

Abstract
The cone snails belong to the superfamily Conoidea, comprising ∼10,000 venomous marine gastropods. We determined the complete mitochondrial DNA sequence of Conus textile. The gene order is identical in Conus textile, Lophiotoma cerithiformis (another Conoidean gastropod), and the neogastropod Ilyanassa obsoleta, (not in the superfamily Conoidea). However, the intergenic interval between the coxI/coxII genes, was much longer in C. textile (165 bp) than in any other previously analyzed gastropod.
We used the intergenic region to evaluate evolutionary patterns. In most neogastropods and three conidean families the intergenic interval is small (<30 nucleotides). Within Conus, the variation is from 130-170 bp, and each different clade within Conus has a narrower size distribution. In Conasprella, a subgenus traditionally assigned to Conus, the intergenic regions vary between 200-500 bp, suggesting that the species in Conasprella are not congeneric with Conus. The intergenic region was used for phylogenetic analysis of a group of fish-hunting Conus, despite the short length resolution was better than using standard markers. Thus, the coxI/coxII intergenic region can be used both to define evolutionary relationships between species in a clade, and to understand broad evolutionary patterns across the large superfamily Conoidea.
Keywords: Conus textile, coxI-coxII intergenic sequence, evolution, Superfamily Conoidea, Conasprella
Introduction
The cone snails (Conus) are a species-rich genus of venomous marine gastropods. In the fossil record, the earliest Conus species are found in the Eocene (Kohn, 1990) several major radiations have given rise to the impressive present day biodiversity that makes Conus one of the largest genera of marine invertebrates (∼700 species) (Bouchet and Rocroi, 2005; Ponder and Lindberg, 1997; Ponder and Waren, 1988; Taylor et al., 1993). All Conus species are venomous; individual Conus species prey on fish, other molluscs or marine worms (Rockel et al., 1995). The venom of at least one fish-hunting species, the geography cone, Conus geographus, is sufficiently potent to cause a high frequency (∼70%) of human fatality in the absence of medical intervention (Cruz and White, 1995; Fegan and Andresen, 1997). The venoms of cone snails are being systematically analyzed in a number of laboratories, and a compound from a cone snail venom was recently approved as a commercial drug for alleviating severe pain (Czerwiec et al., 2006; Miljanich, 2004; Olivera, 2000, 2006; Terlau and Olivera, 2004).
However, an evolutionary framework for the 700 species in the genus is not yet available, and the relationship of Conus sp. to the other venomous snails belonging to the superfamily Conoidea is poorly understood. Most taxonomists place all of the venomous neogastropod taxa in the single superfamily, Conoidea (Duda and Kohn, 2005; Kohn, 1998; Taylor et al., 1993). The groundbreaking work of Bouchet and co-workers has established that the biodiversity of venomous mollusks is much larger than previously suspected (probably >10,000 species) (Bouchet et al., 2002).
Here, we report the complete mitochondrial DNA sequence of Conus textile (Linné, 1758), the “cloth-of-gold-cone”. This is the first such analysis for any cone snail. As will be reported below, the gene order in the mitochondrial genome of Conus textile and of the turrid Lophiotoma cerithiformis (Powell, 1964), the only other species in the superfamily for which a complete mitochondrial DNA sequence is available (Bandyopadhyay et al., 2006), is conserved, suggesting that gene order is not useful for phylogenetic analysis of Conoidean gastropods.
However, a large change in an intergenic region between coxI and coxII was revealed by a comparison of the two conoidean mitochondrial DNA sequences. In this paper we present data suggesting that both the length of, and the rapid sequence divergence of the coxI and coxII intergenic region are useful for evaluating evolutionary relationships, both broadly within the superfamily Conoidea, as well as between closely-related Conus species.
Methods
The cone snails used in this paper were collected in the Phillipines. High molecular weight DNA was isolated from hepato-pancreas of the snails. Genomic DNA was prepared from 20mg of frozen tissue from each species using the Gentra PUREGENE DNA Isolation Kit (Gentra Systems, Minneapolis, MN) according to the manufacturer's standard protocol (Sambrook and Russel, 2001). Segments of mitochondrial DNA were amplified by polymerase chain reaction (PCR) using degenerate primers described in (Bandyopadhyay et al., 2006). Primers were designed from amino acid sequences conserved in aligned mitochondrial protein and RNA sequences (Table I). PCRs were carried out using BD Advantage™2 PCR enzyme system (Clontech, CA, USA) in a PTC 200 thermocycler (MJ Research, MA, USA). The PCR cycling profile consisted of initial denaturation at 94°C for 2min, then 39 cycles of conditions were 94°C for 45s, 45°C for 45s, 68°C for 1-5 min (depending on the length of the expected DNA fragment) and a final elongation step of 10min at 68°C. The amplified DNA was purified by agarose gel electrophoresis, isolated using Qiagen kit (QIAGEN Inc. CA, USA) and cloned into pGEM®-T Easy vector(Promega, WI,USA). The nucleotide sequence of the insert was determined using oligonucleotide primers corresponding to T7 and SP6 promoter sequences and in cases where the sequences did not overlap, by primer walking. Specific primers designed from the sequences were used to amplify the rest of the mitochondrial genome and their sequence determined as described above. Amplification of mitochondrial DNA from within coxI to rrnL was carried out using Txmt3 and Lr283r; cob to nad4 using TxCytb e.1 and nad 4-2; coxIII to coxI using coxIII-1 and Txmt2; nad4 to coxIII using ND4CO3F and CO3r.2; and rrnL to cob using 187S.1 and cob-2 (see Table I for sequences of primers). The PCR cycling profile consisted of initial denaturation at 94°C for 2min followed by 39 cycles of 94°C for 45sec, 55°C for 45sec and elongation at 68°C for 1-6min depending on the expected length of the fragment and a final elongation of 10min at 68°C. The Expand Long Template PCR System from Roche Molecular Biochemicals (Roche Diagnostics Corporation, Indianapolis,IN, USA) was used for these experiments.
Table 1. Primers used for the amplification of mitochondrial DNA by PCR.
| BarCOI(+): | 5′ACN AAY CAY AAR GAY ATH GG 3′ |
| COI3(-): | 5′CCT CCT GCT GGA TCA AAR AAD GC 3′ |
| 12s(+)a: | 5′TGC CAG CAG YCG CGG TTA 3′ |
| 12s(-): | 5′AGA GYG RCG GGC GAT GTG T 3′ |
| rrnL-1(+): | 5′CCG GTC TGA ACT CAG ATC ACG T 3′ |
| rrnL-2(-)b: | 5′GTT TAC CAA AAA CAT GGC TTC 3′ |
| cob-1(+): | 5′CCN TGR GGN CAR ATR WSN TWY TGR GGN GC3′ |
| cob-2(-): | 5′GCN CCY CAR WAN SWY ATY TGN CCY CAN GG 3′ |
| nad4-1(+): | 5′TGR GGN TAY CAR CCN GAR CG 3′ |
| nad4-2(-): | 5′CGY TCN GGY TGR TAN CCY CA 3′ |
| nad5-1(+): | 5′TGR YTN CCN GCN GCN ATR GC 3′ |
| nad5-2(-): | 5′GCY ATN GCN GCN GGN ARY CA 3′ |
| coxIII-1(+): | 5′GCN GCN GCN TGR TAY TGR CA 3′ |
| coxIII-2(-) | 5′TGY CAR TAY CAN GCN GCN GC 3′ |
| Txmt3: | 5′GCATCTTGCTGGGGTTTCTTCTATTTTAGGAGC 3′ |
| Lr283r: | 5′GGCTTTACTTGCTCCTCGGTTGCC 3′ |
| TxCytb e.1: | 5′GTTGGTGGACTTCTAGTTATG 3′ |
| Txmt2: | 5′CCGCCG(A/G)CATGCGCTAAATTTCCTGC 3′ |
| ND4CO3F: | 5′CGCGGCCGCCTGCAGGTCGAC 3′ |
| CO3r.2: | 5′CTACCTACTAGTAACTAAAACTTAGC 3′ |
| 187S.1: | 5′ACG GTTGGTCTG TTC GAC 3′ |
(+) and (-) refers to coding and non-coding strands and (N=A,G,C,T; R=A,G; Y=C,T; W=A,T; S=C,T;H=A,C,T; D=A,G,T).
The complete sequence was assembled using SeqMan(DNASTAR, Inc. WI USA). The nucleotide sequence have been submitted to GenBank(accession no. NC_008797). Protein genes were identified by comparing the sequence of the encoded proteins to products of previously identified mt-protein genes (Bandyopadhyay et al., 2006; Boore and Brown, 1994a; Clary and Wolstenholme, 1985a; Maynard et al., 2005).
The ribosomal RNA genes were identified by homology to other mt-rRNA genes and hence are not defined by their actual 5′ and 3′ ends. Transfer RNA (tRNA) genes were identified by their ability to fold into the clover leaf structure characteristic of metazoan mt-tRNA, from the tri-nucleotide in the anticodon position and using tRNAscan-SE Search server (www.genetics.wustl.edu/eddy/tRNAscan-SE) or by eye.
Intergenic sequences between coxI and coxII were determined from PCR amplified product from total genomic DNA using oligonucleotide primers close to the C-terminus of coxI (COI-Ct421: 5′GGI ATR CCI CGI CGI TAY WSI GAY TAY CC 3′) and NH2- terminus of coxII (COII-24r: 5′ATI GCR TGR TCR TGR AAR AAR AT 3′). I=inosine and the other single letter codes are described in Table 1. The amplified DNA was cloned into pGEM T-Easy and the nucleic acid sequence of five clones from each species were determined. Nucleotide sequences for the 16s ribosomal RNA for the same species were also determined in a similar manner except 16s rRNA specific primers (16s fw.: 5′CCG GTC TGA ACT CAG ATC ACG T 3′ and 16s rev.: 5′GTT TAC CAA AAA CAT GGC TTC 3′) (Espiritu et al., 2001) were used for the PCR amplification. The PCR cycling profile were as follows: for 16s- initial denaturation (95°C, 60sec); followed by 40 cycles of denaturation (95°C, 20sec); annealing (55°C, 20 sec); and extension (72°C, 30 sec); for coxI- coxII intergenic- initial denaturation (95°C, 2min; followed by 40 cycles of denaturation (95°C, 30sec); annealing (55°C, 30 sec); and extension (72°C, 30 sec).
Nucleotide sequences have been submitted to GenBank. The accession numbers of the 16s sequences are EU078935-EU078945 and EU078946-EU078956 for the intergenic region.
Phylogenetic analysis was carried out using the nucleic acid sequences. Final nucleotide alignments were done by visual inspection (Table 3). Both trees were created from two independent runs each using the software program MrBayes (Huelsenbeck and Ronquist, 2001; Kumar et al., 2004; Ronquist and Huelsenbeck, 2003). 1,000,000 trees were made in each run, 10,000 of which were saved. The first 500 of each of those 10,000 were discarded as “burn-in”. The two independent runs were combined into a single tree where branches were preserved if they were found in 75% of those trees not discarded. The average standard deviation of split frequencies after 1,000,000 generations for the intergenic data was 3.031 × 10-3, and 2.556 × 10-3 for the ribosomal data.
Table 3. Alignment of intergenic sequences between coxI-coxII.
| Species | 1 | 11 | 21 | 31 | 41 |
|---|---|---|---|---|---|
| C. monachus | --------TT | AAAGTTAATT | ---------- | GTAATCTA-G | GTTT---AAT |
| C. magus | --------TT | AAAGTTAATT | ---------- | GTGATTTA-- | GTTT---AAT |
| C. consors | --------TT | AAAGTTAATT | ---------- | ATAATTTA-G | GTTC---AAT |
| C. stercusmuscarum | --------TT | GG-ACTACCT | ---------- | GTGATTAA-G | GTCC---AAT |
| C. circumcisus | --------TT | GA-ATTAATT | ---------- | ATGGTCTA-G | GTTC---AAT |
| C. aurisiacus | --------TT | GA-ATTAATT | ---------- | ATGGTCTA-G | GTTT---AAT |
| C. gaugini | --------TT | GA-ATTAATT | ---------- | ATGGTCTA-G | GTTC---AAT |
| C. striatus | --------TT | AA-GTTGAT- | ---------- | GTAGTCT--G | TTTCT--AAT |
| C. dalli | CTAGTGTGTT | AT-TTTATCT | AAGGGCATGT | ATAGTGGA-G | GCCCTGGTAT |
| C. textile | ----TGTATT | AT-TTTATCT | AAAGGCATAC | ATAGCGAA-G | GCCCTAGTAT |
| C. episcopatus | ----TAC-TC | GC-TTTATTT | AGGGATATAA | ATAGTTATTA | ATTC---TA- |
| Species | 51 | 61 | 71 | 81 | 91 |
| C. monachus | AA-TCGGAGT | GA-A------ | ----TTTGTA | TA-A------ | G-TTGT-TGT |
| C. magus | AS-TCGTAGT | GA-G------ | ----TTTATA | TA-G------ | G-TTGT-TGT |
| C. consors | AA-TTGCAGT | GA-A------ | ----TTTATA | TA-G------ | G-TTGT-TAT |
| C. stercusmuscarum | AA-CTACAGT | GA-G------ | ---AGGGATA | TAGATTATA- | GTTTAT-TGT |
| C. circumcisus | AATTAGCAAT | AA-G------ | ---AACTATA | TAGATTA--- | GTTTGT-TGT |
| C. aurisiacus | AA-TCGCAAT | AA-G------ | ---AACTATA | TAGATTA--- | GTTTGT-TGT |
| C. gaugini | AA-TCGCAAT | AA-G------ | ---AACTATA | TAGATTA--- | GTTTGT-TGT |
| C. striatus | AT-TCGTAAT | AA-G------ | ----TTTAGA | TAGACTGT-G | GCTTGT-TAT |
| C. dalli | AA-TGCTATT | CTTG-TTTGG | TTAGTTTAAT | TGCTTAGTAG | ATTT-CAAGT |
| C. textile | AA-TGCTGTT | -TTG-TTTAA | TTAGTTTAGT | TGCATAGTAG | GTTT-TAAGT |
| C. episcopatus | AA-TAGGACT | GTCTATTTAA | TTGATTTAGT | TTTCTAATAG | ATTT-TAAAT |
| Species | 101 | 111 | 121 | 131 | |
| C. monachus | TTGATATTTA | AATTGGTTAT | AAAGCT-TAA | GGTATATGCG | |
| C. magus | TTATTGTTTA | AATTGGTTAT | AAAGCT-TAA | AGTATATGCG | |
| C. consors | TTGTTGTTTA | AATTGGTTAT | AAAACT-TAA | AGTATATGCA | |
| C. stercusmuscarum | TTGATATTTA | AGTGAGTTAT | ACAGCT-TAA | AGTATATGCA | |
| C. circumcisus | TTGATGTTTA | AGTGAGTCAT | ACAGCT-TAA | AGTATATACA | |
| C. aurisiacus | TTGATGTTTA | AGTGAGTTAT | ACAGCTATAA | AGTATATACA | |
| C. gaugini | TTGATGTTCA | AGTGAGTTAT | ACAGCT-TAA | AGTATATACA | |
| C. striatus | TTGTTGAATA | AGTTGGTTAT | AAAGCT-TAA | AGTATATACG | |
| C. dalli | TT-ATTTTT- | GAT-GGTTAT | AAAACC-CAA | GGTGCACGCG | |
| C. textile | TT-ATTTTT- | GGT-GGTTAT | AAAACC-TAG | GGTGCATACA | |
| C. episcopatus | CTTATTTAT- | AGT-GGTTAT | AAAGCT-TAA | AGTACATGCA | |
| Species | 141 | 151 | 161 | 171 | |
| C. monachus | TGCAAGTATA | TAAATATAT- | AG---TTAAA | TATATTACTT | |
| C. magus | TGTAAGTATA | TAAATATAT- | AG---TTAAA | TATATTACTT | |
| C. consors | TGTAAGTGTA | TAAATATAT- | AG---TTAAA | TATATTACTT | |
| C. stercusmuscarum | TGTTAGTATA | TAAATATAT- | AA---ATAAA | TATATTACTT | |
| C. circumcisus | TGTTAGTATA | TAAATATAT- | AA-------- | ---------- | |
| C. aurisiacus | TGTTAGTATA | TAAATATAT- | AA---ATAAA | TATATTACTT | |
| C. gaugini | TGTTAGTATA | TAAATATAT- | AA---ATAAA | TATATTACTT | |
| C. striatus | TGCAAGTATA | TTAATATAT- | AA---ATGAA | TATATTACTT | |
| C. dalli | TGAGAGTGTG | TAAATATATT | AGCAAATA-T | TATATTATTT | |
| C. textile | TGAGAGTATG | AAAATATATT | AACAGATA-T | TATATCACTT | |
| C. episcopatus | TGAGAGTGTG | TGGATATATt | AATTGATA-T | TATATTACTT | |
A general time reversible (GTR) model was used, with the rate variation of some sites being invariable and the remaining rates drawn from a gamma distribution.
Results
The organization and gene content of Conus textile mitochondrial genome is shown in Figure 1. The size of the C.textile genome, 15,562 base pairs, is similar to the 15,380 base pairs for L. cerithiformis (Bandyopadhyay et al., 2006) the only other conoidean gastropod whose sequence has been determined, and the genome of the non-conoidean neogastropod, Ilyanassa obsoleta which is 15,263 base pairs (Simison et al., 2006).
Figure 1.
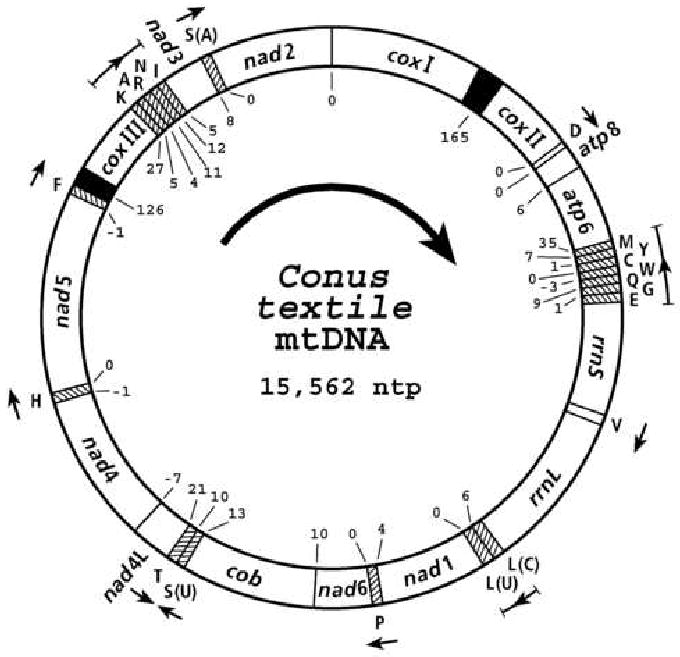
Gene arrangement of Conus textile mitochondrial DNA. Each tRNA gene is identified by the one letter amino acid code. The serine and leucine tRNA genes are identified by the first letter of the codon family (in parenthesis) that their transcription products recognize. The central arrow indicates the direction of transcription of proteins and rRNA genes. The direction of transcription of the tRNA genes are indicated by arrows. Numbers at gene boundaries indicate apparently non-coding nucleotides between genes. Overlap of genes are indicated by a minus sign preceding the numbers. The solid black sectors indicate the longest non-coding regions.
Genes
The genome encodes the expected 13 proteins, 2 ribosomal RNAs and 22 tRNAs. The mitochondrial genome encoded proteins, cytochrome oxidase subunits 1-3, (cox I-III); subunits 6 and 8 of ATPase (atp6 and atp8); subunits 1-6 and 4L of NADH dehydrogenase (nad 1-6 and nad 4L); cytochrome b (cob); tRNAs for D, V, L(1), L(2), P, S(1), S(2), H, F, K, A, R, N, I and small and large subunits of ribosomal RNA (rrnS, rrnL) are transcribed from the same DNA strand while tRNAs for M, Y, C, W, Q, G, E, and T are encoded in the complementary strand (Figure 1.). The intergenic unassigned spaces vary between 0 and 35 nucleotides with two relatively large regions of 165 and 126 nucleotides between coxI and coxII and tRNAPhe and coxIII respectively. The coding sequences of NAD4 and NAD4L overlap by 7 nts, NAD4 and tRNAHis by 1nt, NAD5 and tRNAPhe by 1nt, tRNATrp and tRNAGln by 3nts.
The sizes of the predicted mitochondrial proteins are similar to that reported for other gastropods.
Ribosomal RNA and transfer RNA genes
Ribosomal RNA genes were identified by homology to known ribosomal RNAs. The rrnS is located between 3′ end tRNAGlu (nt3851) and 5′ end of tRNAVal (nt4808). The rrnL is located between 3′ end of tRNAVal (nt4875) and 5′ end of tRNALeu (nt6254). Since the 5′ and 3′ termini of the RNAs have not been experimentally determined the assignments are tentative. The rrnS and rrnL thus defined are 958nt and 1380nt respectively.
Transfer RNA genes were identified on the possibility of these sequences to fold into secondary structures characteristic of metazoan mt-tRNAs. 22 putative tRNAs were identified.
Comparison of gene order in mitochondrial genomes of gastropod molluscs
The mitochondrial gene order of the neogastropods Conus textile, (this study) Lophiotoma cerithiformis (Bandyopadhyay et al., 2006) and Ilyanassa obsoleta (Simison et al., 2006) is conserved and is similar to that of Littorina saxatilis, a Littorinamorpha. (A partial sequence of L. saxitalis has been published (Wilding et al., 1999). The gene arrangement in the neogastropods can be derived from that of the polyplacophoran species Katharina tunicata (Boore and Brown, 1994a) and the vetigastropoda species, Haliotis rubra (Maynard et al., 2005) mainly by two major rearrangements as described previously for L. cerithiformis (Bandyopadhyay et al., 2006).
Unassigned intergenic sequences
Genes coding for tRNAs are interspersed between rRNA and protein encoding genes except for coxI/coxII, atp8/atp6, nad6/cob, nad4L/nad4, nad2/coxI. The tRNAs may serve as signals for processing larger transcripts (Battey and Clayton, 1980; Ojala et al., 1981). In addition, it has been suggested that in the absence of tRNAs, short non-coding sequences with putative secondary structure may serve as alternative signals for transcript processing (Bibb et al., 1981).
Two relatively large non-coding regions are present in Conus textile, a 126nt region between tRNAPhe and coxIII and a 165 nt region between coxI and coxII. The intergenic region between tRNAPhe and coxIII in the two other neogastropods, Lophiotoma cerithiformis and Ilyanassa obsoleta, that have been sequenced are 139 and 56 nt respectively. The secondary structures of the intergenic region between tRNAPhe and coxIII of the C. textile, L. cerithiformis, and I. obsoleta sequences include potential 23nt, 38nt and 14nt inverted repeats respectively. The longest non-coding region in Lophiotoma cerithiformis and Ilyanassa obsoleta mitochondrial genomes, is between tRNAPhe and coxIII and probably represents the control regions for replication and transcription. By analogy we suggest that this region plays a similar role in C. textile.
coxI/coxII intergenic sequences are useful as phylogenetic markers
The length of the coxI/coxII intergenic region in the C. textile mitochondrial genome (165nt) was unexpected. The intergenic sequences between coxI and coxII are considerably shorter for the other neogastropods for which a complete mt DNA sequence is available (12nt for L. cerithiformis and 28nt for I. obsoleta). We have determined the nucleotide sequence of this region from 20 cone snail species and 10 non-conus species generally assigned to the superfamily Conoidea. The intergenic region is longer in all cone snails analyzed: Table 2 shows the lengths of this intergenic region together with the lengths from two non-conoidean species obtained from the GenBank (I.obsoleta, DQ 238598; L.saxatilis, AJ 132137).
Table 2. Comparison of intergenic lengths between coxI and coxII.
| A. | |
|---|---|
| Non-conoideans | |
| Family Nassariidae | |
| Ilyanassa obsoleta | 28 bp |
| Family Littorinidae | |
| Littorina saxatilis | 30 bp |
| B. | |
| Superfamily Conoidea | |
| Family Turridae | |
| Genus Turris | |
| T. garnonsii | 23 bp |
| Genus Lophiotoma | |
| L. cerithiformis | 12 bp |
| Genus Gemmula | |
| G. speciosa | 0 bp |
| Family Terebridae | |
| Genus Hastula | |
| H. lanceata | 33 bp |
| Subgenus Impages | |
| H. (Impages) hectica | 28 bp |
| Genus Terebra | |
| T. guttatta | 23 bp |
| Family Drillidae | |
| D. regius | 32 bp |
| Family undetermined | |
| Genus Typhlosyrinx | |
| T. matsukumai | 75 bp |
| Genus Thatcheria | |
| T. mirabilis | 88 bp |
| Family Conidae | |
| Genus Conus | |
| Subgenus Pionoconus | |
| C. magus | 131 bp |
| C. consors | 132 bp |
| C. monachus | 132 bp |
| C. achatinus | 132 bp |
| C. aurisiacus | 138 bp |
| C. catus | 138 bp |
| C. consors | 132 bp |
| C. stercusmuscarum | 130 bp |
| C. striolatus | 137 bp |
| C. gaugini | 137 bp |
| C. striatus | 137 bp |
| Subgenus Gastridium | |
| C. obscurus | 149 bp |
| C. tulipa | 153 bp |
| Subgenus Rhizoconus | |
| C. capitaneus | 153 bp |
| C. vexillum | 157 bp |
| C. miles | 160 bp |
| Subgenus Cylinder | |
| C. textile | 165 bp |
| C. dalli | 165 bp |
| C. episcopatus | 164 bp |
| Subgenus Conasprella | |
| (see discussion) | |
| C. vimineus | 197 bp |
| C. memiae | 217 bp |
| C. arcuatus | 316 bp |
| C. orbignyi | 322 bp |
| C. ione | 429 bp |
| C. mahogani | 456 bp |
| C. jaspideus | 504 bp |
Four discrete size classes of intergenic sequences are apparent from the data in Table 2: < 40 nts, 70-90 nts, 130-170 nts and 190-510 nts. The intergenic region of the majority of the Conus species are between 130-170 nts with a narrower distribution within clades of related species in the genus. Species analyzed in the Pionoconus clade that comprise fish-hunting species widely distributed across the Indo-Pacific, had intergenic region between 130-140 nts in length and the three species in Cylinder (to which Conus textile belong) are between 165-170 nts. As shown in Table 2B, one group of species conventionally assigned to Conus have an extremely broad range of intergenic lengths, 190-510 nts (see Discussion below).
We examined if the intergenic sequences of different Conus species within a clade could be used to clarify their evolutionary relationships. The alignment of the intergenic sequences is shown in Table 3. Phylogenetic relationships for the same series of species were also deduced using 16s rRNA sequences. Figure 2 shows a comparison of the results of the analysis using the two data sets; the trees obtained in both cases are similar. The mollusc-hunting species C. textile, C. dalli, and C. episcopatus in the Cylinder clade are well resolved from the fish-hunting species C. monachus, C. magus, C. consors, C. stercusmuscarum, C. circumcisus, C. aurisiacus, C. gaugini, and C. striatus from the Pionoconus clade. However, the analysis using the intergenic region appears to yield somewhat better resolution, separating C. magus and C. consors from C. monachus in one sub-branch of Pionoconus branch, and C. textile and C. dalli from C. episcopatus in the Cylinder branch.
Figure 2.
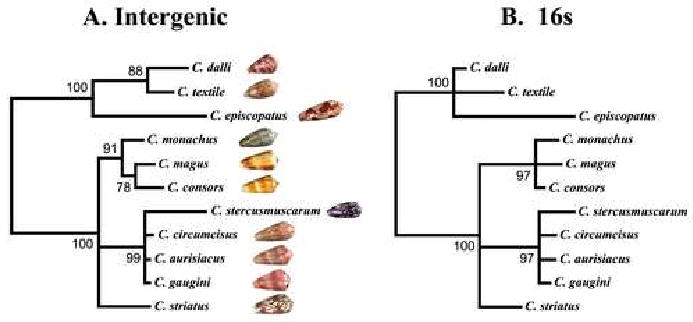
Comparison of phylogenetic trees constructed using MrBayes and coxI-coxII intergenic sequences (A) and 16s mitochondrial DNA sequences (B). (Only those branches that were found in 75% or more of the trees were retained. Shells for each species are also shown in A.
Discussion
When this study was initiated, one goal was to assess whether changes in mitochondrial gene order could be utilized to evaluate the phylogeny of Conoidean molluscs, a large and taxonomically complex group of predatory venomous snails (recently estimated to exceed 10,000 species). However, it is likely that gene order in the entire order Neogastropoda (to which Conoidean molluscs belong) is the same; this probably extends even to other groups of gastropods such as the Littorinamorphs (e.g. Littorina saxitalis). The conoidean gene order can be readily derived from the polyplacophoran Katharina tunicata by only two rearrangements, (Bandyopadhyay et al., 2006; Boore and Brown, 1994a). Thus, the gene order described above for Conus textile appears to be the major pattern in many gastropod lineages, although some groups, such as the vermetid species within the genus Dendropoma (Caenogastropoda, Littorinamorpha,Vermetidae), exhibit striking changes in gene order that are estimated to have occurred only in the last 38-48 million years (Rawlings et al., 2001). The present results suggest that such gene order changes have probably not occurred extensively in Conoideans, or in other neogastropod groups, although the possibility that a small clade of species may have undergone gene order changes remains a possibility.
The most notable and useful discovery resulting from the determination of the complete Conus textile mitochondrial DNA sequence is based on the striking difference found between the coxI – coxII intergenic of C. textile and that of Lophiotoma cerithiformis. Since the C. textile intergenic region was unexpectedly large, a larger survey of a number of diverse Conoidean taxa was carried out, including species in the families Turridae, Terebridae, Drillidae and additional Conoidea, as well as some problematic taxa.
The non-Conoideans, and all of the species in the families Turridae, Terebridae and Drillidae had relatively short intergenic regions, <40 bp in length. All of the Conus species examined had a longer intergenic region; for most species, this generally varied in length from 130–170 bp, with the exceptions discussed below. Notably, one group of conoideans comprising two different genera, Thatcheria, and Typhlosyrinx, had an intergenic region of intermediate length, from 70-90 bp. This is a potentially significant result, because these genera are somewhat problematic in their placement within the superfamily. Conoideans in these taxa have traditionally been included in the large group known as “turrids”, and in the conventional phylogeny, were all included in the family Turridae. However, these genera are part of a group of “turrids” that were recently proposed to be transferred from their traditional assignment in the family Turridae to the family Conidae (Taylor et al., 1993); they are regarded as “Daphnellid genera”, and included in a subfamily group referred to as Daphellinae or Raphitominae by various workers. Their intergenic regions are clearly longer than Lophiotoma cerithiformis and other related taxa, but not as long as in all of the species of Conus examined. The intermediate length of the intergenic region of these taxa this provides an additional molecular guidepost for the assessment of where these groups should be placed within the superfamily Conoidea.
Among cone snails (Conus), the species we analyzed fall into five different clades, generally given subgeneric rank within the genus. For four of the clades, there is a small variation in the length of the intergenic region observed; thus, fish-hunting Conus species belonging to the Pionoconus clade have a smaller intergenic region (∼135 bp.) than the molluscivorous species that belong to the Cylinder clade (∼165 bp.). The four clades all have coxI-coxII intergenic regions between 130-170 bp in length.
The most surprising and unexpected result was the discovery that a group of Conus species had the largest coxI-coxII intergenic regions so far discovered in any animal mitochondra; a species in this group has a coxI-coxII region >500 bp in length. These are mostly species traditionally assigned to the subgenus Conasprella. Repeated sequence motifs are not observed in the long intergenic regions. In addition, the alignment of the short intergenic regions to the longer ones occur in short dispersed regions. As shown in Table 2, not only are the intergenic regions unusually long but they are also extremely variable in length (and always longer than any other group within the genus Conus, or in the whole superfamily Conoidea).
Recently, this group of species was shown to be surprisingly distant phylogenetically from all other Conus clades; similar results were previously reported by us (our own data is shown in Figure 3) and by others (Kohn et al., 1999; Rolan and Raybaudi Massilia, 1994a, 1994b). The discovery that there is a qualitative difference in the intergenic region length of these species from other clades in Conus, as well as the unexpectedly great phylogenetic divergence suggested by the 16S data in Figure 3 all indicate that these group of species are not congeneric with other Conus species. The intergenic region provides a defining genetic marker that makes this group of species qualitatively distinguishable from all of the other taxa in the superfamily Conoidea. On the basis of the data presented above, we suggest that this group of species, for which Conasprella appears to be the earliest available name, be regarded as a separate genus from Conus. The new genus, includes not only species traditionally assigned to Conasprella but all the other Conus species with expanded intergenic regions. This suggestion is consistent with anatomical studies on the radular teeth of Conus/Conasprella spp (Kohn et al., 1999; Rolan and Raybaudi Massilia, 1994a, 1994b).
Figure 3.
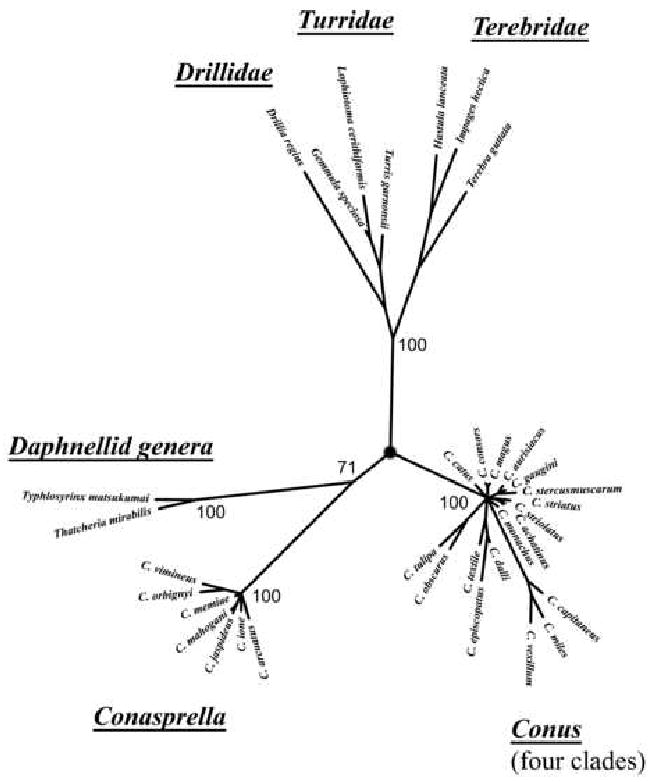
Phylogenetic relationship of Conoideans constructed using MrBayes and 16s mitochondrial rRNA sequences. Members of the Drillidae, Turridae, Terebridae, Conidae (four clades), Conasprella and the “Daphellid genera” (see text) are represented.
In summary, the intergenic region between coxI and coxII is a useful genetic marker for molecular evolution studies of the superfamily Conoidea. The length of the region can be used to assess broad evolutionary trends in the superfamily, while direct comparison of sequences of this region can be used to help clarify how closely related species in a clade are related to each other.
Our results raise a number of intriguing questions: why has there been an expansion in this intergenic region in some conoidean lineages but not in others? In Figure 3 we indicated the demarcation between the lineages in which the intergenic region remains <40 nucleotides, vs. the lineages where it has amplified.
The most remarkable lineage where an intergenic expansion has been taking place are the species in Conasprella. Using 16S data (shown in Fig 2) as well as 12S data (not shown), all of the species with intergenic regions >190 bp appear to be closely related to each other (see Fig 3). Not only are the intergenic regions of all Conasprella longer than in any other Conoideans, but the variability observed in the expanded region has resulted in an enormous range in the length of these sequences; Conus jaspideus, with >500 nucleotides has reached an apogee in Conoidean intergenic amplification. Why the length of the intergenic interval appears to be constrained over a narrow range in most clades but unconstrained in Conasprella, is a phenomenon for which we strain to come up with even the most speculative of conjectures at this time.
Metazoan mtDNA usually has a single large non-coding region. This region referred to as the control region (CR) contain signals for the initiation of transcription and replication(Boore, 1999; Taanman, 1999; Wolstenholme, 1992). In mollusks the length of the longest non-coding region varies and in some cases they are considerably short. In the absence of experimental evidence it is not possible to ascertain what features in the non-coding region (other than being A-T rich and secondary structure) constitute signals for initiation of replication and transcription. Though a single CR is most common, duplicate CRs have also been observed. In such cases the two CRs have identical or highly similar nucleotide sequences. However, in birds duplication has been reported in which one of the copies of the CR is a degenerate control region. Shao et al. (Shao et al., 2005) suggested that the two CRs may evolve independently and that duplicate CRs may be useful markers at the genus or family level.
In the case of the neogastropods Ilyanasa obsoleta, Lophiotoma cerithiformis and Conus textile whose gene-orders are known, the longest non-coding region occur at comparable locations (between tRNAPhe and coxIII) and may indeed be functional CRs. The sequence of the intergenic region between coxI and coxII in C. textile is 30% similar to that between tRNAPhe and coxIII and is unlikely to be a duplicate CR. At present we do not have an explanation for the wide variation in intergenic length. However, in analogy to trinucleotide expansions a plausible scenerio would be that the region between coxI and coxII might have been prone to expansion due to presence of short repeats in the ancestral sequence. Different degrees of expansion and subsequent mutations have resulted in the heterogeneity of observed lengths.
Acknowledgments
We thank the University of Utah core facilities for DNA sequencing and oligonucleotide synthesis (supported by NCI CA42014). This work was supported by a grant from the National Institutes of Health, GM48677.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- Bandyopadhyay PK, Stevenson BJ, Cady MT, Olivera BM, Wolstenholme DR. Complete mitochondrial DNAn sequence of a Conoidean gastropod, Lophiotoma (Xenuroturris): Gene order and gastropod phylogeny. Toxicon. 2006;48:29–43. doi: 10.1016/j.toxicon.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Battey J, Clayton DA. The transcription map of human mitochondrial DNA implicates transfer RNA excision as a major processing event. J Biol Chem. 1980;255:11599–11606. [PubMed] [Google Scholar]
- Bibb MJ, van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26:167–180. doi: 10.1016/0092-8674(81)90300-7. [DOI] [PubMed] [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL, Brown WM. Complete DNA sequence of the mitochondrial genome of the black chiton, Katharina tunicata. Genetics. 1994a;138:423–443. doi: 10.1093/genetics/138.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchet P, Lozouet P, Maestrati P, Heros V. Assessing the magnitude of species richness in tropical marine environments: high numbers of molluscsat a New Caledonia site. Biological Journal of the Linnean Society. 2002;75:421–436. [Google Scholar]
- Bouchet P, Rocroi JP. Classification and nomenclator of Gastropod families. 2005;47:1–397. [Google Scholar]
- Clary DO, Wolstenholme DR. The mitochondrial DNA molecule of Drosophila yakuba: nucleotide sequence, gene organization, and genetic code. J Mol Evol. 1985a;22:252–271. doi: 10.1007/BF02099755. [DOI] [PubMed] [Google Scholar]
- Cruz LJ, White J. Clinical toxicology of Conus snail stings. CRC Press; Boca Raton, FL: 1995. [Google Scholar]
- Czerwiec E, Kalume DE, Roepstorff P, Hambe B, Furie B, Furie BC, S J. Novel gamma-caboxyglutamic acid- containing peptides from the venom of Conus textile. FEBS J. 2006;273:2779–2788. doi: 10.1111/j.1742-4658.2006.05294.x. [DOI] [PubMed] [Google Scholar]
- Duda TFJ, Kohn AJ. Species-level phylogeography and evolutionary history of the hyperdiverse marine gastropod genus Conus. Mol Phylogenet Evol. 2005;34:257–272. doi: 10.1016/j.ympev.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Espiritu DJD, Watkins M, Dia-Monje V, Cartier GE, Cruz LJ, Olivera BM. Venomous cone snails: molecular phylogeny and generation of toxin diversity. Toxicon. 2001;39:1899–1916. doi: 10.1016/s0041-0101(01)00175-1. [DOI] [PubMed] [Google Scholar]
- Fegan D, Andresen D. Conus geographus envenomation. The Lancet. 1997;349:1672. doi: 10.1016/S0140-6736(05)62639-6. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogeny. Bioinformatics. 2001;17:753–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Kohn AJ. Tempo and mode of evolution in Conidae. Malacologia. 1990;32:55–67. [Google Scholar]
- Kohn AJ. Superfamily Conoidea. In: Beesley PL, Ross GJB, Wells A, editors. Mollusca: The Southern Synthesis. Fauna of Australia. Part B Viiii. CSIRO Publishing; Melbourne: 1998. pp. 846–854. [Google Scholar]
- Kohn AJ, Nishi M, Pernet B. Snail Spears And Scimitars: A Character Analysis Of Conus Radular Teeth. J Moll Stud. 1999;65:461–481. [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA 3: Integrated software for olecular Evolutionary Genetics Analysis and sequence alignment. Briefings in Bioinformatics. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Maynard BT, K LJ, McKiernan JM, Jansen ES, Hanna PJ. Mitochondrial DNA sequence and gene organization in Australian blacklip abalone, Haliotis rubra (leach) Mar Biotechnol (NY) 2005;7:645–658. doi: 10.1007/s10126-005-0013-z. [DOI] [PubMed] [Google Scholar]
- Miljanich G. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr Med Chem. 2004;11:3029–3040. doi: 10.2174/0929867043363884. [DOI] [PubMed] [Google Scholar]
- Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–474. doi: 10.1038/290470a0. [DOI] [PubMed] [Google Scholar]
- Olivera BM. From Marine Snail Venom to Analgesic Drug. Karger; Basel: 2000. [Google Scholar]
- Olivera BM. Conus Peptides: Biodiversity-based Discovery and Exogenomics. Journal of Biological Chemistry. 2006;281:31173–31177. doi: 10.1074/jbc.R600020200. [DOI] [PubMed] [Google Scholar]
- Oliverio M, Mariottini P. A molecular framework for the phylogeny of Coralliophila and related muricoids. The Journal of Molluscan Studies. 2001;67:215–224. [Google Scholar]
- Ponder WF, Lindberg DR. Towards a phylogeny of gastropod molluscs: an analysis using morphological characters. Zoological Journal of the Linnean society. 1997;119:83–265. [Google Scholar]
- Ponder WF, Waren A. Classification of the Caenogastropoda and Heterostropha- A list of the family-group names and higher taxa. Malacological Review. 1988 4:288–328. [Google Scholar]
- Rawlings TA, Collins TM, Bieler R. A major mitochondrial gene rearrangement among closely related species. Mol Biol Evol. 2001;18:1604–1609. doi: 10.1093/oxfordjournals.molbev.a003949. [DOI] [PubMed] [Google Scholar]
- Rockel D, Korn W, Kohn AJ. Manual of Living Conidae. Verlag Christa Hemmen; Wiesbaden, Germany: 1995. [Google Scholar]
- Rolan E, Raybaudi Massilia G. New investigations on the radular teeth of Conus. Part I. Argonauta. 1994a;8(16):6–59. [Google Scholar]
- Rolan E, Raybaudi Massilia G. New investigations on the radular teeth of Conus. Part II. Argonauta. 1994b;8(79):9–68. [Google Scholar]
- Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russel DW. Molecular Cloning A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- Shao R, Barker SC, Mitani H, Aoki Y, Fukunaga M. Evolution of duplicate control regions in the mitochondrial genomes of metazoa: a case study with Australasian Ixodes ticks. Mol Biol Evol. 2005;22:620–629. doi: 10.1093/molbev/msi047. [DOI] [PubMed] [Google Scholar]
- Simison WB, Lindberg DR, Boore JL. Rolling circle amplification of metazoan mitochondrial genomes. Molecular Phylogenetics and Evolution. 2006;39:562–567. doi: 10.1016/j.ympev.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410:103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- Taylor JD, Kantor YI, Sysoev AV. Foregut anatomy, feeding mechanisms, relationships and classification of the Conoidea (=Toxoglossa) (Gastropoda) Bulletin of the Natural History Museum, Zoology Series. 1993;59:125–170. [Google Scholar]
- Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- Wilding CS, Mill PJ, Grahame J. Partial Sequence of the Mitochondrial genome of Littorina saxatilis: relevance to gastropod phylogenetics. J Mol Evol. 1999;48:348–359. doi: 10.1007/pl00006479. [DOI] [PubMed] [Google Scholar]
- Wolstenholme DR. Animal mitochondria DNA: structure and evolution. Int Rev Cytol. 1992;141:173–216. doi: 10.1016/s0074-7696(08)62066-5. [DOI] [PubMed] [Google Scholar]