixtures of several compounds related to 2 and 4, and these fractions were not purified further due to lack of material.
5.5.1. Isolissoclinotoxin B (1)
Light yellow foam; UV (MeOH) λmax (log ε) 356 (3.39), 218 (4.09); IR (neat film) νmax 3324, 2919, 2851, 1675, 1476, 1201, 1066, 721 cm−1; 1H NMR (CD3OD), See Table 1; 13C NMR (CD3OD), See Table 1; HRES-TOFMS (positive ion) m/z 353.9781 ([M+H]+ calcd for C11H16NO2S5: 353.9785), 290.0356 ([M−S2+H]+ calcd for C11H16NO2S5: 290.0343).
Table 1.
NMR Data for Compounds 1, 4–5 (CD3OD).
| 1 | 4 | 5 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pos. | δC | δH | δC | δH | δC | δH | |||
| 1 | 133.94 | 132.67 | 8.34 | d, 8.0, 1H | 119.27 | 7.82 | d, 8.4, 1H | ||
| 2 | 131.54 | 133.32 | 7.99 | t, 7.7, 1H | 136.45 | 7.76 | t, 7.5, 1H | ||
| 3 | 150.18 | 131.39 | 7.90 | t, 7.7, 1H | 125.04 | 7.38 | t, 7.5, 1H | ||
| 4 | 151.58 | 124.93 | 8.80 | d, 7.5, 1H | 126.16 | 8.20 | d, 8.4, 1H | ||
| 4a | 123.10 | 116.19 | |||||||
| 4b | 139.08 | 150.76 | |||||||
| 5 | 115.37 | 7.00 | s, 1H | 122.01 | 8.90 | d, 5.0, 1H | 105.08 | 7.45 | d, 6.6, 1H |
| 6 | 136.52 | 150.89 | 9.14 | d, 4.4, 1H | 143.95 | 8.13 | d, 6.6, 1H | ||
| 7 | 32.06 | 3.30 | m, 1H | ||||||
| 3.32 | m, 1H | ||||||||
| 7a | 147.71 | 124.67 | |||||||
| 8 | 59.56 | 3.29 | m, 2H | 181.05 | 136.60 | ||||
| 9 | 144.73 | 139.78 | |||||||
| 10 | 57.02 | 3.96 | s, 3H | 150.21 | 108.43 | ||||
| 10a | 150.72 | 133.79 | |||||||
| 10b | 118.76 | 120.40 | |||||||
| 11a | 147.07 | 141.86 | |||||||
| 12 | 43.58 | 2.98 | s, 6H | 29.09 | 3.89 | t, 7.4, 2H | 29.30 | 3.29* | m, 2H |
| 13 | 40.17 | 3.37 | t, 7.2, 2H | 38.07 | 3.24 | m, 2H | |||
| 14 | 17.87 | 2.68 | s, 3H | 78.88 | 6.11 | s, 2H | |||
Signal overlapped with CD2HOD peak; detected by HSQC analysis
5.5.2. Diplamine B (4)
Dark orange film; UV (MeOH) λmax (log ε) 208 (4.39), 265 (3.70), 379 (3.05); IR (neat film) νmax 2924, 2853, 1675, 1202, 1131, 723 cm−1; 1H NMR (CD3OD), See Table 1; 13C NMR (CD3OD), See Table 1; HRES-TOFMS (positive ion) m/z 322.1005 ([M+H]+ calcd for C18H16N3OS: 322.1014).
5.5.3. Lissoclinidine B (5)
Purple film; UV (MeOH) λmax (log ε) 214 (4.37), 265 (3.98), 298 (3.81), 309 (3.78), 389 (3.15), 477 (3.10); IR (neat film) νmax 2923, 2852, 1681, 1204, 1135, 723 cm−1; 1H NMR (CD3OD), See Table 1; 13C NMR (CD3OD), See Table 1; HRES-TOFMS (positive ion) m/z 322.1003 ([M+H]+ calcd for C18H16N3OS: 322.1014).
5.5.4. Preparation of O-acetyl-isolissoclinotoxin B (7)
To a 4 mL screw-cap vial was added 0.5 mg 1, 0.2 mL pyridine, and 0.2 mL acetic anhydride. This mixture was stirred for 24 hours at room temperature, after which 1.0 mL of MeOH was added and the solution dried under a stream of nitrogen without further purification to afford 7 (0.5 mg): yellow-green film; 1H NMR (CD3OD), δ 2.30 (s, 3H, CH3CO), 2.86 (s, 6H, H-11 and H-12), 3.18 (m, 2H, H-8), 3.91 (s, 3H, H-10); 7.20 (s, 1H, H-6); LRESIMS (positive ion) m/z 396.0 ([M+H]+ calcd for C13H18NO3S5: 396.0).
5.6. Biological Evaluation of Compounds
5.6.1. Hdm2 biochemical assay procedure
The Hdm2 assay was performed as described previously.13 Briefly, the activity of Hdm2 was measured by detecting polyubiquitylated Hdm2 by electrochemiluminescence using a ruthenium-labeled polyubiquitin antibody. Cell lysates containing a GST-Hdm2 fusion protein were added to glutathione-coated 96-well plates (MesoScale Discovery, Gaithersburg, MD) and incubated at room temperature for 1 h. Separately, cell lysates containing rabbit E1 (MesoScale Discovery) and E2 (UbcH5B; MesoScale Discovery) were incubated with a solution containing ubiquitin, phosphate buffered saline solution (PBS), ATP, and DTT for 30 min to pre-charge the E2 ligases for ubiquitin transfer.
Assay samples and the pre-charged E1/E2 solution were then simultaneously added to the plate wells containing Hdm2 and allowed to react for 30 min at room temperature. The reaction was then stopped by washing the plate with PBS. A ruthenium labeled poly-ubiquitin antibody in a Tris-buffered solution of tripropylamine and a surfactant was then added, and the plate was incubated for 1 h at room temperature. The luminescence of the wells was recorded using a MesoScale Discovery SECTOR™ PR100 plate reader. On each plate, the ubiquitylation reaction was performed in the absence of added inhibitors to give a 0% inhibition threshold, while the reaction was also performed in the presence of EDTA to establish a 100% inhibition threshold; luminescence data collected were normalized based on this scale for each plate. The compounds were assayed at concentrations of 100, 20, 4, 0.8, and 0.02 µg/mL. Each purified compound was assayed in quadruplicate.
5.6.2. Western blot
Cells were lysed in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM iodoacetamide and Complete Mini (Roche, Mannheim, Germany)) and centrifuged at 14,000 g for 20 min at 4°C. Equal amount of post-nuclear supernatants were resolved by SDS-PAGE, transferred to PVDF membranes (Millipore, Billerica, MA) and immunoblotted with specific antibodies using standard procedures.30 Bands corresponding to specific proteins were visualized with horseradish peroxidase-labeled secondary antibodies and chemiluminescence agents (Pierce, Rockford, IL).
5.6.3. Luciferase assay
U2OS-pG13 cells were lysed with reporter lysis buffer (Promega, Madison, WI). The resultant lysates were centrifuged at 14,000 g for 20 min. Luciferase activity in the supernatant was determined according to the manufacturer’s instructions (Promega).
5.6.4. Trypan blue exclusion
Staining was performed by 1 min bath perfusion of 0.4% Trypan Blue Stain (Cambrex Bio Science, Walkersville, MD). Under a microscope, non-viable cells are stained and viable cells excluded the stain.
Supplementary Material
1H NMR, 13C NMR spectra for 1, 4, and 5 and 1H NMR spectra for 2 and 3 are provided with the online version of the manuscript. This material is available via the Internet at http://sciencedirect.com.
Acknowledgement
We thank Cheryl Thomas, and Tanya Johnson (MTDP) for performing the Hdm2 assay, John Lloyd (NIDDK, NIH) for collecting MS data, Dave Newman (Natural Products Branch, NCI) and Pat Colin (Coral Reef Research Foundation) for collections, Tom McCloud (SAIC-Frederick) for extraction of the animal material, and Kentaro Takada (MTDP) for helpful discussions. We are also grateful to the NIH Fellows Editorial Board for editorial assistance. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. J.K. is a fellow of Japanese Society for the Promotion of Science. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- NCI
National Cancer Institute
- CCR
Center for Cancer Research
- Mdm2
mouse double minute 2
- Hdm2
human ortholog of Mdm2
- Mdm2
Mdm2 gene
- Hdm2
Hdm2 gene
- E1
ubiquitin-activating enzyme
- E2
ubiquitin conjugating enzyme
- E3
ubiquitin ligase
- p53
tumor suppressor
- p53
p53 gene
- RF
ring finger, a structural motif that characterizes one class of E3 enzymes
- ESI-TOF MS
electrospray ionization time-of-flight mass spectrometry
- HRESI-TOF MS
high resolution, electrospray ionization time-off-flight mass spectrometry
- RPE
retinal pigment epithelial cells
- ALLN
N-acetyl-leucyl-leucyl-norleucinal
- MEF
mouse embryo fibroblast
- CMV
cytomegalovirus
- U2OS
human osteosarcoma cell line
- HCT116
human colon carcinoma cell line
- U2OS-pG13
U2OS cell line with stably expressed p53 responsive luciferase reporter pG13
- Adr
adriamycin
- RLU
relative light units
- RPE-EIA
RPE cells transformed with the adenovirus EIA
- Ha-ras
ras gene from Harvey murine sarcoma
- PARP
poly(ADP-ribose)polymerase
- A9
p53 deficient mouse embryo fibroblast
- C8
mouse embryo fibroblast with wild- type p53
- SPE
solid phase extraction
- PBS
phosphate buffered saline
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- DTT
dithiothreitol
- EDTA
ethylene diamine tetraacetic acid
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel
- PVDF
polyvinylidene fluoride
- Rb
retinoblastoma
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aylon Y, Oren M. Cell. 2007;130:597. doi: 10.1016/j.cell.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ, Hu W, Feng Z. Cell Death Differ. 2006;13:1027. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- 3.Vousden KH, Lane DP. Nat. Rev. Mol. Cell Biol. 2007;8:275. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 4.Yang Y, Li CC, Weissman AM. Oncogene. 2004;23:2096. doi: 10.1038/sj.onc.1207411. [DOI] [PubMed] [Google Scholar]
- 5.Fang S, Weissman AM. Cell Mol. Life Sci. 2004;61:1546. doi: 10.1007/s00018-004-4129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haupt Y, Maya R, Kazaz A, Oren M. Nature. 1997;387:296. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 7.Kubbutat MH, Jones SN, Vousden KH. Nature. 1997;387:299. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 8.Honda R, Tanaka H, Yasuda H. FEBS Lett. 1997;420:25. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 9.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. J. Biol. Chem. 2000;275:8945. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- 10.Jones SN, Roe AE, Donehower LA, Bradley A. Nature. 1995;378:206. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 11.Montes de Oca Luna R, Wagner DS, Lozano G. Nature. 1995;378:203. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 12.Momand J, Jung D, Wilczynski S, Niland J. Nucleic Acid Res. 1998;26:3453. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasiela CA, Stewart DH, Kitagaki J, Safiran YJ, Yang Y, Weissman AM, Oberoi P, Davydov IV, Goncharova E, Beutler JA, McMahon JB, O’Keefe BR. J. Biomol. Screen. 2008;13:229. doi: 10.1177/1087057108315038. [DOI] [PubMed] [Google Scholar]
- 14.Ireland C, Scheuer PJ. J. Am. Chem. Soc. 1980;102:5688. [Google Scholar]
- 15.Ireland CM, Durso AR, Jr, Newman RA, Hacker MP. J. Org. Chem. 1982;47:1807. [Google Scholar]
- 16.Perez LJ, Faulkner DJ. J. Nat. Prod. 2003;66:247. doi: 10.1021/np0204601. [DOI] [PubMed] [Google Scholar]
- 17.Uddin MJ, Kokubo S, Ueda K, Suenaga K, Uemura D. J. Nat. Prod. 2001;64:1169. doi: 10.1021/np010066n. [DOI] [PubMed] [Google Scholar]
- 18.Molinski TF, Ireland CM. J. Org. Chem. 1989;54:4256. [Google Scholar]
- 19.Appleton DR, Pearce AN, Lambert G, Babcock RC, Copp BR. Tetrahedron. 2002;58:9779. [Google Scholar]
- 20.Searle PA, Molinski TF. J. Org. Chem. 1994;59:6600. [Google Scholar]
- 21.Biard J-F, Roussakis C, Kornprobst J-M, Gouiffes-Barbin D, Verbist J-F, Cotelle P, Foster MP, Ireland CM, Debitus C. J. Nat. Prod. 1994;57:1336. doi: 10.1021/np50112a002. [DOI] [PubMed] [Google Scholar]
- 22.Degnan BM, Hawkins CJ, Lavin MF, McCaffrey EJ, Parry DL, Watters DJ. J. Med. Chem. 1989;32:1354. doi: 10.1021/jm00126a035. [DOI] [PubMed] [Google Scholar]
- 23.Davidson BS, Molinski TF, Barrows LR, Ireland CM. J. Am. Chem. Soc. 1991;113:4709. [Google Scholar]
- 24.Compagnone RS, Faulkner DJ, Carte BK, Chan G, Freyer A, Hemling MA, Hofmann GA, Mattern MR. Tetrahedron. 1994;50:12785. [Google Scholar]
- 25.Davis RA, Sandoval IT, Concepcion GP, da Rocha RM, Ireland CM. Tetrahedron. 2003;59:2855. [Google Scholar]
- 26.Liu H, Fujiwara T, Nishikawa T, Mishima Y, Nagai H, Shida T, Tachibana K, Kobayashi H, Mangindaan REP, Namikoshi M. Tetrahedron. 2005;61:8611. [Google Scholar]
- 27.Ford PW, Narbut MR, Belli J, Davidson BS. J. Org. Chem. 1994;59:5955. [Google Scholar]
- 28.Makarieva TN, Stonik VA, Dmitrenok AS, Grebnev BB, Isakov VV, Rebachyk NM. J. Nat. Prod. 1995;58:254. doi: 10.1021/np50116a015. [DOI] [PubMed] [Google Scholar]
- 29.Charyulu GA, McKee TC, Ireland CM. Tetrahedron Lett. 1989;30:4201. [Google Scholar]
- 30.Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV, Safiran YJ, Oberoi P, Kenten JH, Phillips AC, Weissman AM, Vousden KH. Cancer Cell. 2005;7:547. doi: 10.1016/j.ccr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 31.Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Science. 1991;252:1708. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- 32.Lowe SW, Ruley HE, Jacks T, Housman DE. Cell. 1993;74:957. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 33.Blint E, Phillips AC, Kozlov S, Stewart CL, Vousden KH. Proc. Natl. Acad. Sci. U.S.A. 2002;99:3529. doi: 10.1073/pnas.062491899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poyurovsky MV, Prives C. Genes Dev. 2006;20:125. doi: 10.1101/gad.1397506. [DOI] [PubMed] [Google Scholar]
- 35.Chen L, Lu W, Agrawal S, Zhou W, Zhang R, Chen J. Mol. Med. 1999;5:21. [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang R, Wang H, Agrawal S. Curr. Cancer Drug Targets. 2005;5:43. doi: 10.2174/1568009053332663. [DOI] [PubMed] [Google Scholar]
- 37.Chene P. Nat. Rev. Cancer. 2003;3(2):102–109. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- 38.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 39.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LGGC, Masucci M, Pramanik A, Selivanova G. Nat. Med. 2004;10:1321. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 40.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PR, Wang S. J. Med. Chem. 2006;49:3432. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 41.Li WD, Wang MJ, Ding F, Yin DL, Liu ZH. World J. Gastroenterol. 2005;11:2927. doi: 10.3748/wjg.v11.i19.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davydov IV, Woods D, Safiran YJ, Oberoi P, Fearnhead HO, Fang S, Jensen JP, Weissman AM, Kenten JH, Vousden KH. J. Biomol. Screen. 2004;9:695. doi: 10.1177/1087057104267956. [DOI] [PubMed] [Google Scholar]
- 43.Newman DJ, Cragg GM, Snader KM. J. Nat. Prod. 2003;66:1022. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 44.Duncan SJ, Grüschow S, Williams DH, McNicholas C, Purewal R, Hajek M, Gerlitz M, Martin S, Wrigley SK, Moore M. J. Am. Chem. Soc. 2001;123:554. doi: 10.1021/ja002940p. [DOI] [PubMed] [Google Scholar]
- 45.Zhao J, Wang M, Chen J, Luo A, Wang X, Wu M, Yin D, Liu Z. Cancer Lett. 2002;183:69. doi: 10.1016/s0304-3835(02)00084-8. [DOI] [PubMed] [Google Scholar]
- 46.Stoll R, Renner C, Hansen S, Palme S, Klein C, Belling A, Zeslawski W, Kamionka M, Rehm T, Muehlhahn P, Schumacher R, Hesse F, Kaluza B, Voelter W, Engh RA, Holak TA. Biochemistry. 2001;40:336. doi: 10.1021/bi000930v. [DOI] [PubMed] [Google Scholar]
- 47.Chen J, Lin J, Levine AJ. Mol. Med. 1995;1:142. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H NMR, 13C NMR spectra for 1, 4, and 5 and 1H NMR spectra for 2 and 3 are provided with the online version of the manuscript. This material is available via the Internet at http://sciencedirect.com.