

Abstract
Introduction
Beta-adrenergic blockade ameliorates the hypermetabolism and catabolism in severe burn injury. Despite the salutary effects of beta-adrenergic blockade, the immunological responses that accompany beta blockade are not known. We have shown that burn sepsis is associated with increased sympathetic activation leading to altered monocytopoiesis and cytokine release in MØ. Recent evidence suggests that murine MØ expressing F4/80+Gr1+ are the inflammatory phenotype. Here we report that propranolol given after burn sepsis modulates the number and function of myeloid cells in circulation.
Method
B6D2F1 male mice were divided into sham (S), burn (B) and burn sepsis (BS) groups. Dorsal hair was shaved from S, B & BS; B & BS received 15% scald burn; BS was inoculated with Pseudomonas aeruginosa (PA 14, 4–5K CFU) at the burn site. Mice from each group were then subjected to two different treatment regimens. One set received subcutaneous injections of propranolol (5mg/Kg body wt.) at 24h and 48h after the injury while the control groups received saline. Blood was collected by cardiac puncture at 72h. The distribution of total F4/80+ monocyte population was determined by flow cytometry. Inflammatory monocyte subset was gated on Gr-1+ expression in the F4/80+ fraction. LPS stimulated intracellular TNF-α (ic-TNF) was also measured as an indicator of inflammatory response.
Results
The total F4/80+ monocyte fraction was significantly increased in BS (45±0.8%) vs. S and B (10±0.8%; 9.5±0.6%). Propranolol treatment for 2 days reduced the number of circulating monocytes by 60% in BS. The mean fluorescent intensity (MFI) of ic-TNF produced per cell (F4/80+Gr1+ MØ) was significantly decreased in B and BS (S: 3043±213, B: 1638±343, BS: 1463±67). Of importance, propranolol treatment partially restored the MFI of ic-TNF (2177±114) and increased the percentage of inflammatory monocyte subset (F4/80+Gr1+) in BS by 70% compared to saline treatment. In contrast, beta-blockade after BS increased the percentage of granulocytes in circulation (28.4±3.6% in BS propranolol vs. 15.4±0.3% in BS Saline; p<0.05) and augmented their TNF production (MFI = 903±102 in BS propranolol vs. 644±5 in BS Saline; p<0.05).
Conclusion
Propranolol reverses burn sepsis-induced monocytosis and simultaneously increases the number of granulocytes and enhances the inflammatory potential of the granulocytes and inflammatory monocyte subsets in circulation suggesting that monitoring MØ subsets and granulocytes in blood is a reliable biomarker to predict the efficacy of beta blockade.
Keywords: Peripheral blood, monocytes, F4/80/Gr1, granulocytes, burn-sepsis, propranolol, beta-blockade, TNF
INTRODUCTION
Macrophages (MØ) are versatile cells that are essential in orchestrating immune response through multiple functions. They act as effectors for both innate and adaptive immune responses. Aberrant cytokine production by activated macrophages is considered central to the pathophysiology of trauma and sepsis1. To date, much research has focused on activation of tissue macrophages and peripheral blood monocytes under injury conditions and how they contribute to the dysregulated cytokine and growth factor production1–4. More recently, the concept has emerged that subsets of monocytes and MØ exist in the periphery and in tissues respectively, based on their ability to produce cytokines and growth factors in response to specific stimuli1–5. The heterogeneity of such subsets could explain in part why there is often great variation in patient cytokine values following critical injury.
Critical trauma is associated with sympathetic activation both in humans6–11 and animal models leading to the dynamic release of catecholamines12–16. Increased efferent nerve-stimulated release of norepinephrine within the bone marrow compartment and a simultaneous increase in monocytopoiesis based on the clonogenic potential of bone marrow progenitors is demonstrated during experimental sepsis12,13,16.
When norepinephrine is depleted prior to thermal injury and sepsis, monocytopoiesis is significantly attenuated12. Nevertheless, beta-adrenergic blockade has been shown to ameliorate burn-induced hyper metabolism and catabolism17. Our reports demonstrate that thermal injury and sepsis alters the affinity and density of β2-adrenergic receptors expressed on late monocyte progenitors18 and that bone marrow progenitor derived MØ obtained from septic animals possesses differential cytokine response compared to sham animals19 suggesting the importance of adrenergic regulation in sepsis.
Circulating blood monocytes and tissue MØ are derived from the bone marrow through specific commitment of myeloid progenitors into monocytic lineage20–23. Recent reports from Geissmann’s group postulate two functionally distinctive subsets of monocytes based on their chemokine expression and migration properties24,25. In relevance to this classification we have recently documented that F4/80+ monocytes expressing Gr1+ antigen (inflammatory subset) produced the majority of LPS mediated TNF-α in comparison to F4/80+ Gr1− monocytes (resident subset). Moreover, critical injury such as burn sepsis diminished the cytokine production by monocytes compared to normal steady state26. It is not clear, if the altered bone marrow milieu in critical injury and sepsis drives the production of the reported monocyte subsets or if the subsets arise only in the circulation and tissues because of local environmental factors. While both scenarios are plausible, our prior study demonstrated a temporal relationship between circulating monocyte subsets and bone marrow monocyte progenitor derived MØ subsets in a murine model of thermal injury and sepsis26. Our results indicated an increase in total circulating monocytes at 72 hours after bun injury and sepsis. What drives monocytosis in burn injury and sepsis remains unclear. Based on the experimental evidence demonstrating burn sepsis-induced monocytosis26 and abrogation of bone marrow monocytopoiesis in sympathectomized animals13, we framed our hypothesis that blocking beta-adrenergic receptors following burn injury and sepsis should similarly reverse monocytosis.
Here we report that administering a non-selective beta-adrenergic blocker (propranolol) after burn injury and sepsis reduces the percentage of monocytes and augments that of granulocytes in circulation while improving the inflammatory potential of circulating inflammatory monocyte subset as well as TNF-α production by the granulocytes.
METHODS
Materials
Cytofix/Cytoperm Plus kit, rat anti mouse anti-F4/80, anti-Gr-1, and anti-TNF-α antibodies were purchased from BD biosciences, San Diego, CA. Easy-Lyse Whole Blood Erythrocyte Lysing Kit was bought from Leinco Technologies, Inc., St. Louis, MO. Propranolol was obtained from Sigma-Aldrich Inc., St. Louis, MO. ELISA kits for TNF-α and IL-6 were bought from BioSource, Camarillo, CA.
Animals
B6D2F1 male mice weighing 25 to 30 grams were purchased from Jackson Laboratories (Barr Harbor, ME). Prior to the start of the experiments, mice were allowed to acclimatize for seven days following arrival at our Comparative Medicine Facility under a controlled temperature (20–22° C) and humidity (20–40%) environment with a 12hour light – dark cycle. All experimental protocols were approved by the IACUC (Institutional Animal Care and Use Committee) of Loyola University Medical Center.
Bacteria
Pseudomonas aeruginosa (PA 14) a clinical isolate originally obtained from a burn patient that displays similar pathogenicity in a mouse model was used to induce burn wound sepsis27.
Burn Injury And Sepsis
A dorsal scald burn injury was induced in mice similar to the well established model of burn sepsis with modifications to the methods described by Walker and Mason in rats28. The animals were randomized into Sham (S), Burn (B) and Burn Sepsis (BS) groups. All mice were anesthetized using IP ketamine/xylazine (100mg/kg; 2.5mg/kg, respectively) and the dorsal hair removed. Animals in both B and BS group received a 15% total body surface area deep partial thickness scald burn on their back by immersion in a 100° C water bath for 7 seconds as previously published29. Animals in the BS group were inoculated with 4–5K Colony Forming Units (CFU) of Pseudomonas aeruginosa (PA 14) at the burn wound site. All animals were resuscitated with 2ml IP injection of normal saline. Approximately 25–30% mortality was noticed in the BS group between 48 and 72h. Mice were euthanized by gaseous CO2 overdose at 72 hours following injury. Blood was collected by cardiac puncture in heparin-coated syringes. Spleens were removed from BS groups.
In vivo beta-adrenergic blockade: Mice from each S, B and BS group as mentioned in the thermal injury and sepsis model were subjected to two different treatment regimens. One set of animals was administered pan beta-blocker propranolol (5mg/Kg body weight) subcutaneously30–36 at 24h and 48h while the other set of animals were given saline injection as control.
Survival studies
Fifteen animals each from the BS group with and without propranolol treatments given at 24 and 48 hours after injury were followed up to 10 days for survival. Animals were checked daily for any visible changes in weight.
Systemic dissemination
Spleens from BS group were harvested aseptically, weighed and homogenized in 10 ml Luria broth for incubation at 37° C overnight. Aliquots of the spleen suspension were seeded in cetrimide agar plates for 18h at 37° C to determine the bacterial burden in spleen that is expressed as CFU per mg tissue.
Isolation of Blood Monocytes
The composition of monocyte subsets in blood was determined by labeling the cells with FITC conjugated anti-F4/80 and perCP-CY5.5 conjugated anti-Gr-1 Abs before lysing the erythrocytes with Easy-Lyse Whole Blood Erythrocyte Lysing Kit. Isotype controls conjugated to corresponding fluorochromes were stained simultaneously to set the gates for non-specific staining. The granulocyte populations were gated on Gr-1+ fraction not expressing F4/80. F4/80 is a specific marker for macrophages. The distribution of total monocyte population was based on F4/80+ cells. The inflammatory subset was gated on Gr-1+ fraction and is represented as the percentage of F4/80+ subset also expressing Gr-1+.
Cytokine measurements
100µl of peripheral blood was stimulated with LPS (200ng) at 37° C for 6 hours in the presence of the protein transport inhibitor brefeldin (1µg/ml) followed by labeling with cell surface antigens anti-F4/80 and anti-Gr1. The cells were then fixed and permeabilised using a Cytofix/Cytoperm Plus with GolgiPlug kit and the intracellular cytokine was measured using PE- conjugated anti TNF-α following the instructions provided by the manufacturer. TNF-α and IL-6 levels in plasma were measured by ELISA technique using commercially available kits.
STATISTICAL ANALYSIS
All experiments were done with six to eight animals per group and the results are expressed as mean ± SEM. The experiments were repeated two times. For comparison between groups, multivariate analysis was conducted using the ANOVA statistics, followed by Tukey’s test for significance. Comparison between two groups was determined by student’s t test for unpaired data with equal variance. Statistical significance was set at p < 0.05.
RESULTS
Beta-blockade Attenuates Circulating F4/80+ Monocyte Numbers in Burn Sepsis
Our recent studies demonstrated an increase in total F4/80+ circulating monocytes or monocytosis at 72h in response to burn sepsis26. Sympathetic axotomy in burn septic animals was shown to reverse bone marrow progenitor derived monocytopoiesis12. Here we tested our hypothesis that administering beta-adrenergic blockade following burn injury and sepsis would reduce the total F4/80+ circulating monocytes at 72h in response to burn sepsis. Peripheral blood samples were harvested at 72h from sham (S), burn (B) and burn infected (BS) animals given injections of propranolol or saline following injury. Total monocytes in blood were identified by the cell surface expression of F4/80 using flow cytometry. Animals in burn sepsis group subjected to saline treatment had elevated circulating total F4/80+ monocytes in comparison to sham and burn groups (Sham Saline: 9.5 ± 0.6%, Burn Saline: 10 ± 0.8%, BS Saline: 45 ± 0.8%; p<0.05) demonstrating monocytosis in BS. However, administration of propranolol to burn infected animals attenuated circulating F4/80+ monocytes by 60% (BS Propranolol: 29±4%; p<0.05 vs. BS Saline). The data is represented as bar graphs in Fig 1 panel A. Panel B is a representative histogram illustrating total F4/80+ monocytes on the X-axis against number of cells on the Y-axis. Interestingly, beta-blockade administration in sham and burn animals (S and B) did not affect the circulating total F4/80+ monocytes (data not shown).
Figure 1.
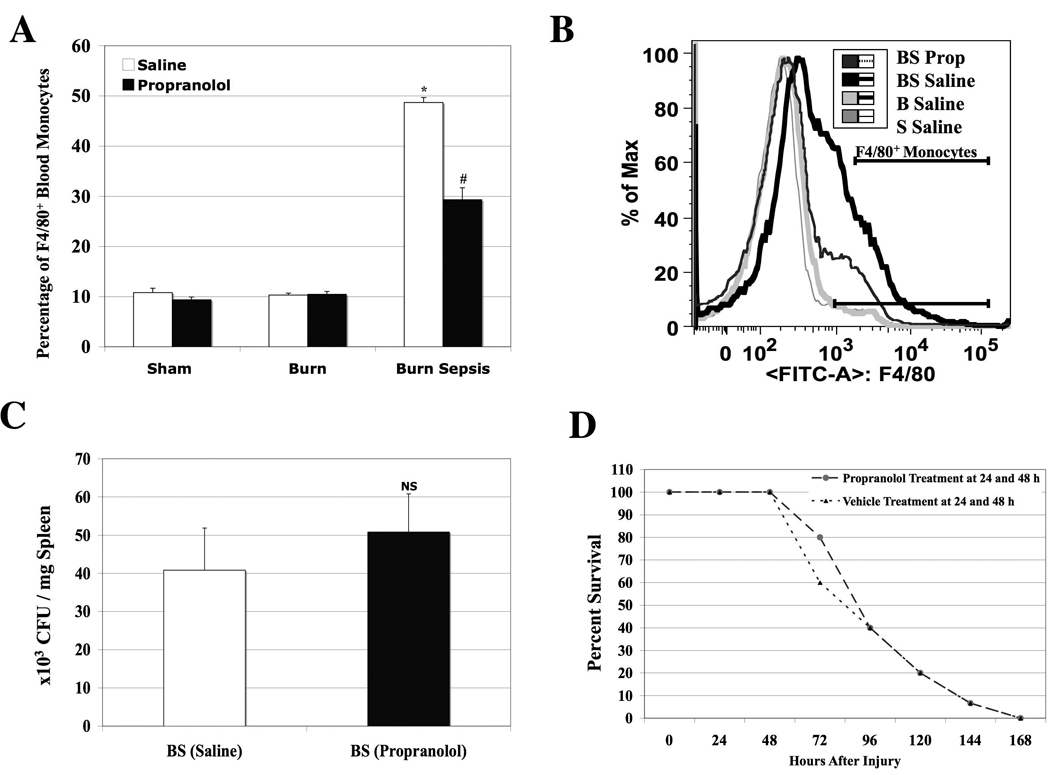
Panel A: A bar graph illustrating the MFI of total F4/80+ monocytes in blood isolated at 72h from sham (S), burn (B) and burn sepsis (BS) animals with (Black bars) or without beta-blockade (Open bars). Percentage of F4/80+ monocytes in circulation was significantly increased in BS with saline treatment; * p<0.05 vs. S and B whereas, beta -blockade significantly reduced the percentage of F4/80+ monocytes in BS (29±4% vs. 45 ± 0.8%; # p<0.05). n=6. Panel B: A representative histogram illustrating the MFI of total F4/80+ monocytes on the X-axis and number of cells on the Y-axis. Panel C: Bar graph illustrating CFU’s of Pseudomonas aerugenosa per mg spleen from saline or propranolol treated BS animals from quantitative cultures. NS: Not significant. n=6. Panel D: Percentage of surviving animals followed up to 168 hours after BS with saline or propranolol treatments at 24 and 48 hours after injury. There were no significant changes in overall survival between the treatment groups. Sham and burn groups did not have any mortalities. n =15.
Quantitative cultures of spleen from the BS group demonstrated comparable CFU’s of Pseudomonas aerugenosa per mg spleen (BS Saline: 41±12 ×103; BS Propranolol: 51±10 ×103). This confirms equal systemic spread of the inoculated bacteria in both propranolol and saline treated animals (Fig 1 panel C). Panel D is a representative graph showing percent survival after BS. Administration of beta-blockade or saline was started at 24 h after burn sepsis and given again at 48 hours. All animals were followed for mortality on a daily basis. We did not notice any significant changes in the overall survival with and without propranolol treatment. 100% mortality was noted by day 7 in both groups. However, we noticed repeatedly at the time of harvest (72 h post injury) that the mortality rate was significantly lower in propranolol treated as compared to the saline treated BS animals (16±3% vs. 44±3%, P<0.05). There were no mortalities in the sham and burn groups.
Propranolol Augments Plasma Cytokine Levels and Improves the Inflammatory Potential of F4/80+ Gr1+ Monocyte Subsets in Peripheral Blood
Heparinized blood samples obtained at 72 h after burn injury and sepsis were analyzed for cytokine expression (Table-1). Levels of plasma IL-6 were significantly higher following beta-blockade in BS group compared to vehicle treatment (4,174 ± 823 vs. 518 ± 199 pg/ml, p< 0.05). While TNF-α levels were below detection limits following vehicle treatment, beta-blockade increased TNF-α levels to 47±12 pg/ml (well within the standard range) in the BS group. TNF-α and IL-6 levels were below detection limits (15.6 pg/ml) in sham and burn groups irrespective of beta-blockade or vehicle treatment (data not included). Plasma cytokines, which is a steady state level, is an indication of systemic inflammatory response. However, this represents the production by a heterogeneous population of different immune cell types. Results from our previous section indicated that beta-blockade attenuates circulating F4/80+ monocyte numbers in burn sepsis, therefore we investigated the effect of beta-blockade on the functionality of monocyte population.
Table-1.
Plasma cytokine levels at 72h post injury. ND-Not detectable.
| Plasma Cytokine (pg/ml) | Burn Sepsis (Saline) | Burn Sepsis (Propranolol) |
|---|---|---|
| TNF-α | ND | 47±12 |
| IL-6 | 518±199 | *4,174±823 |
p<0.05 vs. Saline treatment. N=6.
A fraction of F4/80+ monocytes express Ly6C antigen that binds to Gr1 antibody37,38. Peripheral blood cells binding to both F4/80 and Gr1 are called inflammatory monocytes (F4/80+/Gr1+) and those that do not express Ly6C are the resident monocytes (F4/80+Gr1−) according to their migration potential24. We have previously reported that inflammatory monocytes contribute to the majority of LPS induced cytokine production26. Because propranolol administered following burn sepsis significantly reduced the circulating monocytes in circulation, we investigated LPS induced intracellular TNF-α production by the circulating monocyte subsets under these conditions that will delineate the inflammatory potential of monocyte subsets. Peripheral blood samples collected at 72h from all treatment groups were labeled with fluorochrome conjugated anti-F4/80 and anti-Gr1 antibodies followed by determination of LPS induced intracellular TNF-α (i TNF-α) by flow cytometry. The bar graph in Fig 2 shows the mean fluorescent intensity (MFI) of ic-TNF-α produced per monocyte in circulation with (black bars) and without (white bars) propranolol treatment. Panels A and B represent ic-TNF-α produced by F4/80+Gr1− resident monocytes and F4/80+Gr1+ inflammatory monocytes respectively.
Figure 2.
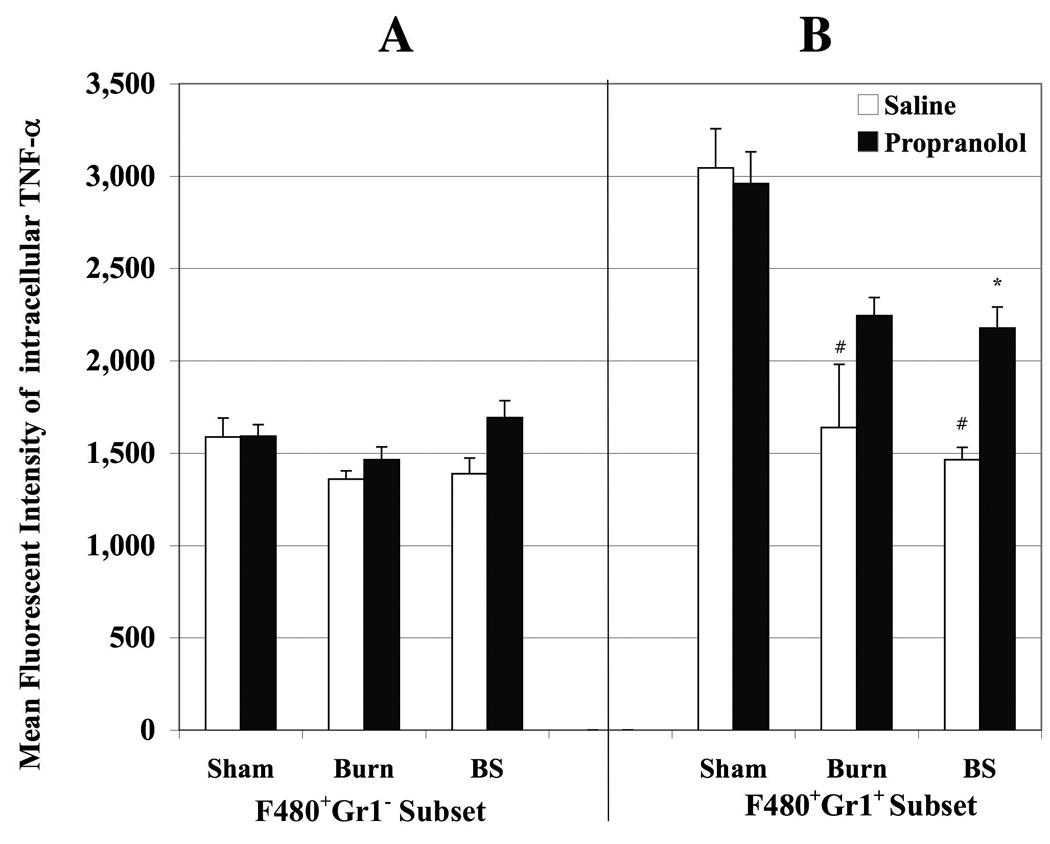
Bar graph delineating MFI of intracellular TNF-α produced by monocytes obtained from peripheral blood of sham (S), burn (B) and burn sepsis (BS) animals with saline (open bars) and propranolol (black bars) treatments. Panels A and B represent ic-TNF-α produced by F4/80+Gr1− resident monocytes and F4/80+Gr1+ inflammatory monocytes respectively. Burn and burn sepsis significantly reduced ic- TNF-α production by Gr1+F4/80+ monocyte subsets compared to sham (# p< 0.05 vs. S and B). Propranolol restored the inflammatory potential of Gr1+F4/80+ monocyte subsets in BS group (* p < 0.05 vs. saline treatment in BS). n= 4 in S and B, n=6 in BS.
There were no significant changes in TNF-α production per resident monocyte fraction between different groups. Conversely, TNF-α produced per inflammatory monocyte was significantly reduced in burn and burn sepsis groups compared to sham following saline treatment (S: 3043±213; B: 1638±343; BS: 1463±67; p<0.05 vs. sham). Beta-blockade after burn sepsis increased the TNF-α production only in the inflammatory monocyte fraction (2177±114 vs. 1463±67; p<0.05). However, beta-blockade had no effect on TNF-α produced by either monocyte subset in sham group and did not reach statistical significance in burn group. Since we observed a significant reduction in ic-TNF-α in spite of an increase in circulating total F4/80+ monocyte population in burn sepsis and that beta-blockade abrogated these responses, the logical next step to interrogate was the distribution of F4/80+ monocyte subsets.
Figure 3 is a representation of (N=6) dot plots gated by selecting all F4/80+ peripheral blood cells isolated at 72h after burn sepsis. Panels A and B correspond to saline and propranolol treated animals respectively. X- and Y-axes represent the mean fluorescent intensities (MFI) of F/480 and Gr1 respectively. The upper right quadrant (R1) shows the distribution of inflammatory monocytes (F4/80+Gr1+) and the lower right quadrant (R2) represents resident monocyte subset (F4/80+Gr1−). There were no significant changes in the percentage of cells represented in R1 and R2 fractions between the two treatment groups. However, beta-blockade for two days after burn sepsis significantly augmented Gr1high expression (indicated by arrow) in the inflammatory monocyte fraction by 60% over saline treatment (Panels A and B). Similarly, LPS induced ic-TNF-α production per inflammatory monocyte was significantly increased following beta-blockade following burn sepsis (Figure 3C). Figure 3D is a representative histogram demonstrating the mean fluorescent intensity of ic-TNF-α on the X-axis and the number of cells on the Y-axis.
Figure 3.
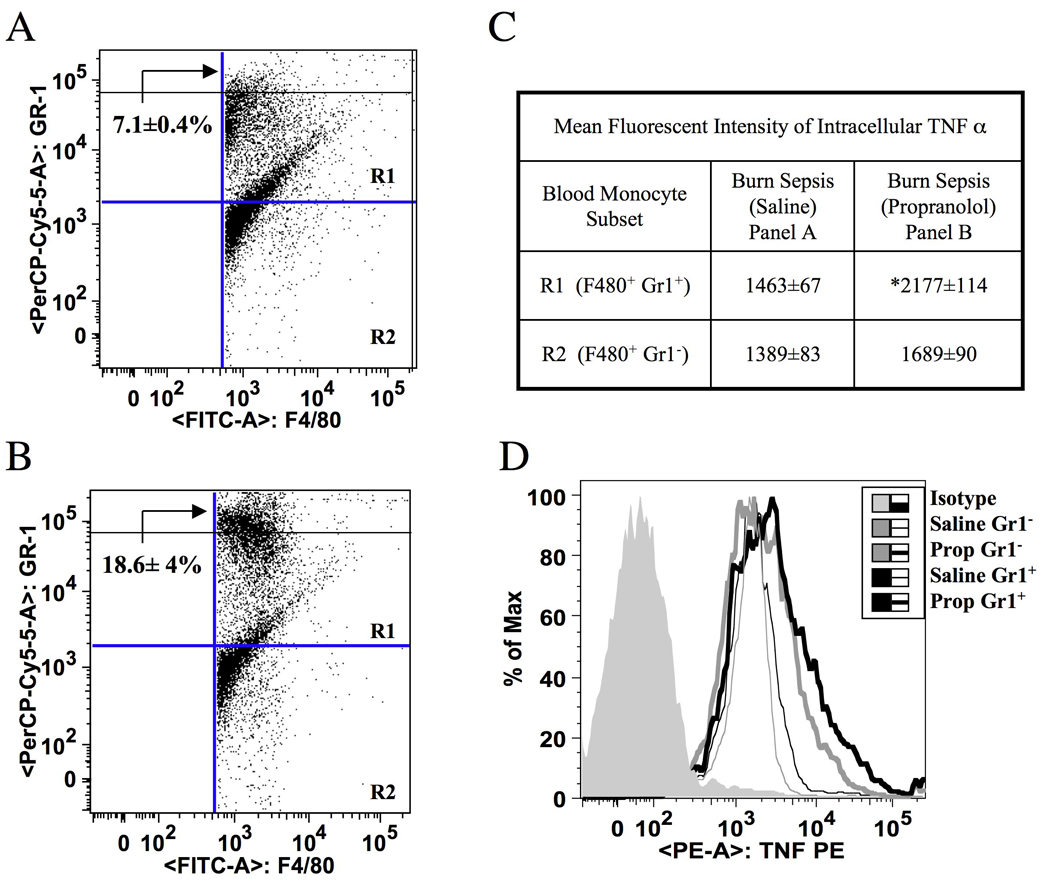
Beta-blockade in BS augments TNF-α in circulating Gr1+ F4/80+ inflammatory monocytes. Panels A and B; Representative dot plots of dual color flow cytometry, defining Gr1 (Y-axis) gated on F4/80+ monocytes (X-axis) from saline and propranolol treatment groups respectively following burn-sepsis (BS). Beta-blockade significantly augmented Gr1high expression (indicated by arrows) in the inflammatory monocyte fraction by 60% over saline treatment. Panel C represents MFI of ic-TNF-α expressed by the monocyte subsets in R1 and R2 regions from panels A and B ( * p< 0.05 vs. saline treatment). Panel D: Representative histogram illustrating the MFI of ic-TNF-α on the X-axis and number of cells on the Y-axis. n=6.
Results from this study demonstrate that critical trauma such as burn sepsis blunts the TNF-α response in the circulating inflammatory monocyte subset at 72h, and this effect can be reversed by the administration of pharmacological beta-blockers soon after the injury.
Beta-blockade Increases Circulating F4/80−/Gr1+ Granulocytes in Burn Sepsis
While macrophages express F4/80 antigen, which is a specific marker for tissue macrophages and blood monocytes39, subpopulations of monocytes also express Ly6C antigen. Anti granulocyte receptor-1 (Gr1) monoclonal antibody binds both Ly6C, which is present on monocytes and Ly6G, which is present on neutrophils40. Although Gr1 is widely accepted as a granulocyte marker, those Gr1+ cells that do not express F4/80 (F4/80−/Gr1+) are true granulocytes. Similarly, a fraction of F4/80+ monocytes also bind to Gr1 antibody. Using these criteria, we explored the role of beta-adrenergic blockade on circulating granulocytes following BS. Peripheral blood samples were harvested from saline treated and propranolol treated animals at 72h following sham, burn and burn sepsis. Granulocytes were identified by flow cytometry using cell surface markers anti-Gr1 and anti- F4/80.
Section I in Fig 4 represents dot plots of peripheral blood cells expressing F4/80 on the X axis and Gr1 on the Y axis. Q1 shows the percentage of peripheral blood cells expressing only Gr1 (granulocytes), whereas Q2 represents monocytes expressing both F4/80 and Gr1. Panels a, c and e represent S, B, and BS in that order with saline treatment and panels b, d, and f represent the corresponding propranolol treatment groups. Table in section II enumerates the percentage of peripheral blood cells (Mean ± SEM) expressing only Gr1 (Q1) or both Gr1 and F4/80 (Q2). The percentages of granulocytes (Q1) remained unaltered in saline treated burn and burn sepsis groups compared to sham (n=4). Beta-blockade in burn and sham were comparable to the corresponding saline treated groups. In contrast, propranolol administration after burn sepsis significantly increased the percentage of granulocytes (28.4±3.6% vs. 15.4±0.3%; p<0.05) in peripheral blood. The Q2 fraction-representing monocytes were augmented by burn sepsis in the saline treated group as compared to sham and burn groups. Beta-blockade following burn sepsis was shown to significantly reduce this monocyte population (23.93±4.5% vs. 37.3±4.7%). All experimental observations were made at 72h post injury.
Fig 4.
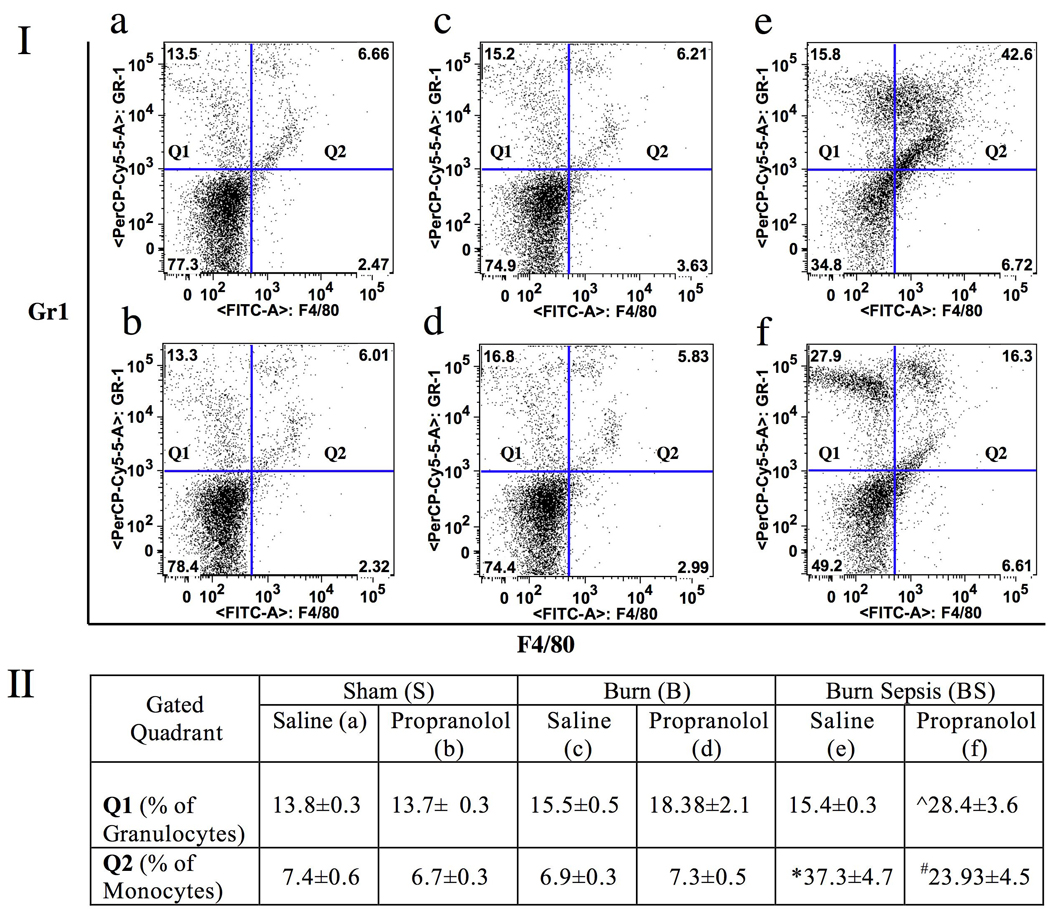
Section I: Representative dot plots of peripheral blood cells expressing F4/80 on the X-axis and Gr1 on the Y-axis. Q1 shows the percentage of peripheral blood cells expressing only Gr1 (granulocytes) and Q2 represents monocytes expressing both F4/80 and Gr1. Panels a, c and e represent S, B, and BS respectively with saline treatment while panels b, d, and f represent the corresponding propranolol treatment groups. Section II: Table representing the percentage of those peripheral blood cells (Mean ± SEM) that express only Gr1 (Q1) or both Gr1 and F4/80 (Q2). Beta-blockade in BS significantly increased the percentage of cells in Q1 (granulocytes) while reducing the monocyte (Q2) population. * vs. S and B, ˆ vs. S, B and BS saline treatment,# vs. BS saline treatment; P<0.05. n = 4 in S and B, n=6 in BS.
Propranolol Administration Following Burn Sepsis Augments the Inflammatory Potential of Circulating F4/80−Gr1+ Granulocytes
Beta-blockade administered for two days after burn injury reduced burn sepsis- induced monocytosis and restored the inflammatory potential of the circulating monocytes while augmenting the number of granulocytes. Activated granulocytes are capable of cytokine production albeit to a lesser degree than the monocytes41. Here we investigated the effect of beta-blockade on the inflammatory potential of the granulocytes. Peripheral blood samples were harvested at 72h from sham, burn and burn infected animals following either saline or propranolol treatments. MFI of LPS stimulated intracellular TNF-α expressed per granulocyte (F4/80− Gr1+) was determined by flow cytometry. Granulocytes were identified as Gr1+ cells that do not express F4/80.
LPS stimulated TNF-α produced by the granulocyte fraction is presented in Fig 5. There were no significant differences in TNF-α levels produced by granulocytes isolated from saline treated animals subjected to sham, burn or burn sepsis (S: 520±28, B: 592± 63, BS: 644±5). Similarly, beta-blockade after sham and burn had very little effect on TNF-α production (S Prop: 494±25, B Prop: 711±40). However, propranolol administration after burn sepsis augmented TNF-α production in granulocytes in comparison to saline treatment (MFI: 903±102 vs. 644±5; p<0.05).
Fig 5.
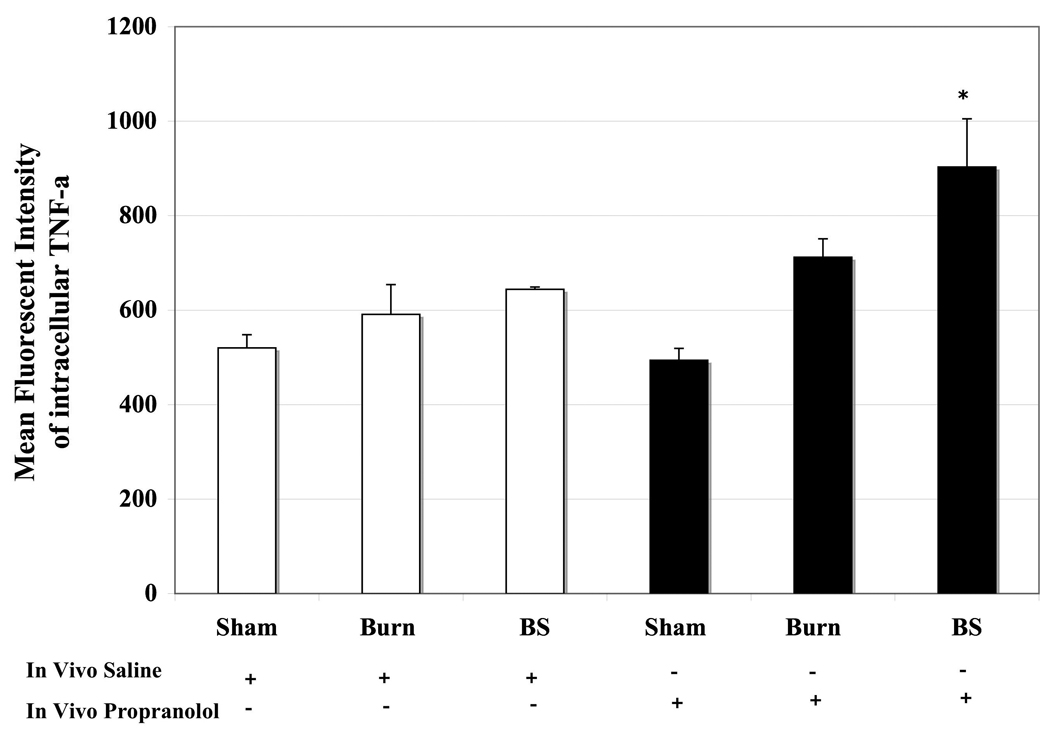
Bar graphs denoting MFI of intracellular TNF-α produced per granulocyte from peripheral blood of sham (S), burn (B) and burn sepsis (BS) animals. The first half represents saline treatment and the other half shows the result of beta-blockade. Propranolol augments TNF-α production in circulating Gr1+ granulocytes in BS group. * vs. BS saline; p<0.05. n = 4 in S and B, n=6 in BS.
Therefore, our results indicate that beta-blockade following burn sepsis promotes activated granulocytes in circulation aside from increasing the number of circulating granulocytes.
DISCUSSION
Results gleaned from the present study demonstrate that burn injury and sepsis mediates sympathetic stimulation that drives monocytosis while suppressing the inflammatory response of the monocytes. Systemic blockade of the beta-adrenergic receptors after burn sepsis using the pharmacologic agent propranolol ameliorates burn sepsis induced monocytosis. Further, propranolol treatment after burn sepsis also improves the inflammatory potential of the inflammatory monocyte subset while simultaneously augmenting granulocytosis and TNF-α production by the circulating granulocytes.
As a primordial response to critical trauma such as thermal injury and sepsis, plasma catecholamines are increased over ten fold indicating the sympathetic activation of nerve terminals and the adrenal medulla6–11. Beta-blockade in pediatric burn patients has been validated to ameliorate hyper-metabolism and reverses muscle-protein catabolism17,42,43. Although the salutary effect of beta-adrenergic blockade in pediatric burn patients is well documented, its potential influence on immunological responses in thermal injury and sepsis is unclear44. Several reports emerging from other labs and our own shows evidence for neuro-immune modulation of host defense13,15,45–48. Therefore, we have tested the premise that beta-adrenergic blockade will influence peripheral blood monocyte distribution and function.
Tang et al have shown direct evidence for enhanced bone marrow norepinerhrine release and monocytopoiesis in animal model of thermal injury and sepsis using clonal expansion of bone marrow progenitors12. Here we have characterized monocytes from peripheral blood by flow cytometry using F4/80 antigen to document monocytosis at 72h post burn sepsis. In contrast, administration of propranolol after the injury paradigm for 2 days significantly reduced the percentage of circulating monocytes (Fig 1 A). This observation is in line with Tang et al’s evidence for reversal of monocytopoiesis with norepinerhrine depletion by chemical sympathectomy prior to thermal injury and sepsis12,16.
Despite monocytosis and monocytopoiesis, monocyte deactivation and dysregulated cytokine responses are hallmark of severe sepsis4,49–52. Using a murine model of burn injury and sepsis we have previously demonstrated two distinct subsets of monocytes based on Gr1 expression. F4/80+ monocytes that express Gr1 produced majority of LPS stimulated TNF-α26. The seminal work of Geissman et al using genetically engineered CX3CR1gfp mice, has led to the classification of Gr1+ (Ly-6Chigh) subset of monocytes as inflammatory based on their migration properties24,37. Results from the present study (Fig 2) demonstrate that the TNF-α produced by the inflammatory (F4/80+Gr1+) monocyte subset is reduced by burn sepsis and that beta-blockade restores the inflammatory potential of the inflammatory monocyte subset. Interestingly, monocytes from sham and burn group were resistant to beta-blockade in sham and burn animals indicating lack of significant sympathetic stimulus. A lack of effect in burn animals may be due to a relatively small size of the burn wound in this model (15% total body-surface area burn), which may not be a severe enough perturbation to invoke profound adrenergic stimulus. The small size burn is one of the limitations in the present model that may have failed to produce similar changes normally seen after burn injury in clinical situations such as a 40 to 60 % third degree burn. Unfortunately, a larger burn such as >30% in this model is not feasible and routinely results in an unacceptable mortality rate due to the inability to overcome the profound hemodynamic disturbances53.
Several in vitro and in vivo studies have documented adrenergic regulation of cytokine responses. The majority of those studies involved adherent macrophages19,54–56. In this study we describe that beta-blockade ameliorates burn sepsis induced suppression of TNF-α in a subset of monocytes in circulation. Hence, monitoring the number and function of monocyte subsets in blood could be considered a reliable biomarker to predict the efficacy of beta-blockade.
Increased circulating Ly-6Chigh monocytes are reported in sepsis. Ly-6Cmed-high monocytes are preferentially recruited to the site of peritoneal inflammation37. However these studies involved acute sepsis with Lysteria monocytogenes or chronic sepsis with Leishmania major. Yet, the effect of propranolol treatment had not been evaluated in any sepsis model. In our burn sepsis model with Pseudomonas aerugenosa, beta-blockade enhanced Gr1 (Ly6-C) expression in F4/80+ monocytes at 72h post injury with a log order shift in the MFI of Gr1 expression. Additionally, TNF-α expressed by the inflammatory subset of monocytes was increased by propranolol. Taken together this signifies the possibility of enhanced migration of inflammatory monocytes to the site of sepsis with beta-blockade treatment given in burn injury.
Both monocytes and granulocytes contribute to the majority of innate and adaptive immune cells of the myeloid lineage. Several clinical and experimental evidences suggest adrenergic stimulation increases circulating leukocyte counts and neutrophil sequestration in in vivo model as well as in pulmonary sepsis models57–59. A reduction in G-CSF responsive clonal expansion of bone marrow myeloid progenitors was reported in burn injury and sepsis53,60. Since beta-blockade triggered a reversal of burn-sepsis induced monocytosis shifting the commitment pattern of myeloid cells, it is logical to investigate the influence of beta-blockade on the granulocyte arm. Our results indicated a parallel increase in granulocytes and a concomitant boost in the TNF-α production with propranolol treatment following burn sepsis. While there is evidence for catecholamine-induced demargination of granulocytes59 our current data demonstrating an increase in Gr1+ granulocytosis resulting from beta-blockade following burn sepsis may be due to an efflux from the bone marrow and not demargination. Further investigations on beta-blockade mediated myeloid commitment of bone marrow progenitors are warranted to test this thesis.
Our current report describes for the first time, that beta-blockade in critical burn sepsis could manipulate immunologic aspects of the myeloid cells in circulation. Much of the previous studies were carried out in tissue samples such as bone marrow whereas, in our present study, this is illustrated in the most easily accessible compartment -the blood- paving the way for future clinical studies to predict the immunologic consequence of beta-blockade. This study did not demonstrate significant changes in monocyte and granulocyte phenotypes in propranolol treated burn animals with a relatively small burn. However, it does not preclude significant propranolol-induced phenotypic changes in larger burns. Nonetheless, our approach makes it feasible to follow peripheral blood monocyte and granulocyte phenotypes in burn patients.
ACKNOWLEDGEMENTS
This work was supported by Dr. Ralph and Marion C. Falk Medical Research Trust and the NIH grant R01 GM 42577.
REFERENCES
- 1.Ayala A, Chaudry IH. Immune dysfunction in murine polymicrobial sepsis: mediators, macrophages, lymphocytes and apoptosis. Shock. 1996;6 Suppl 1:S27–S38. [PubMed] [Google Scholar]
- 2.Faist E, Schinkel C, Zimmer S, Kremer JP, Von Donnersmarck GH, Schildberg FW. Inadequate interleukin-2 synthesis and interleukin-2 messenger expression following thermal and mechanical trauma in humans is caused by defective transmembrane signalling. J Trauma. 1993;34:846–853. doi: 10.1097/00005373-199306000-00016. discussion 853–844. [DOI] [PubMed] [Google Scholar]
- 3.Miller CL, Baker CC. Changes in lymphocyte activity after thermal injury. The role of suppressor cells. J Clin Invest. 1979;63:202–210. doi: 10.1172/JCI109290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller-Graziano CL, Szabo G, Kodys K, Griffey K. Aberrations in post-trauma monocyte (MO) subpopulation: role in septic shock syndrome. J Trauma. 1990;30:S86–S96. [PubMed] [Google Scholar]
- 5.Faist E. The mechanisms of host defense dysfunction following shock and trauma. Curr Top Microbiol Immunol. 1996;216:259–274. doi: 10.1007/978-3-642-80186-0_12. [DOI] [PubMed] [Google Scholar]
- 6.Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med. 2001;29:S109–S116. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 7.Benedict CR, Grahame-Smith DG. Plasma noradrenaline and adrenaline concentrations and dopamine-beta-hydroxylase activity in patients with shock due to septicaemia, trauma and haemorrhage. Q J Med. 1978;47:1–20. [PubMed] [Google Scholar]
- 8.Frayn KN. Hormonal control of metabolism in trauma and sepsis. Clin Endocrinol (Oxf) 1986;24:577–599. doi: 10.1111/j.1365-2265.1986.tb03288.x. [DOI] [PubMed] [Google Scholar]
- 9.Goodall M, Stone C, Haynes BW., Jr Urinary output of adrenaline and noradrenaline in severe thermal burns. Ann Surg. 1957;145:479–487. doi: 10.1097/00000658-195704000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilmore DW, Aulick LH. Metabolic changes in burned patients. Surg Clin North Am. 1978;58:1173–1187. doi: 10.1016/s0039-6109(16)41685-3. [DOI] [PubMed] [Google Scholar]
- 11.Wilmore DW, Long JM, Mason AD, Jr, Skreen RW, Pruitt BA., Jr Catecholamines: mediator of the hypermetabolic response to thermal injury. Ann Surg. 1974;180:653–669. doi: 10.1097/00000658-197410000-00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang Y, Shankar R, Gamboa M, Desai S, Gamelli RL, Jones SB. Norepinephrine modulates myelopoiesis after experimental thermal injury with sepsis. Ann Surg. 2001;233:266–275. doi: 10.1097/00000658-200102000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Y, Shankar R, Gamelli R, Jones S. Dynamic norepinephrine alterations in bone marrow: evidence of functional innervation. J Neuroimmunol. 1999;96:182–189. doi: 10.1016/s0165-5728(99)00032-6. [DOI] [PubMed] [Google Scholar]
- 14.Jones SB, Kovarik MF, Romano FD. Cardiac and splenic norepinephrine turnover during septic peritonitis. Am J Physiol. 1986;250:R892–R897. doi: 10.1152/ajpregu.1986.250.5.R892. [DOI] [PubMed] [Google Scholar]
- 15.Marino F, Cosentino M, Bombelli R, et al. Measurement of catecholamines in mouse bone marrow by means of HPLC with electrochemical detection. Haematologica. 1997;82:392–394. [PubMed] [Google Scholar]
- 16.Cohen MJ, RS J, Stevenson R, Fernandez RL, Gamelli, Jones SB. Bone marrow norepinephrine mediates development of functionally different macrophages following thermal injury and sepsis. Annals of Surgery. 2004 doi: 10.1097/01.sla.0000130724.84914.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herndon DN, Hart DW, Wolf SE, Chinkes DL, Wolfe RR. Reversal of catabolism by beta-blockade after severe burns. N Engl J Med. 2001;345:1223–1229. doi: 10.1056/NEJMoa010342. [DOI] [PubMed] [Google Scholar]
- 18.Muthu K, Deng J, Gamelli R, Shankar R, Jones SB. Adrenergic modulation of cytokine release in bone marrow progenitor-derived macrophage following polymicrobial sepsis. J Neuroimmunol. 2005;158:50–57. doi: 10.1016/j.jneuroim.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Muthu K, Deng J, Romano F, et al. Thermal injury and sepsis modulates beta-adrenergic receptors and cAMP reponses in monocyte-committed bone marrow cells. J Neuroimmunol. 2005;165:129–138. doi: 10.1016/j.jneuroim.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto T, Naito M, Moriyama H, et al. Repopulation of murine Kupffer cells after intravenous administration of liposome-encapsulated dichloromethylene diphosphonate. Am J Pathol. 1996;149:1271–1286. [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi K, Naito M, Takeya M. Development and heterogeneity of macrophages and their related cells through their differentiation pathways. Pathol Int. 1996;46:473–485. doi: 10.1111/j.1440-1827.1996.tb03641.x. [DOI] [PubMed] [Google Scholar]
- 22.del Hoyo GM, Martin P, Vargas HH, Ruiz S, Arias CF, Ardavin C. Characterization of a common precursor population for dendritic cells. Nature. 2002;415:1043–1047. doi: 10.1038/4151043a. [DOI] [PubMed] [Google Scholar]
- 23.Martinez del Hoyo G, Martin P, Arias CF, Marin AR, Ardavin C. CD8alpha+ dendritic cells originate from the CD8alpha-dendritic cell subset by a maturation process involving CD8alpha, DEC-205, and CD24 up-regulation. Blood. 2002;99:999–1004. doi: 10.1182/blood.v99.3.999. [DOI] [PubMed] [Google Scholar]
- 24.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 25.Robben PM, LaRegina M, Kuziel WA, Sibley LD. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J Exp Med. 2005;201:1761–1769. doi: 10.1084/jem.20050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muthu K, He LK, Melstrom K, Szilagyi A, Gamelli RL, Shankar R. Perturbed bone marrow monocyte development following burn injury and sepsis promote hyporesponsive monocytes. J Burn Care Res. 2008;29:12–21. doi: 10.1097/BCR.0b013e31815fa499. [DOI] [PubMed] [Google Scholar]
- 27.Jander G, Rahme LG, Ausubel FM. Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J Bacteriol. 2000;182:3843–3845. doi: 10.1128/jb.182.13.3843-3845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker HL, Mason AD., Jr A standard animal burn. J Trauma. 1968;8:1049–1051. doi: 10.1097/00005373-196811000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Cohen MJ, Carroll C, He LK, et al. Severity of burn injury and sepsis determines the cytokine responses of bone marrow progenitor-derived macrophages. J Trauma. 2007;62:858–867. doi: 10.1097/01.ta.0000222975.03874.58. [DOI] [PubMed] [Google Scholar]
- 30.Hargrove DM, Bagby GJ, Lang CH, Spitzer JJ. Adrenergic blockade does not abolish elevated glucose turnover during bacterial infection. Am J Physiol. 1988;254:E16–E22. doi: 10.1152/ajpendo.1988.254.1.E16. [DOI] [PubMed] [Google Scholar]
- 31.Hargrove DM, Bagby GJ, Lang CH, Spitzer JJ. Adrenergic blockade prevents endotoxin-induced increases in glucose metabolism. Am J Physiol. 1988;255:E629–E635. doi: 10.1152/ajpendo.1988.255.5.E629. [DOI] [PubMed] [Google Scholar]
- 32.Kabbaj M, Piazza PV, Simon H, Le Moal M, Maccari S. Opposite effects on hippocampal corticosteroid receptors induced by stimulation of beta and alpha 1 noradrenergic receptors. Neuroscience. 1995;66:539–545. doi: 10.1016/0306-4522(94)00620-k. [DOI] [PubMed] [Google Scholar]
- 33.Klein I. Thyroxine-induced cardiac hypertrophy: time course of development and inhibition by propranolol. Endocrinology. 1988;123:203–210. doi: 10.1210/endo-123-1-203. [DOI] [PubMed] [Google Scholar]
- 34.Plank DM, Yatani A, Ritsu H, et al. Calcium dynamics in the failing heart: restoration by beta-adrenergic receptor blockade. Am J Physiol Heart Circ Physiol. 2003;285:H305–H315. doi: 10.1152/ajpheart.00425.2002. [DOI] [PubMed] [Google Scholar]
- 35.Nielson KA, Czech DA, Laubmeier KK. Chronic administration of propranolol impairs inhibitory avoidance retention in mice. Neurobiol Learn Mem. 1999;71:248–257. doi: 10.1006/nlme.1998.3873. [DOI] [PubMed] [Google Scholar]
- 36.Miyamoto M, Tomaki M, Lotvall J, Linden A. Beta-adrenoceptor stimulation and neutrophil accumulation in mouse airways. Eur Respir J. 2004;24:231–237. doi: 10.1183/09031936.04.00035204. [DOI] [PubMed] [Google Scholar]
- 37.Sunderkotter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 38.Drevets DA, Dillon MJ, Schawang JS, et al. The Ly-6Chigh monocyte subpopulation transports Listeria monocytogenes into the brain during systemic infection of mice. J Immunol. 2004;172:4418–4424. doi: 10.4049/jimmunol.172.7.4418. [DOI] [PubMed] [Google Scholar]
- 39.Leenen PJ, de Bruijn MF, Voerman JS, Campbell PA, van Ewijk W. Markers of mouse macrophage development detected by monoclonal antibodies. J Immunol Methods. 1994;174:5–19. doi: 10.1016/0022-1759(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 40.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 41.Mandi Y, Endresz V, Krenacs L, Regely K, Degre M, Beladi I. Tumor necrosis factor production by human granulocytes. Int Arch Allergy Appl Immunol. 1991;96:102–106. doi: 10.1159/000235479. [DOI] [PubMed] [Google Scholar]
- 42.Baron PW, Barrow RE, Pierre EJ, Herndon DN. Prolonged use of propranolol safely decreases cardiac work in burned children. J Burn Care Rehabil. 1997;18:223–227. doi: 10.1097/00004630-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Breitenstein E, Chiolero RL, Jequier E, Dayer P, Krupp S, Schutz Y. Effects of beta-blockade on energy metabolism following burns. Burns. 1990;16:259–264. doi: 10.1016/0305-4179(90)90136-k. [DOI] [PubMed] [Google Scholar]
- 44.Jeschke MG, Norbury WB, Finnerty CC, Branski LK, Herndon DN. Propranolol does not increase inflammation, sepsis, or infectious episodes in severely burned children. J Trauma. 2007;62:676–681. doi: 10.1097/TA.0b013e318031afd3. [DOI] [PubMed] [Google Scholar]
- 45.Besedovsky HO, del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev. 1996;17:64–102. doi: 10.1210/edrv-17-1-64. [DOI] [PubMed] [Google Scholar]
- 46.Madden KS, Felten SY, Felten DL, Hardy CA, Livnat S. Sympathetic nervous system modulation of the immune system. II. Induction of lymphocyte proliferation and migration in vivo by chemical sympathectomy. J Neuroimmunol. 1994;49:67–75. doi: 10.1016/0165-5728(94)90182-1. [DOI] [PubMed] [Google Scholar]
- 47.Madden KS, Sanders VM, Felten DL. Catecholamine influences and sympathetic neural modulation of immune responsiveness. Annu Rev Pharmacol Toxicol. 1995;35:417–448. doi: 10.1146/annurev.pa.35.040195.002221. [DOI] [PubMed] [Google Scholar]
- 48.Maestroni GJ. Adrenergic regulation of haematopoiesis. Pharmacol Res. 1995;32:249–253. doi: 10.1016/s1043-6618(05)80012-x. [DOI] [PubMed] [Google Scholar]
- 49.Simpson SQ, Modi HN, Balk RA, Bone RC, Casey LC. Reduced alveolar macrophage production of tumor necrosis factor during sepsis in mice and men. Crit Care Med. 1991;19:1060–1066. doi: 10.1097/00003246-199108000-00015. [DOI] [PubMed] [Google Scholar]
- 50.Volk HD, Reinke P, Krausch D, et al. Monocyte deactivation--rationale for a new therapeutic strategy in sepsis. Intensive Care Med. 1996;22 Suppl 4:S474–S481. doi: 10.1007/BF01743727. [DOI] [PubMed] [Google Scholar]
- 51.Volk HD, Thieme M, Heym S, et al. Alterations in function and phenotype of monocytes from patients with septic disease--predictive value and new therapeutic strategies. Behring Inst Mitt. 1991:208–215. [PubMed] [Google Scholar]
- 52.Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88:1747–1754. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santangelo S, Gamelli RL, Shankar R. Myeloid commitment shifts toward monocytopoiesis after thermal injury and sepsis. Ann Surg. 2001;233:97–106. doi: 10.1097/00000658-200101000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bergmann MaS T. Immunomodulatory effects of vasoactive catecholamines. Wiener Klinische Wochenschrift. 2002;114:752–761. [PubMed] [Google Scholar]
- 55.Deng J, Muthu K, Gamelli R, Shankar R, Jones SB. Adrenergic modulation of splenic macrophage cytokine release in polymicrobial sepsis. Am J Physiol Cell Physiol. 2004;287:C730–C736. doi: 10.1152/ajpcell.00562.2003. [DOI] [PubMed] [Google Scholar]
- 56.van der Poll T, Coyle SM, Barbosa K, Braxton CC, Lowry SF. Epinephrine inhibits tumor necrosis factor-alpha and potentiates interleukin 10 production during human endotoxemia. J Clin Invest. 1996;97:713–719. doi: 10.1172/JCI118469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morken JJ, Warren KU, Xie Y, Rodriguez JL, Lyte M. Epinephrine as a mediator of pulmonary neutrophil sequestration. Shock. 2002;18:46–50. doi: 10.1097/00024382-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 58.Abraham E, Kaneko DJ, Shenkar R. Effects of endogenous and exogenous catecholamines on LPS-induced neutrophil trafficking and activation. Am J Physiol. 1999;276:L1–L8. doi: 10.1152/ajplung.1999.276.1.L1. [DOI] [PubMed] [Google Scholar]
- 59.Benschop RJ, Rodriguez-Feuerhahn M, Schedlowski M. Catecholamine-induced leukocytosis: early observations, current research, and future directions. Brain Behav Immun. 1996;10:77–91. doi: 10.1006/brbi.1996.0009. [DOI] [PubMed] [Google Scholar]
- 60.Shoup M, Weisenberger JM, Wang JL, Pyle JM, Gamelli RL, Shankar R. Mechanisms of neutropenia involving myeloid maturation arrest in burn sepsis. Ann Surg. 1998;228:112–122. doi: 10.1097/00000658-199807000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]