

Abstract
The progression of atherosclerosis is associated with leukocyte infiltration within lesions. We describe a technique for the ex vivo imaging of cellular recruitment in atherogenesis which utilizes quantum dots (QD) to color-code different cell types within lesion areas. Spectrally distinct QD were coated with the cell-penetrating peptide maurocalcine to fluorescently-label immunomagnetically isolated monocyte/macrophages and T lymphocytes. QD–maurocalcine bioconjugates labeled both cell types with a high efficiency, preserved cell viability, and did not perturb native leukocyte function in cytokine release and endothelial adhesion assays. QD-labeled monocyte/macrophages and T lymphocytes were reinfused in an ApoE−/− mouse model of atherosclerosis and age-matched controls and tracked for up to four weeks to investigate the incorporation of cells within aortic lesion areas, as determined by oil red O (ORO) and immunofluorescence ex vivo staining. QD-labeled cells were visible in atherosclerotic plaques within two days of injection, and the two cell types colocalized within areas of subsequent ORO staining. Our method for tracking leukocytes in lesions enables high signal-to-noise ratio imaging of multiple cell types and biomarkers simultaneously within the same specimen. It also has great utility in studies aimed at investigating the role of distinct circulating leukocyte subsets in plaque development and progression.
1. Introduction
Atherosclerosis is a complex disease exacerbated by multiple cellular and molecular participants. A key contributor to atherogenesis is the dysfunction of vascular endothelium, which presents molecular cues promoting leukocyte infiltration into arterial intima [34, 35]. Leukocytes which localize within atherosclerotic plaques include monocytes [59, 34] and lymphocytes, primarily of the CD4+ subset [56, 65]. Monocytes migrate toward lesions and accumulate lipids in a process that transforms them into macrophages and macrophage-derived foam cells [32]. These lipid-laden macrophages secrete reactive oxygen species and proteinases which induce a positive-feedback monocyte recruitment cascade, eventually leading to plaque instability and rupture [13]. Lymphocytes also contribute to the progression of atherosclerosis via the secretion of the proinflammatory cytokine interferon gamma (IFN-γ), which contributes to leukocyte recruitment into plaques and to plaque instability [56, 21, 40, 52]. Other cell types, such as neutrophils [49] and dendritic cells [7, 8], also play a role in potentiating atherosclerotic complications via their activation proximal to inflammatory plaque surfaces. Quantitative imaging of inflammatory cells such as macrophages [48] and T lymphocytes [22] within atherosclerotic lesions has been reported and is useful for elucidating biological processes and response to therapies in vascular disease. Given the diversity of cell types involved in lesion biology, multiplexing techniques for visualizing spatial and temporal dynamics for a variety of cellular mediators are needed to identify their specific roles in the different stages of atherosclerosis.
Advances in understanding the pathobiology of atherosclerosis have identified a number of key cellular and molecular mediators involved in early and late stages of disease [11, 12]. Furthermore, innovations in both imaging instrumentation and in vivo imaging reagents complement these therapeutic and diagnostic targets for developing strategies to visualize vascular disease in vivo [26, 27]. Current work has enabled the in vivo visualization of macrophages in lesions using magnetic resonance imaging [3, 37]. Other modalities facilitating studies of atherosclerosis include optical imaging [44, 62, 14], and positron emission tomography (PET) [50], through approaches targeting either intracellular or cell surface biomarkers. However, PET is limited in resolution due to scattering artifacts and is limited in the number of applicable tracers. For optical imaging purposes, many cell-labeling optical tracers such as calcein-AM (CAM) and acridine orange have low photostability, may leak out of cells once internalized, and have emission spectra that significantly overlap with the autofluorescence of tissues [46, 47]. Encoding cells with fluorescent proteins has been successfully utilized for bioimaging applications, but requires in vitro or ex vivo transfections, and fluorescent protein emissions may overlap with the autofluorescence window of tissues and atherosclerotic plaques [54]. Magnetic resonance imaging and other clinically relevant imaging modalities such as ultrasound and immunoscintigraphy cannot detect distinct ‘colors’ within tissue, complicating multiplexed approaches [41]. Recently, we investigated a strategy for the color-coding of circulating leukocytes and cell surface biomolecules with fluorescing semiconducting nanocrystals, or quantum dots (QD), for multiplexed in vivo vascular imaging in rodent models of ocular inflammation [29]. The size-tunable emission spectra of QD enable the simultaneous in vivo and ex vivo tracking of multiple cells and biomarkers within the vasculature of the same animal with only one excitation source. Furthermore, QD are amenable to bioconjugation of ligands for cell targeting, and feature high quantum yields and photostability for long-term imaging applications [2, 19, 6, 17, 24, 28].
In this report, we demonstrate the utility of a method based on the optical properties of QD to perform ex vivo imaging of atherosclerotic cellular components. The technique uses QD coated with the cell-penetrating peptide maurocalcine [15] to fluorescently label leukocytes. This peptide has been shown to translocate fluorescent cargoes across membranes in a variety of live cells such as hippocampal neurons in vitro without adverse functional effects or toxicity [9, 15, 38]. We isolated monocyte/macrophages and T lymphocytes based on the biomarkers CD11b and TCR, respectively, for QD labeling. Both populations have been strongly associated with the pathogenesis of lesion formation [35]. These populations were labeled with distinct QD emission wavelengths so that they could be simultaneously detected in tissue. This approach was used to visualize leukocyte subtype recruitment to aortic lesions in the ApoE−/− mouse model of atherosclerosis [45], and is a promising strategy for the multiplexed imaging of cells and biomolecules in vascular processes.
2. Methods
2.1. Immunomagnetic isolation of leukocytes
All animal procedures were approved by the Institutional Animal Care and Usage Committee at Vanderbilt University. Spleens from wild-type and ApoE−/− mice aged from 7 months to 1 year were collected in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 1% streptomycin-Fungizone. Splenocytes were prepared by disrupting the spleen with a syringe plunger on a nylon mesh filter. Collected cells were washed once in RPMI medium. Lymphocytes were isolated using Lympholyte M density gradient medium (Cedarlane Laboratories, Burlington, NC) according to manufacturer's instructions. Lympholyte M removes granulocytes and erythrocytes from the sample, leaving a supernatant consisting of mononuclear cells for the subsequent enrichment of monocyte/macrophages and lymphocytes [1, 25, 53]. Monocyte/macrophage and T lymphocyte-rich leukocyte fractions were enriched using anti-CD11b-coated paramagnetic microbeads (Miltenyi Biotec, Auburn, CA, # 130-049-601) and biotin-anti-T cell receptor antibody (TCR β chain, BD Biosciences, San Jose, CA) coated paramagnetic microbeads (Miltenyi Biotec # 130-091-256) for CD11b+ cells and T lymphocytes, respectively. Each population was characterized by flow cytometry. Antibodies used were phycoerythrin (PE) anti-CD11b (BD Biosciences # 557397) for validation of immunomagnetic isolations, Alexa Fluor 488 anti-CD68 (AbD Serotec, Raleigh, NC, # MCA1957A488) for monocyte/macrophages, and PE anti-CD4 (BD # 553049) and PE anti-CD8a (BD # 553032) for detection of T lymphocytes (double positive staining). In all cases, the purity was >90% (n = 4). Data was analyzed using a FACSCalibur flow cytometer (BD Biosciences) and FlowJo 7.2 (Treestar Software, Ashland, OR).
2.2. Quantum dot labeling of leukocytes
QD coated with 6–8 streptavidins/nanoparticle emitting at 585 and 655 nm (QD585 and QD655) were purchased from Invitrogen Corporation (Carlsbad, CA). Biotinylated maurocalcine peptide was synthesized by Biomatik Corporation according to previously published protocols [15]. A 35-fold molar excess of peptide was mixed with 1 molar equivalent of QD585 or QD655 in 100 mM PBS, pH = 7.4, for 30 min at room temperature. QD–maurocalcine was then separated from excess peptide using size-exclusion chromatography over a Superdex 200 column according to the manufacturer's instructions (GE Healthcare, UK). Elution fractions were measured using a BioTek Synergy HT microplate spectrophotometer (BioTek, Vermont, USA) configured for QD fluorescence at 585 or 655 nm, and peptide fluorescence as measured by fluorescamine assay (Sigma Chemical, St Louis, MO). This process typically yielded QD with 12–17 peptides per nanocrystal.
For each cell-labeling experiment, 1× 106 cells ml−1 were labeled with 100 nM QD–maurocalcine in PBS, pH = 7.4, at 37 °C for 30 min. In some cases, leukocytes were labeled ex vivo with 1 μM calcein-AM for 30 min according to the manufacturer's instructions (Invitrogen). Cells were rinsed with PBS via centrifugation at 200g for 10 min. The fraction of viable cells was determined by the LIVE/DEAD single color fixable green kit according to the manufacturer's instructions (Invitrogen) on a FACSCalibur flow cytometer. Live cell population was then gated to determine the fraction of QD-labeled cells. Unlabeled cells subjected to the same rinsing and centrifugation steps served as controls.
2.3. Monocyte adhesion assay
A bEnd.3 mouse endothelial cell line was a gift from Dr Jack Virostko of the Vanderbilt University Institute for Imaging Science. Cells were cultured in DMEM supplemented with 10% fetal bovine serum to confluence on 24-well tissue culture grade microplates. Cells were then treated with 500 ng ml−1 Salmonella Typhimurium lipopolysaccharide (LPS, Sigma) or untreated in complete media overnight. Monocyte/macrophages (1 × 106 cells ml−1) were incubated with 1 μM calcein-AM (CAM, Invitrogen) for 30 min. From this suspension, aliquots containing 100000 monocyte/macrophages were either incubated in PBS or PBS containing 100 nM QD–maurocalcine for 30 min at 37 °C. After rinsing with PBS, cells were plated onto LPS-stimulated or untreated bEnd.3 monolayers and incubated for 1 h at 37 °C. At the end of the experiment, monolayers were rinsed three times with PBS to remove non-adherent cells, and the microplate was measured for calcein-AM fluorescence (490 nm excitation/525 nm emission). Results were compared by t-test and plotted using Sigmaplot 9 (SYSTAT, San Jose, CA).
2.4. Cytokine ELISA of QD-labeled leukocytes
Cytokine secretion by QD-labeled cells in the presence or absence of stimulating agents was investigated by ELISA. CD11b+ and TCR+ immunomagnetically purified cells were labeled with QD–maurocalcine, and incubated with or without lipopolysaccharide (LPS for CD11b+) or Concanavalin A (ConA, TCR+) to stimulate cytokine secretion. Following exposure, cell supernatants was assayed for cell-specific cytokine secretion using mouse IL-2 and TNFα ELISA kits according to the manufacturer's instructions (eBioscience, San Diego, CA). Concentrations and durations used for stimuli were: ConA, 2 μg ml−1 for 22 h, and LPS: 50 ng ml−1 for 6 h. Experiments were performed in triplicate.
2.5. Analysis of QD-labeled leukocytes in ApoE−/− mouse aorta
ApoE−/− and wild-type mice ranging from 5 months to 1 year of age fed on a chow diet were injected with 1 × 106 QD–maurocalcine-labeled or CAM-labeled monocyte/macrophages or T lymphocytes via the retro-orbital plexus. In some cases, a solution of 100 nM QD–maurocalcine in 100 μl PBS was injected. Animals were euthanized 2, 3, 5, or 28 days post-injection, and both aortic sinus and whole aorta were harvested, sectioned, and prepared as described previously [57]. 5 μm frozen sections were stained for immunofluorescence with Alexa Fluor 488-conjugated anti-CD68 or anti-TCR and mounted in VECTASHIELD with DAPI (Vector Laboratories, Burlingame, CA). Sections were analyzed by fluorescence microscopy on a Nikon TE2000U Eclipse inverted microscope (Nikon Instruments, Melville, NY) using DAPI, Alexa Fluor 488/calcein-AM, QD585, and QD655-specific bandpass filter sets in conjunction with a Hamamatsu C7780 cooled CCD camera (Hamamatsu Corp., Bridgewater, NJ). Image analysis was conducted using Image Pro Plus 5.1 (Media Cybernetics, Bethesda, MD).
2.6. Ex vivo imaging
En face aortas from ApoE−/− and wild-type mice were imaged for QD fluorescence using an IVIS 200 fluorescence imaging system configured for spectrally distinct detection of QD585 and QD655 using bandpass emission filters (Caliper Life Sciences, Hopkinton, MA). For QD imaging, GFP excitation settings were used, and for ORO, Texas Red excitation settings were used. Following QD imaging, aortas were stained with Oil Red O (ORO) and the percentage of atherosclerotic lesions (ORO positive area) was quantified using a previously described technique [16]. The IVIS 200 was then used to image ORO-stained areas via analysis of dye-induced fluorescence [60, 64] with QD fluorescence subtracted from the image. QD585 lymphocytes, QD655-monocyte/macrophages, and ORO could be colocalized within the same tissue due to their distinct spectra. For quantification of lesion fluorescence intensities, ORO+ lesions in 12 month old ApoE−/− en face aortas were identified and encircled using the region of interest (ROI) function in Image Pro Plus. The same ROI's were then placed in matched regions on the age-matched wild-type mice and younger ApoE−/− mouse (5 mos.) aortas. For these matched ROI's (n = 6 areas per aorta), mean fluorescence intensities in the QD585, QD655, and ORO-specific channels were measured.
3. Results
3.1. Loading of leukocytes with QD–maurocalcine bioconjugates
The attachment of maurocalcine cell-penetration peptides to QD enabled their efficient loading into T lymphocytes and monocyte/macrophages ex vivo, with no appreciable effects on cell viability and function. Immunomagnetic isolation procedures using anti-TCR and anti-CD11b beads yielded distinct leukocyte subpopulations as demonstrated by flow cytometric analysis (figures 1(A) and (B)). Both cell subsets were distinctly identified via characteristic markers CD11b/CD68 (monocyte/macrophage) and CD4/CD8 (T lymphocyte). Labeling of these populations separately with QD585-maurocalcine and QD655-maurocalcine was more efficient than labeling with QD-streptavidin alone (figures 1(C) and (D)).
Figure 1.
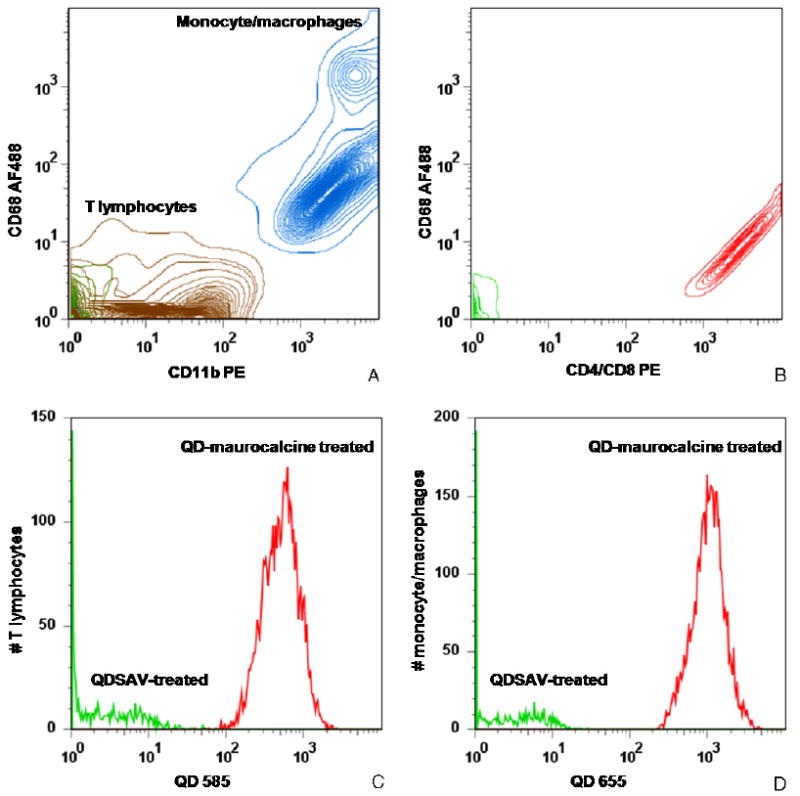
FACS contour analysis of immunomagnetically isolated leukocyte subpopulations (A) and (B) and QD–maurocalcine labeling efficiency of T lymphocytes and monocyte/macrophages (C) and (D). (A) TCR-enriched cells (brown) exhibited weak or absent CD11b expression and were negative for CD68, a monocyte/macrophage marker. CD11b-enriched cells were positive for both CD11b and CD68 markers (blue). Two subpopulations can be identified based on relative CD68 expression, which indicate macrophages (high expression) and monocytes (relatively lower expression). Unlabeled lysed whole blood fluorescence is labeled green. (B) TCR-enriched cells are positive for both CD4 and CD8a (red), indicating that TCR-enriched cells bear T lymphocyte markers. Unstained TCR-enriched cells are labeled green. (C) and (D) T lymphocytes (C) and monocyte/macrophages (D) were incubated with either QD–maurocalcine (red) or QD-streptavidin not conjugated to peptide (green). QD–maurocalcine efficiently labeled both cell populations.
(This figure is in colour only in the electronic version)
3.2. Effects of QD labeling on leukocyte function
Flow cytometric viability assays did not reveal significant differences in cell viability between unlabeled cells and cells labeled with QD–maurocalcine, for either cell population (figure 2(A)). Furthermore, monocyte/macrophages labeled with QD adhered to LPS-stimulated bEnd.3 mouse endothelium at a statistically similar efficiency to cells loaded only with CAM cell tracking dye (figure 2(B)). In cytokine release assays, monocyte/macrophages loaded with QD–maurocalcine released <20 pg ml−1 TNF-α, statistically similar to untreated cells. T lymphocytes secreted less than 0.05 ng ml−1 IL-2, similar to untreated cells.
Figure 2.
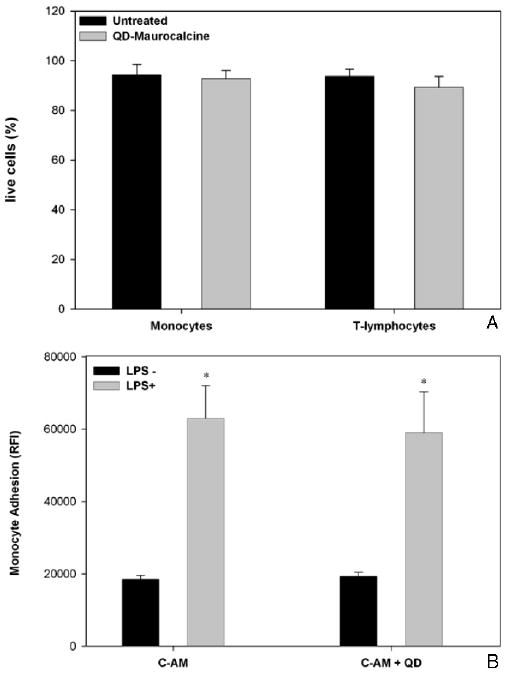
Analysis of QD655+-CD11b+ and QD585+-TCR+ cell viability (A) and QD655+-CD11b+ adhesion on endothelial monolayers in vitro (B). (A) Cell viability as reported for 20 000 cells. (B) Adhesion of 1 × 105 calcein-AM (CAM)-labeled or QD–maurocalcine and CAM-labeled CD11b+ cells to bEnd.3 mouse endothelial cell monolayers, as detected by CAM fluorescence intensity. Reported as mean ± S.D, n = 3. * − P < 0.05.
3.3. Localization of QD-labeled leukocyte subsets in atherosclerotic lesions
QD-labeled monocyte/macrophages were observed in atherosclerotic lesions as early as two days post-injection and as late as four weeks (last time point of the study) in ApoE−/− mice. Figure 3 shows QD-labeled monocyte/macrophages (QD655-CD11b+) within aortic root lesions of 7 (figures 3(A)–(C)) and 12 month old (figures 3(D)–(F)) ApoE−/− mice, representing early and late atherosclerotic lesions. In very early atherosclerosis (5 month old ApoE−/−), however, only minimal QD655-CD11b+ accumulation were detectable by fluorescence microscopy in thin sections of immature lesions (figure 3(H)). In all three groups of ApoE−/− mouse lesions, infused monocyte/macrophages appeared to have infiltrated endothelium to take up residence in deep regions of the lesion, together with pre-existing macrophages, as indicated by colocalization of QD655-CD11b+ cells with AF488-CD68, a monocyte/macrophage-specific marker (figures 3(C), (F), (H)). The extent of accumulation of both resident and reinfused cells in the lesions of younger animals was clearly lower than the accumulation in 1 year old ApoE−/− mouse lesions. Our observations confirmed that plaque monocyte/macrophage accumulation was proportional to the extent of lesion progression as determined by lesion size and ORO staining. QD655-CD11b+ cells were observed to accumulate within the same regions in plaques as CAM-labeled CD11b+ cells as indicated by colocalization of the two cell populations, indicating that QD labeling procedure did not affect leukocyte homing capabilities (figure 3(I)). Additionally, a solution of QD–maurocalcine systemically injected at the same volume and concentration used for cell loading did not accumulate within plaque regions which accumulated CAM-labeled CD11b+ monocyte/macrophages (figure 3(J)). QD signal was readily distinguishable from autofluorescence of tissue sections (figure 3(L)).
Figure 3.
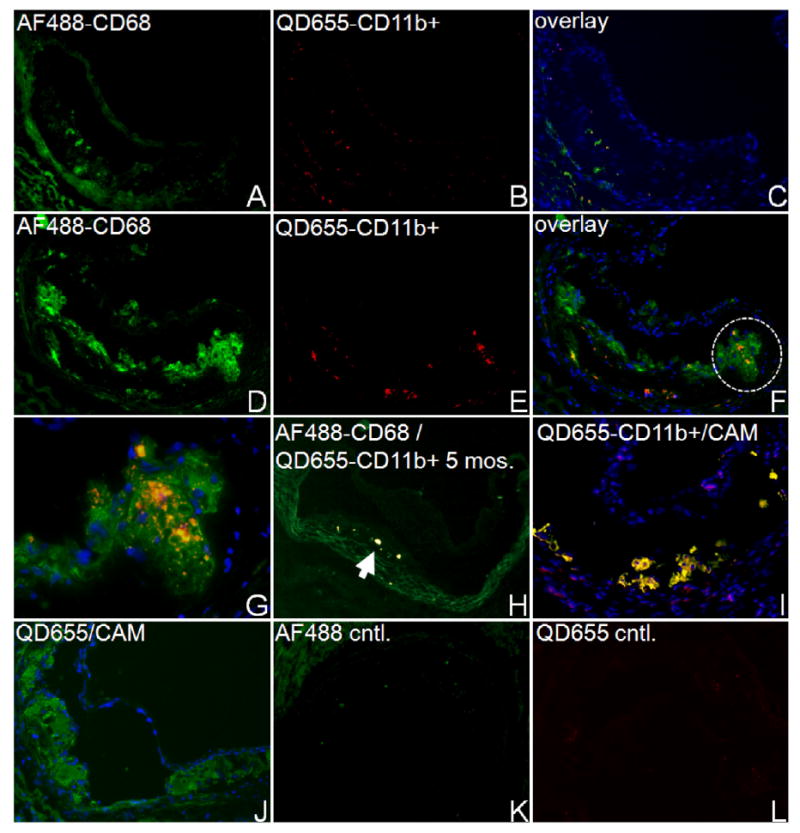
Representative fluorescence micrographs of aortic root tissue lesions of ApoE−/− mice, 5 days post-injection of 1 × 106 QD655-CD11b+ cells. Nuclei were stained with DAPI (blue). (A)–(F) Comparison of QD-labeled monocyte/macrophage accumulation in lesions of 7 (A)–(C) and 12 month old (D)–(F) ApoE−/− mice (magnification 50×). Resident and reinfused monocyte/macrophages were labeled with Alexa Fluor 488 (AF488)-labeled CD68 (A, D). (B, E) Visualization of QD655-CD11b+ cells in the same lesion. (C, F) Areas of colocalization between QD655-CD11b+ cells (red) and AF488-anti-CD68 (green) appear orange-yellow. (G) Magnified region circled in (F) (200×) indicating colocalization of infused QD655-CD11b+ cells with AF488-CD68+. (H) Lesions in 5 month old ApoE−/− mice show minimal accumulation of QD655-CD11b+ cells and colocalization with AF488-CD68+ regions (arrowheads) in sections. (I) QD655-CD11b+ monocyte/macrophages colocalized with CAM-labeled monocyte/macrophages within 12 month old ApoE−/− mouse lesions (colocalization in yellow), whereas QD–maurocalcine injected as a solution was not detected in regions of CAM-labeled cell accumulation (J). (K) and (L) Micrographs of unstained lesions (No QD, CAM, AF488) show minimal autofluorescence in relevant emission channels under identical image acquisition settings (magnification 50×).
QD-labeled T lymphocytes (QD585-TCR+) cells were observed in aortic lesions of ApoE−/− mice as early as two days and as late as four weeks after injection. QD585-labeled cells were detected within lesions as small clusters (yellow) colocalizing with AF488-labeled anti-TCR antibody (AF488-TCR, figure 4(A)), confirming the phenotype of the infiltrate. QD585-TCR+ were detected in CD68-rich areas (yellow) or areas of low or absent CD68 expression (red, figure 4(B)). As with QD-labeled monocyte/macrophages, the high quantum efficiency of QD enabled high signal-to-background ratio imaging of T lymphocytes in lesion areas (figure 4(C)).
Figure 4.
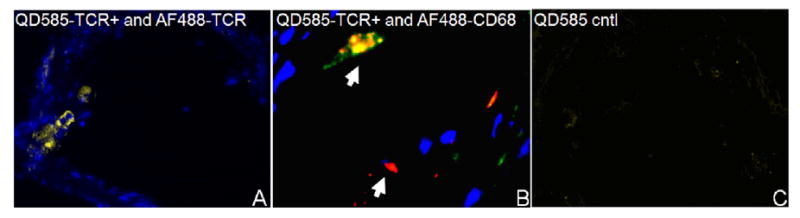
Representative fluorescence micrographs of QD585-TCR+ T lymphocyte recruitment to lesions. (A) Injected QD585-TCR+ cells colocalized with AF488-anti-TCR (yellow) within lesions of a 5 month old ApoE−/− mouse lesion. (B) QD585-TCR+ lymphocytes within a lesion of a 12 month old ApoE−/− mouse aorta. Arrowheads indicate lymphocytes in regions colocalized with AF488-anti-CD68 (yellow), or in distinct areas (red). (C) Lesions from 12 month old ApoE−/− mice not injected with QD585-TCR+ lymphocytes show minimal autofluorescence under the same image acquisition settings. (Magnification 50×.)
The circulation time allotted for QD-labeled cells to home to lesions generally did not affect relative levels of leukocyte accumulation in lesions, as 4 week post-injection time-points exhibited similar accumulation of leukocytes as 2–5 day post-injection time-points (data not shown). Generally, QD were not observed to fade even for up to a month following sample preparation, nor did extended circulation time of QD-labeled cells adversely affect photostability and photointensity of QD within lesions.
3.4. Correlation of leukocyte accumulation with ORO staining by en face analysis
Macroscopic fluorescence imaging of en face preparations of ApoE−/− (5, 12 months) and wild-type (12 months) mouse aortas was indicative of QD585-TCR+ and QD655-CD11b+ cell accumulation in ORO-stained lipid regions throughout the aortic stem (figures 5(A)–(C)). No cells were observed in wild-type aortas (WT), indicating that QD-labeled cells homed specifically to lesion areas. The fluorescence spectra of ORO did not overlap with that of QD655 and QD585, due to the use of distinct excitation and emission filters and the serial nature of sample preparation, whereby QD images were acquired prior to ORO staining to allow for digital image background correction. Quantitative image analysis indicated that QD and ORO signal intensity in aortas was proportional to mouse atherosclerotic lesion burden, as 5 month old ApoE−/−mice had substantially lower intensities that the 12 month old ApoE−/− mice injected and stained under the same conditions (figure 5(D)). QD labeling afforded as high as 90% signal intensity enhancement of lesion area over wild-type aortic background. Thus, macroscopic fluorescence imaging in conjunction with QD-labeled cells and ORO can be used to quantify both cell and lipid content of lesions.
Figure 5.
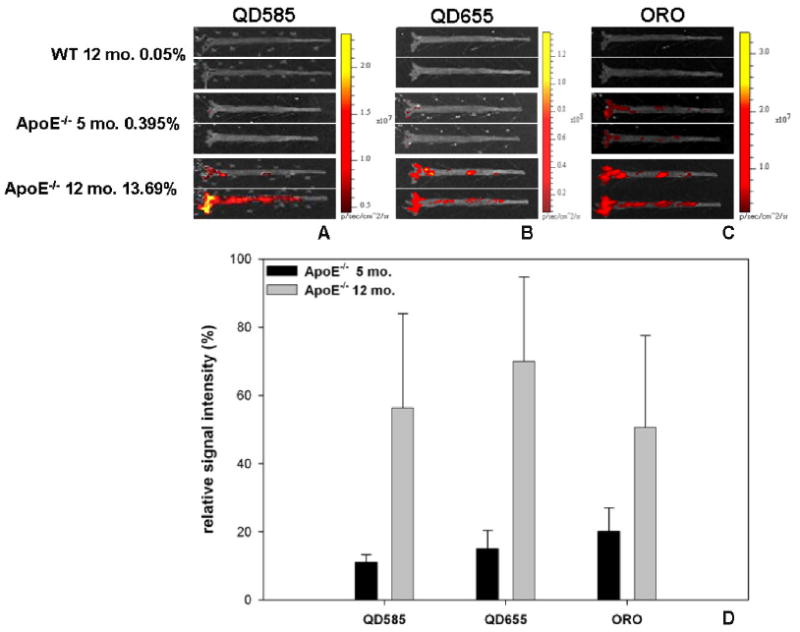
Ex vivo imaging of en face aorta preparations using macroscopic fluorescence imaging of QD585-TCR+ cells (A), QD655-CD11b+ cells (B), and oil red O (ORO) regions (C) in wild-type (12 months) and ApoE−/− (5 and 12 months) mice. ORO staining was conducted following QD imaging of tissue in order to optimize background subtraction procedures. %ORO+ lesion area is indicated next to each animal group (mean of n = 2). (D) % signal intensity enhancement in each of the three fluorescence channels (QD585, QD655, ORO) of 5 month old and 12 month old lesions regions relative to matched regions on wild-type aortas (n = 6 regions, reported as mean ± S.D.).
4. Discussion
QD are versatile reagents for imaging disease in preclinical models, due to the flexibility of ligands (nucleic acids, antibodies, peptides) that can be accommodated, high photostability and emission intensity, size-tunable emission wavelengths, and imaging of multiple QD within tissue with only one excitation wavelength [18]. QD have been loaded into (cytoplasm) or onto (cell surface) multiple cell types for imaging of dynamic processes in vivo, including tracking of cancer metastasis [63] and leukocyte-endothelial interactions during inflammatory vascular disease [29]. The multispectral imaging capabilities exhibited by QD make them attractive reagents for visualizing dynamic processes consisting of multiple cellular and biomolecular mediators in vivo.
In this work, we have adapted QD for monitoring atherogenesis in the ApoE−/− mouse model, with particular attention given to the potential utility of this approach for imaging of leukocyte subset accumulation within the atheroma. The latter is of particular interest since leukocytes which home to lesions, namely monocytes, which differentiate into macrophages within the lesion and contribute to plaque rupture, can also be genetically engineered to induce regression of lesions [58]. Therefore, for both reasons, a technique to monitor their biodistribution is particularly desirable. Flow cytometric analysis indicates that QD–maurocalcine bioconjugates were internalized efficiently within leukocyte subtypes (figures 1(C) and (D)), with no significant effects on cell viability (figure 2(A)), homing to inflammatory endothelium (figure 2(B)), or cytokine release observed. These in vitro studies collectively indicate that QD are suitable for studies of leukocyte function in vascular disease.
Although further cytotoxicity studies are required to ensure that QD do not perturb leukocyte function for long-term studies of disease, we monitored essential leukocyte subset functions to verify that basic cell functions, such as response to stimuli and endothelial adhesion capacity, were not apparently altered by QD–maurocalcine labeling. Several in vivo studies have incorporated QD for molecular imaging applications without acute toxicity over several months [5, 42]. It is important to note that QD used in this study are composed of cadmium which has been known to cause toxicity in multiple organs. The composition of QD may preclude clinical in vivo applications without the synthesis of QD with alternative cores, or extensive surface modifications to minimize adverse effects on tissues [23, 36, 51, 61]. To this end, renal filtration and urinary excretion of QD with small hydrodynamic diameters has been reported [10], making in vivo application a future possibility. Potential applications for QD in vivo might include their conventional use as targeted optical imaging agents, or alternative purposes such as photodynamic therapy [30, 43].
Homing of both cell types to lesions was observed (figures 3–5), with varying degrees of penetration and colocalization with resident macrophages. Therefore, QD may be a useful tool to study differential localization on a spatial or temporal scale for several cell types within the same tissue. The multiplexing capability afforded by the use of spectrally distinct QD makes the simultaneous detection of multiple cell types a possibility, even in combination with the use of conventional immunofluorescence markers, in this case Alexa Fluor 488 anti-CD68 and the nuclear stain DAPI, without spectral bleed-through. Although organic dyes were used in conjunction with QD-labeled cells in this application, QD-antibody conjugates could be substituted in order to multiplex immunofluorescence analysis as well to increase the breadth of information available in lesion analysis.
Macroscopic imaging of QD within en face aorta preparations exhibited both the specificity of QD-labeled leukocytes toward lesions (figures 5(A)–(C)), as well as the high signal-to-noise ratio of QD in tissue (figure 5(D)). Macroscale fluorescence imaging interrogates tissue-level cellular recruitment using exposure times on the scale of seconds, whereas our microscopy analysis used millisecond acquisition times to image tissue in thin (5 μm) sections with lower amounts of fluorescence signal. Macroscale imaging was advantageous for quantitative purposes, as for observing QD-labeled cellular recruitment to lesions in 5 month old mice, microscopic imaging of thin lesion-bearing aortic sections exhibited very few accumulated QD-labeled monocyte/macrophages (figure 3(H)), whereas macroscopic imaging of whole aortas enabled detection of recruited cells to lesions in 5 month old mice due to the increased interrogation volume of whole tissue and longer exposure intervals (figures 5(A) and (B)).
We selected QD configured for emission above green wavelengths for this study, due to the substantial amount of tissue autofluorescence within lesions in this range of the spectrum. The selection of yellow–red wavelengths provide for high signal-to-noise ratio imaging of atherosclerotic lesions without autofluorescence. QD are optimally excited using ultraviolet to blue excitation wavelengths. Since ORO fluorescence is optimally excited at ∼540–580 nm, with a peak emission near 615 nm (figure 5(C)), macroscopic fluorescence imaging of ORO, QD585 (TCR+), and QD655 (CD11b+) was simultaneously performed in the same tissue, since the QD selected do not emit appreciable fluorescence when excited at these wavelengths. Image background correction and compensation for QD fluorescence prior to ORO imaging further minimized spectral overlap. Therefore, QD emission wavelengths can be selected for optimal multispecies imaging, and can also be combined with conventional lesion analysis tools which use fluorescent dyes or immunohistochemical stains. Our initial studies demonstrated the relative signal enhancement of lesions within 12 month old ApoE−/− mice compared to those of 5 month old ApoE−/− and wild-type mice. Therefore, QD labeling and reinfusion of CD11b+ and TCR+ leukocyte subsets can be used effectively to determine relative lesion burden, for the purpose of elucidating cellular mediators of lesion biology, assessing therapies, and staging vascular disease. However, our technique is not limited to the specific cell types studied. Another example of using QD for multiplexed studies of cellular recruitment in atherosclerosis would be to characterize the homing of natural killer, polymorphonuclear, and dendritic cells to plaque regions throughout the lifespan of the ApoE−/− mouse model, for the purpose of monitoring dynamic changes within the same animal while eliminating animal to animal variances.
The peptide maurocalcine, shown in other reports to translocate imaging or therapy cargoes across the cytoplasm of mammalian cells with minimal cytotoxicity [4, 15, 39], could also be used to label these leukocyte subsets with QD as we have demonstrated for monocyte/macrophages and T lymphocytes, provided no adverse effects on cell functions critical to their participation in atherosclerosis are observed. Peptides have been successfully applied to the study of cancer due to their molecular specificity for cancer-specific or cancer-upregulated biomarkers [2]. Targeting peptides also have great potential in improving our understanding of atherosclerosis. For example, VCAM-1, a cell adhesion molecule upregulated on endothelial cells lining plaques, was successfully targeted by nanoparticulate contrast agents incorporating phage display-derived targeting peptides on their surface [31]. Therefore, peptides may be not only incorporated to internally label cells as in this study, but may also be utilized to detect diagnostically and therapeutically relevant biomarkers in vascular disease.
We have demonstrated the feasibility of adapting QD bioconjugates as useful study tools for the multiplexed analysis of atherosclerosis. Our results are comparable to studies of lesion biology which incorporate other contrast agents. QD-labeled monocyte/macrophage accumulation in ApoE−/−aortas measured ex vivo afforded as high as a 90% increase in signal intensity relative to the same region of interest in wild-type aortas (figure 5(D)). This signal intensity enhancement is comparable to that observed with Gadolinium micelle-enhanced ex vivo MRI of 1 yr old ApoE−/− aortas, in which the mean increase was 81% [37]. QD can be designed to emit in the near-infrared emission windows for optimal imaging of tissues without the scattering and absorption limitations imposed by imaging of shorter wavelengths [20]. Although we incorporated visible light wavelength emitting QD in our studies, improved signal-to-background ratios would be expected by substituting QD–maurocalcine emitting at near-infrared to infrared wavelengths such as 705–850 nm, such as those used for in vivo imaging applications [55]. Furthermore, although the UV-blue excitation light required to optimally excite QD would only have limited penetration in thick tissues due to scattering and absorption phenomena, two-photon excitation instruments may help in addressing these challenges [33] for improved resolution. Image processing techniques such as image deconvolution may also improve the quality of information obtained from QD-mediated imaging in terms of spatial localization of cells and/or biomarkers in lesions. The prospect of using QD-antibody and QD-peptide bioconjugates for labeling cell surface biomarkers or the cytoplasm of specific subtypes of cells, high-resolution imaging instrumentation and image processing methods, may facilitate efforts toward the molecular profiling of atherosclerosis for developing diagnostic assays and disease-customized therapeutic interventions for clinical usage.
5. Conclusion
We have developed a technique for monitoring the recruitment of leukocyte subsets into atherosclerotic lesions. The optical properties of QD can be harnessed to permit high signal-to-noise ratio imaging of multiple cell types without spectral overlap, and can also be combined with conventional immunofluorescence and immunohistochemical reagents used in the study of atherosclerosis. Given the many cell types with a role in the initiation and progression of atheroma, QD can be utilized to study different cells within the same specimen in order to understand the temporal, spatial, and functional inter-relations of atherosclerotic mediators. Future efforts with this technique will include molecular profiling of atheroma in vivo including the monitoring of inflammatory biomarker expression in early and late stages of disease, and the assessment of therapeutic response via the tracking of genetically engineered monocytes infused systemically in order to promote lesion regression in vivo.
Acknowledgments
This work was supported in part by National Institutes of Health grants EY017522 (FRH) and HL65709 (SF). Dr Jayagopal was supported by a training grant from the Vanderbilt Vision Research Center (T32EY07135) and the Vanderbilt University Discovery Grant program. Dr Su was supported by the Diabetes Research and Training Center pilot and feasibility grant at Vanderbilt University (P60DK20593). We thank Elizabeth Dworska and Youmin Zhang for technical assistance, and the Vanderbilt University Institute of Imaging Science (VUIIS) for access to the Caliper IVIS 200 fluorescence imaging system.
Abbreviations
- QD
quantum dots
- ORO
oil Red O
- IFN-γ
interferon gamma
- LPS
lipopolysaccharide
- TCR
T cell receptor
- IL-2
interleukin 2
- MCP-1
monocyte chemoattractant protein 1
References
- 1.Ajuebor MN, Hogaboam CM, Le T, Swain MG. C–C chemokine ligand 2/monocyte chemoattractant protein-1 directly inhibits NKT cell IL-4 production and is hepatoprotective in T cell-mediated hepatitis in the mouse. J Immunol. 2003;170:5252–9. doi: 10.4049/jimmunol.170.10.5252. [DOI] [PubMed] [Google Scholar]
- 2.Akerman ME, Chan WCW, Laakkonen P, Bhatia SN, Ruoslahti E. Nanocrystal targeting in vivo. Proc Natl Acad Sci USA. 2002;99:12617–21. doi: 10.1073/pnas.152463399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amirbekian V, et al. Detecting and assessing macrophages in vivo to evaluate atherosclerosis noninvasively using molecular MRI. Proc Natl Acad Sci USA. 2007;104:961–6. doi: 10.1073/pnas.0606281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aroui S, Ram N, Appaix F, Ronjat M, Kenani A, Pirollet F, De Waard M. Maurocalcine as a non toxic drug carrier overcomes doxorubicin resistance in the cancer cell line MDA-MB 231. Pharm Res. 2008;26:836–45. doi: 10.1007/s11095-008-9782-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Noninvasive imaging of quantum dots in mice. Bioconjug Chem. 2004;15:79–86. doi: 10.1021/bc034153y. [DOI] [PubMed] [Google Scholar]
- 6.Biju V, Itoh T, Anas A, Sujith A, Ishikawa M. Semiconductor quantum dots and metal nanoparticles: syntheses, optical properties, and biological applications. Anal Bioanal Chem. 2008;391:2469–95. doi: 10.1007/s00216-008-2185-7. [DOI] [PubMed] [Google Scholar]
- 7.Bobryshev YV. Dendritic cells in atherosclerosis: current status of the problem and clinical relevance. Eur Heart J. 2005;26:1700–4. doi: 10.1093/eurheartj/ehi282. [DOI] [PubMed] [Google Scholar]
- 8.Bobryshev YV, Lord RS. Co-accumulation of dendritic cells and natural killer T cells within rupture-prone regions in human atherosclerotic plaques. J Histochem Cytochem. 2005;53:781–5. doi: 10.1369/jhc.4B6570.2005. [DOI] [PubMed] [Google Scholar]
- 9.Boisseau S, et al. Cell penetration properties of maurocalcine, a natural venom peptide active on the intracellular ryanodine receptor. Biochim Biophys Acta. 2006;1758:308–19. doi: 10.1016/j.bbamem.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV. Renal clearance of quantum dots. Nat Biotechnol. 2007;25:1165–70. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–62. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cybulsky MI, Lichtman AH, Hajra L, Iiyama K. Leukocyte adhesion molecules in atherogenesis. Clin Chim Acta. 1999;286:207–18. doi: 10.1016/s0009-8981(99)00102-3. [DOI] [PubMed] [Google Scholar]
- 13.Davies MJ, Richardson PD, Woolf N, Katz DR, Mann J. Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. Br Heart J. 1993;69:377–81. doi: 10.1136/hrt.69.5.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eriksson EE, Xie X, Werr J, Thoren P, Lindbom L. Direct viewing of atherosclerosis in vivo plaque invasion by leukocytes is initiated by the endothelial selectins. FASEB J. 2001;15:1149–57. doi: 10.1096/fj.00-0537com. [DOI] [PubMed] [Google Scholar]
- 15.Esteve E, et al. Transduction of the scorpion toxin maurocalcine into cells. Evidence that the toxin crosses the plasma membrane. J Biol Chem. 2005;280:12833–9. doi: 10.1074/jbc.M412521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV., Jr Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest. 2001;107:163–71. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao X, Dave SR. Quantum dots for cancer molecular imaging. Adv Exp Med Biol. 2007;620:57–73. doi: 10.1007/978-0-387-76713-0_5. [DOI] [PubMed] [Google Scholar]
- 18.Gao X, Yang L, Petros JA, Marshall FF, Simons JW, Nie S. In vivo molecular and cellular imaging with quantum dots. Curr Opin Biotechnol. 2005;16:63–72. doi: 10.1016/j.copbio.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Gao XH, Cui YY, Levenson RM, Chung LWK, Nie SM. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol. 2004;22:969–76. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 20.Graves EE, Weissleder R, Ntziachristos V. Fluorescence molecular imaging of small animal tumor models. Curr Mol Med. 2004;4:419–30. doi: 10.2174/1566524043360555. [DOI] [PubMed] [Google Scholar]
- 21.Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99:2752–61. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansson GK, Holm J, Jonasson L. Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol. 1989;135:169–75. [PMC free article] [PubMed] [Google Scholar]
- 23.Hoshino A, Manabe N, Fujioka K, Suzuki K, Yasuhara M, Yamamoto K. Use of fluorescent quantum dot bioconjugates for cellular imaging of immune cells, cell organelle labeling, and nanomedicine: surface modification regulates biological function, including cytotoxicity. J Artif Organs. 2007;10:149–57. doi: 10.1007/s10047-007-0379-y. [DOI] [PubMed] [Google Scholar]
- 24.Iga AM, Robertson JH, Winslet MC, Seifalian AM. Clinical potential of quantum dots. J Biomed Biotechnol. 2007;2007:76087. doi: 10.1155/2007/76087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishibashi M, et al. Critical role of monocyte chemoattractant protein-1 receptor CCR2 on monocytes in hypertension-induced vascular inflammation and remodeling. Circ Res. 2004;94:1203–10. doi: 10.1161/01.RES.0000126924.23467.A3. [DOI] [PubMed] [Google Scholar]
- 26.Jaffer FA, Libby P, Weissleder R. Molecular and cellular imaging of atherosclerosis: emerging applications. J Am Coll Cardiol. 2006;47:1328–38. doi: 10.1016/j.jacc.2006.01.029. [DOI] [PubMed] [Google Scholar]
- 27.Jaffer FA, Weissleder R. Seeing within: molecular imaging of the cardiovascular system. Circ Res. 2004;94:433–45. doi: 10.1161/01.RES.0000119321.18573.5A. [DOI] [PubMed] [Google Scholar]
- 28.Jain KK. Applications of nanobiotechnology in clinical diagnostics. Clin Chem. 2007;53:2002–9. doi: 10.1373/clinchem.2007.090795. [DOI] [PubMed] [Google Scholar]
- 29.Jayagopal A, Russ PK, Haselton FR. Surface engineering of quantum dots for in vivo vascular imaging. Bioconjug Chem. 2007;18:1424–33. doi: 10.1021/bc070020r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juzenas P, Chen W, Sun YP, Coelho MA, Generalov R, Generalova N, Christensen IL. Quantum dots and nanoparticles for photodynamic and radiation therapies of cancer. Adv Drug Deliv Rev. 2008;60:1600–14. doi: 10.1016/j.addr.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelly KA, Allport JR, Tsourkas A, Shinde-Patil VR, Josephson L, Weissleder R. Detection of vascular adhesion molecule-1 expression using a novel multimodal nanoparticle. Circ Res. 2005;96:327–36. doi: 10.1161/01.RES.0000155722.17881.dd. [DOI] [PubMed] [Google Scholar]
- 32.Kruth HS, Huang W, Ishii I, Zhang WY. Macrophage foam cell formation with native low density lipoprotein. J Biol Chem. 2002;277:34573–80. doi: 10.1074/jbc.M205059200. [DOI] [PubMed] [Google Scholar]
- 33.Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Water-soluble quantum dots for multiphoton fluorescence imaging in vivo Science. 2003;300:1434–6. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- 34.Libby P, Geng YJ, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GK, Lee RT. Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol. 1996;7:330–5. doi: 10.1097/00041433-199610000-00012. [DOI] [PubMed] [Google Scholar]
- 35.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 36.Lin P, et al. Computational and ultrastructural toxicology of a nanoparticle, Quantum Dot 705, in mice. Environ Sci Technol. 2008;42:6264–70. doi: 10.1021/es800254a. [DOI] [PubMed] [Google Scholar]
- 37.Lipinski MJ, Amirbekian V, Frias JC, Aguinaldo JG, Mani V, Briley-Saebo KC, Fuster V, Fallon JT, Fisher EA, Fayad ZA. MRI to detect atherosclerosis with gadolinium-containing immunomicelles targeting the macrophage scavenger receptor. Magn Reson Med. 2006;56:601–10. doi: 10.1002/mrm.20995. [DOI] [PubMed] [Google Scholar]
- 38.Lukacs B, et al. Charged surface area of maurocalcine determines its interaction with the skeletal ryanodine receptor. Biophys J. 2008;95:3497–509. doi: 10.1529/biophysj.107.120840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mabrouk K, Ram N, Boisseau S, Strappazzon F, Rehaim A, Sadoul R, Darbon H, Ronjat M, De Waard M. Critical amino acid residues of maurocalcine involved in pharmacology, lipid interaction and cell penetration. Biochim Biophys Acta. 2007;1768:2528–40. doi: 10.1016/j.bbamem.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 40.Mach F, Schonbeck U, Fabunmi RP, Murphy C, Atkinson E, Bonnefoy JY, Graber P, Libby P. T lymphocytes induce endothelial cell matrix metalloproteinase expression by a CD40L-dependent mechanism: implications for tubule formation. Am J Pathol. 1999;154:229–38. doi: 10.1016/S0002-9440(10)65269-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McHale JF, Harari OA, Marshall D, Haskard DO. Vascular endothelial cell expression of ICAM-1 and VCAM-1 at the onset of eliciting contact hypersensitivity in mice: evidence for a dominant role of TNF-alpha. J Immunol. 1999;162:1648–55. [PubMed] [Google Scholar]
- 42.Medintz IL, Mattoussi H, Clapp AR. Potential clinical applications of quantum dots. Int J Nanomed. 2008;3:151–67. doi: 10.2147/ijn.s614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan NY, English S, Chen W, Chernomordik V, Russo A, Smith PD, Gandjbakhche A. Real time in vivo non-invasive optical imaging using near-infrared fluorescent quantum dots. Acad Radiol. 2005;12:313–23. doi: 10.1016/j.acra.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 44.Nahrendorf M, Jaffer FA, Kelly KA, Sosnovik DE, Aikawa E, Libby P, Weissleder R. Noninvasive vascular cell adhesion molecule-1 imaging identifies inflammatory activation of cells in atherosclerosis. Circulation. 2006;114:1504–11. doi: 10.1161/CIRCULATIONAHA.106.646380. [DOI] [PubMed] [Google Scholar]
- 45.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–40. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 46.Nishiwaki H, Ogura Y, Kimura H, Kiryu J, Honda Y. Quantitative evaluation of leukocyte dynamics in retinal microcirculation. Invest Ophthalmol Vis Sci. 1995;36:123–30. [PubMed] [Google Scholar]
- 47.O'Malley JP, Waran MT, Balice-Gordon RJ. In vivo observations of terminal Schwann cells at normal, denervated, and reinnervated mouse neuromuscular junctions. J Neurobiol. 1999;38:270–86. [PubMed] [Google Scholar]
- 48.Patel SS, Thiagarajan R, Willerson JT, Yeh ET. Inhibition of alpha4 integrin and ICAM-1 markedly attenuate macrophage homing to atherosclerotic plaques in ApoE-deficient mice. Circulation. 1998;97:75–81. doi: 10.1161/01.cir.97.1.75. [DOI] [PubMed] [Google Scholar]
- 49.Paulsson J, Dadfar E, Held C, Jacobson SH, Lundahl J. Activation of peripheral and in vivo transmigrated neutrophils in patients with stable coronary artery disease. Atherosclerosis. 2007;192:328–34. doi: 10.1016/j.atherosclerosis.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 50.Rudd JH, et al. Imaging atherosclerotic plaque inflammation with [18F]-fluorodeoxyglucose positron emission tomography. Circulation. 2002;105:2708–11. doi: 10.1161/01.cir.0000020548.60110.76. [DOI] [PubMed] [Google Scholar]
- 51.Ryman-Rasmussen JP, Riviere JE, Monteiro-Riviere NA. Surface coatings determine cytotoxicity and irritation potential of quantum dot nanoparticles in epidermal keratinocytes. J Invest Dermatol. 2007;127:143–53. doi: 10.1038/sj.jid.5700508. [DOI] [PubMed] [Google Scholar]
- 52.Schonbeck U, Mach F, Sukhova GK, Atkinson E, Levesque E, Herman M, Graber P, Basset P, Libby P. Expression of stromelysin-3 in atherosclerotic lesions: regulation via CD40-CD40 ligand signaling in vitro and in vivo. J Exp Med. 1999;189:843–53. doi: 10.1084/jem.189.5.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schreurs MW, Eggert AA, de Boer AJ, Figdor CG, Adema GJ. Generation and functional characterization of mouse monocyte-derived dendritic cells. Eur J Immunol. 1999;29:2835–41. doi: 10.1002/(SICI)1521-4141(199909)29:09<2835::AID-IMMU2835>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 54.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–9. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 55.Smith AM, Duan H, Mohs AM, Nie S. Bioconjugated quantum dots for in vivo molecular and cellular imaging. Adv Drug Deliv Rev. 2008;60:1226–40. doi: 10.1016/j.addr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song L, Leung C, Schindler C. Lymphocytes are important in early atherosclerosis. J Clin Invest. 2001;108:251–9. doi: 10.1172/JCI11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su YR, Dove DE, Major AS, Hasty AH, Boone B, Linton MF, Fazio S. Reduced ABCA1-mediated cholesterol efflux and accelerated atherosclerosis in apolipoprotein E-deficient mice lacking macrophage-derived ACAT1. Circulation. 2005;111:2373–81. doi: 10.1161/01.CIR.0000164236.19860.13. [DOI] [PubMed] [Google Scholar]
- 58.Su YR, Ishiguro H, Major AS, Dove DE, Zhang W, Hasty AH, Babaev VR, Linton MF, Fazio S. Macrophage apolipoprotein A-I expression protects against atherosclerosis in ApoE-deficient mice and up-regulates ABC transporters. Mol Ther. 2003;8:576–83. doi: 10.1016/s1525-0016(03)00214-4. [DOI] [PubMed] [Google Scholar]
- 59.Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci USA. 2006;103:10340–5. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taatjes DJ, Wadsworth MP, Schneider DJ, Sobel BE. Histochem Cell Biol. Vol. 113. 2000. Improved quantitative characterization of atherosclerotic plaque composition with immunohistochemistry, confocal fluorescence microscopy, and computer-assisted image analysis; pp. 161–73. [DOI] [PubMed] [Google Scholar]
- 61.Tang M, Wang M, Xing T, Zeng J, Wang H, Ruan DY. Mechanisms of unmodified CdSe quantum dot-induced elevation of cytoplasmic calcium levels in primary cultures of rat hippocampal neurons. Biomaterials. 2008;29:4383–91. doi: 10.1016/j.biomaterials.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 62.Tsourkas A, Shinde-Patil VR, Kelly KA, Patel P, Wolley A, Allport JR, Weissleder R. In vivo imaging of activated endothelium using an anti-VCAM-1 magnetooptical probe. Bioconjug Chem. 2005;16:576–81. doi: 10.1021/bc050002e. [DOI] [PubMed] [Google Scholar]
- 63.Voura EB, Jaiswal JK, Mattoussi H, Simon SM. Tracking metastatic tumor cell extravasation with quantum dot nanocrystals and fluorescence emission-scanning microscopy. Nat Med. 2004;10:993–8. doi: 10.1038/nm1096. [DOI] [PubMed] [Google Scholar]
- 64.Wadsworth MP, Sobel BE, Schneider DJ, Tra W, van Hirtum H, Taatjes DJ. Quantitative analysis of atherosclerotic lesion composition in mice. Methods Mol Biol. 2006;319:137–52. doi: 10.1007/978-1-59259-993-6_6. [DOI] [PubMed] [Google Scholar]
- 65.Yokota T, Hansson GK. Immunological mechanisms in atherosclerosis. J Int Med. 1995;238:479–89. doi: 10.1111/j.1365-2796.1995.tb01229.x. [DOI] [PubMed] [Google Scholar]