

Abstract
Proteolytic processing of the coronavirus replicase polyproteins is essential for ongoing viral ribonucleic acid (RNA) synthesis. Therefore, the severe acute respiratory syndrome (SARS)-coronaviruses (SARS-CoV) proteases are attractive targets for the development of antiviral drugs to reduce viral replication and pathogenicity. The structure and activity of the coronavirus 3C-like protease (3CLpro) has already been elucidated, and the design of inhibitors to 3CLpro as therapeutics has been proposed. The chapter discusses SARS-CoV 3CLpro inhibitors that include covalent inhibitors, noncovalent inhibitors, and inhibitors from screening. SARS-CoV papain-like protease (PLpro) is considered an equally viable target to 3CLpro for drug design because both are essential for viral replication. However, PLpro has likely not been pursued because of the paucity of structural information. Several compounds have been identified that have shown inhibitory activity against SARS-CoV. However, no information regarding their mechanism of action or the corresponding target is known. Glycyrrhizin showed inhibitory activity for SARS-CoV replication with EC50 = 300 mg/L after virus absorption in Vero cells. Some glycyrrhizin acid derivatives were found to inhibit SARS-CoV replication in vitro with EC50 values ranging from 5 to 50 μM. Unfortunately, these compounds show high cytotoxity.
1. Introduction
Severe acute respiratory syndrome (SARS) was first reported in Guangdong Province, China, in November 2002. Since then, it has spread to other Asian countries, North America and Europe. This outbreak reportedly affected more than 8000 individuals by July 2003, and resulted in 774 deaths. SARS is characterized by high fever, malaise, rigor, headache and non-productive cough or dyspnea and may progress to generalized interstitial infiltrates in the lung, requiring intubation and mechanical ventilation [1]. In an unprecedented response, the World Health Organization (WHO) called upon leading laboratories in the world and set up multi-center research to investigate the etiology of SARS and develop effective diagnostic tests. With the aid of modern information technologies, WHO set up a secured website to share emerging scientific information among several laboratories. The critical results including electron micrographs of viruses, sequences of genetic material, identification and characterization of the virus, virus isolates, samples from patients, and postmortem tissues were shared in real time. The goal was to pin down the causative agent for SARS and to develop diagnostic tools. Within months, a novel coronavirus was isolated from patients with SARS. Then within days, after comparison of the sequences of the coronavirus polymerase gene against all previously characterized strains, scientists concluded that this virus is distinct from all previously known human pathogens. Just over a month after the outbreak of the new illness, WHO announced that a new pathogen, a member of the coronavirus family never seen before in humans, is the cause of SARS [2], [3], [4], [5]. In a remarkable scientific collaboration assembled by the WHO among teams of scientists from 13 laboratories in 10 countries, a novel coronavirus was identified as the etiological agent for SARS. Public health measures, including rapid identification of SARS cases and isolation of contacts, ultimately succeeded in controlling the 2002–2003 SARS epidemic. However, the identification of animal reservoirs for the virus and the possibility of re-emergence of epidemic or pandemic SARS provide strong motivation for the development of antiviral agents to treat this potentially fatal respiratory illness.
2. SARS coronavirus proteases
Coronaviruses are a family of positive strand, enveloped RNA viruses that can cause respiratory, gastrointestinal and neurological diseases. Coronavirus virions are composed of a helical nucleocapsid surrounded by a lipid bilayer envelope studded with virus-specific glycoproteins [6]. The helical nucleocapsid structure contains the single-stranded, positive sense RNA genome surrounded by a nucleocapsid protein (N). The viral envelope contains the membrane glycoprotein (M), the envelope protein (E) and the spike glycoprotein (S).
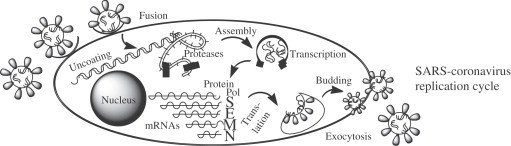
The SARS-CoV replicase is encoded in the 5′-most 21 kb of the ∼29.7 kb viral genomic RNA. The genomic RNA is translated to produce two replicase polyproteins, termed pp1a and pp1ab [6], [7]. The pp1a is a ∼486 kilodalton (kDa) polyprotein that is predicted to contain a single papain-like protease (PLpro) analogous to the second murine coronavirus PLpro domain, a picornavirus 3C-like protease domain (3CLpro, also sometimes noted as Mpro or main protease), three putative membrane proteins, and several additional products of unknown function. The pp1ab (∼790 kDa) is generated by ribosomal frameshifting and extends the pp1a product to include open reading frame 1b, which contains the core RNA polymerase and helical domains, and additional products of unknown function. The pp1a and pp1ab polyproteins are predicted to be processed to generate 16 protein products (termed non-structural proteins, nsp1–nsp16) [8], which assemble to form a membrane-associated viral replication complex.
3. SARS-CoV 3CLpro inhibitors
Proteolytic processing of the coronavirus replicase polyproteins is essential for ongoing viral RNA synthesis. Therefore, the SARS-CoV proteases are attractive targets for the development of antiviral drugs to reduce viral replication and pathogenicity. The structure and activity of the coronavirus 3CLpro has already been elucidated and the design of inhibitors to 3CLpro as therapeutics has been proposed [9], [10]. SARS-CoV 3CLpro has three domains: I (residues 8–101), II (residues 102–184), and III (residues 201–301). Domains I and II, which contain the active site region, are β-barrel domains and III is an α-helical domain. In the active site, a cysteine residue (Cys-145) acts as a nucleophile and a histidine residue (His-41) acts as the general acid base. The X-ray crystal structure of the related enzyme from porcine transmissible gastroenteritis coronavirus (TGEV 3CLpro) and a substrate-analogue hexapeptidyl chloromethyl ketone (CMK) inhibitor 1 (Cbz-Val-Asn-Ser-Thr-Leu-Gln-CMK) has been reported [8]. The sequence of this inhibitor was designed based upon P6 and P1 residues of the N-terminal autoprocessing site of TGEV 3CLpro. The corresponding sequences of SARS-CoV 3CLpro and HCoV-229E 3CLpro are Thr-Ser-Ala-Val-Leu and Tyr-Gly-Thr-Leu-Gln, respectively. The binding mode of this hexapeptidyl Gln inhibitor is similar to that which was observed for related human rhinovirus 3C protease (3Cpro) [9], [11]. AG7088 (2), a prototype inhibitor of human rhinovirus 3Cpro [12] appears to bind to this enzyme in an orientation similar to the peptidyl CMK inhibitor in the binding site of TGEV 3CLpro [9], [11]. Furthermore, substrate specificity of picornavirus 3Cpro for the P1-, P1′- and P4-sites is very similar to that of coronavirus 3CLpro. As a consequence, compounds 1 and 2 have become starting points for the design of SARS-CoV 3CLpro inhibitors.
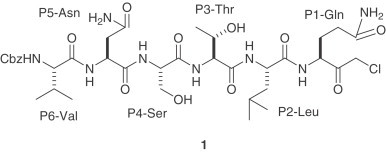
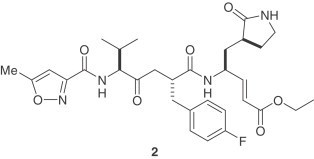
3.1. Covalent inhibitors of SARS-CoV 3CLpro
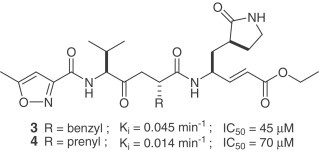
The design and synthesis of two analogues (3,4) of AG7088 (2) was recently reported [13]. Based upon the reported SARS-CoV 3CLpro structure [9], [14], AG7088 was modified by changing the P2 side chain from a p-fluorobenzyl group to the smaller benzyl and prenyl groups. These inhibitors possess a P1/P1′- α,β-unsaturated ester functionality, which can covalently link to the Cys-145. Compound 2 is inactive against SARS-CoV in cell-culture assay. The antiviral activity for 2 was reported to be >100 μg/ml [15]. The modified analogues are not only potent against SARS-CoV 3CLpro (k inact values), but are effective in a SARS-CoV cell assay (IC50 values) as well. No toxicity was observed up to 100 μM. Moreover, an X-ray crystal structure of the SARS-CoV 3CLpro covalently linked with the synthetic small molecule inhibitor (4) was reported. In addition to the important covalent bond formed between 4 and the protease, the X-ray structure also showed crucial hydrogen bonding between 4 and His-164 and Glu-166. It revealed important insight into the molecular recognition of this type of inhibitor by SARS-CoV 3CLpro.
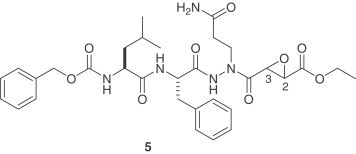
An X-ray crystal structure of a substrate-like aza-peptide epoxide (5) that inhibited the 3CLpro of SARS-CoV was published [16]. While these inhibitors are specific for clan CD cysteine peptidases [17], they are also lead candidates for the SARS-CoV 3CLpro, which has a Cys-145 and His-41 catalytic dyad in the active site. The best inhibition was obtained with the (S,S) diastereomer of compound 5 [k inact/K i=1900(±400) M/s]. The crystal structure revealed a covalent bond formed between the catalytic Cys-145 sulfur atom and the epoxide C3. Modeling studies of the four diastereomers binding to the SARS-CoV 3CLpro before nucleophilic attack by Cys-145 explained the necessity of the (S,S) configuration of the epoxide.
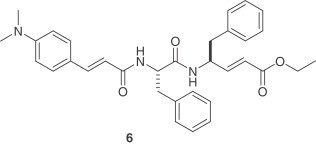
Another important series of covalent inhibitors was recently disclosed [18]. A series of tripeptide α,β-unsaturated esters and ketomethylene isosteres was assayed to target the SARS-CoV 3CLpro. The ketomethylene isosteres and tripeptide α,β-unsaturated esters containing both P1 and P2 phenylalanine residues show modest inhibitory activity (IC50=11–39 μM). The Phe–Phe dipeptide inhibitors were designed on the basis of computer modeling of the enzyme–inhibitor complex. The most potent inhibitor is compound 6 with an inhibition constant of 0.52 μM. The cell-based assays also indicate that this is a non-toxic anti-SARS agent with an EC50 value of 0.18 μM and an IC50 value of 1 μM. The computational study of structure–activity relationships shows that hydrogen bonding with the main chain Glu166 and the side chain Gln189 is crucial for inhibitory potency.
3.2. Non-covalent SARS-CoV 3CLpro inhibitors
Side effects and toxicity often arise with covalently bonded inhibitors which hinder or prevent their development as useful drug therapies [19], [20]. To avoid such pitfalls, it is often desirable to design and develop non-covalent or reversible inhibitors as therapeutic agents.
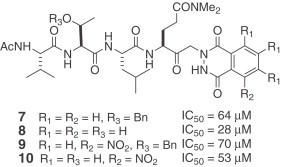
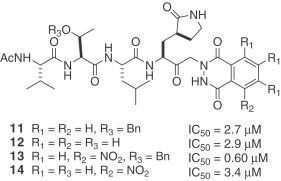
A series of synthetic small molecule, non-covalent inhibitors of SARS-CoV 3CLpro was published in 2004 [21]. These investigators previously reported that keto-glutamine analogues with the phthalhydrazido group at α-position are reversible inhibitors of hepatitis A virus (HAV) 3C proteinase. The IC50 values of the inhibitors were in the low micromolar range [22], [23]. They synthesized a series of keto-glutamine analogues with the phthalhydrazido group at the α-position and attachment of tripeptide (Ac-Val-Thr-Leu) as the inhibitors (7–14) for SARS-CoV 3CLpro. The combined effect of the β and β′ amino groups adjacent to the keto group and intramolecular hydrogen bonding to the carbonyl makes it more electrophilic. As a result, the carbonyl group can form a hemithioacetal with the sulfur of Cys-145. The K i values of these reversible inhibitors remain to be determined.
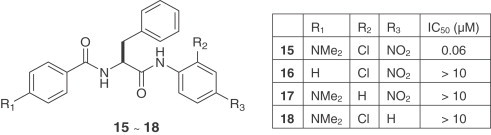
A diversified library of peptide anilides was prepared and their inhibitory activity was examined against the SARS-CoV 3CLpro by a fluorogenic tetradecapeptide substrate [24]. The most potent compound was 15 with a K i value of 0.03 μM. Other analogues (16–18) were examined but showed substantially reduced inhibitory activity. The associated docking study showed that the dimethylamino, chloro and nitro groups that occupied the R1, R2 and R3 positions, respectively, make important interactions with several residues of the SARS-CoV 3CLpro. These interactions are calculated to be responsible for a 9.1 kcal/mol energy difference between compound 15 and its analogues (16–18). Using the same assay methodology, the IC50 values of 15 against trypsin, chymotrypsin and papain were determined to be 110, 200 and 220 μM, respectively. Compound 15 is one of the most potent inhibitors of SARS-CoV 3CLpro reported to date and importantly, it is a competitive inhibitor which does not form covalent bonds with the protease.
A series of synthetic isatin derivatives were also reported as non-covalent SARS-CoV 3CLpro inhibitors [25]. It is known that certain isatin (2,3-dioxindole) compounds are potent inhibitors of rhinovirus 3Cpro [26]. Because SARS-CoV and rhinovirus have similar active sites and catalytic residues, isatin derivatives may be good candidates as SARS-CoV 3CLpro inhibitors. The SARS-CoV 3CLpro inhibition assays were conducted via fluorescence resonance energy transfer (FRET) according to the reported protocol [27]. These isatin derivatives inhibited SARS-CoV 3CLpro in the low micromolar range (0.95–17.5 μM). Among them, compounds 19 and 20 were the most potent inhibitors.
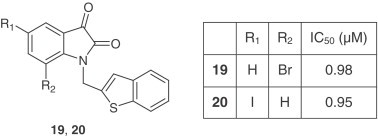
Computer modeling showed that both compounds fit into the active pocket of SARS-CoV 3CLpro. The two carbonyl groups on isatin can form hydrogen bonds with the NH groups on Gly-143, Ser-144, Cys-145 and the His-41 side chain.
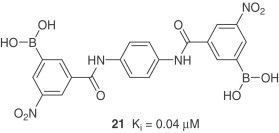
Analysis of the active site of SARS-CoV 3CLpro reveals the presence of a cluster of serine residues (Ser-139, Ser-144, Ser-147). A series of aryl boronic acid derivatives showed high binding affinities and have shown inhibition constants in low micromolar range [28]. Compound 21 with amide group linkages is the most potent.
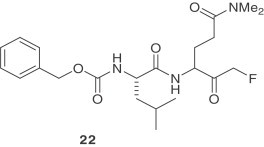
Recently, a series of dipeptidyl fluoromethyl ketones were reported as SARS-CoV 3CLpro inhibitors [29]. The antiviral activity of these compounds was assessed by CPE inhibition in SARS-CoV infected Vero and CaCo-2 cultures. Compound 22 is the most potent inhibitor against SARS-CoV and showed low toxicity in cells. This compound exhibited a cellular-EC50 value of 2.5 μM and a selectivity index of greater than 40.
3.3. SARS-CoV 3CLpro inhibitors from screening
Extensive screening has been carried out in an effort to find structural leads against SARS-CoV 3CLpro from existing drugs. A major advantage is that approved drugs with minimal modifications may have the possibility of gaining accelerated approval by US Food and Drug Administration. It was reported that Kaletra, a mixture of protease inhibitors – Lopinavir and Ritonavir, approved for treating HIV in 2000, shows some effectiveness against the SARS virus [30]. Based on this observation, the binding affinities of six other drugs were investigated against SARS-CoV 3CLpro [31]. These include Lopinavir, Ritonavir, Niclosamide, Promazine and two other HIV inhibitors, PNU and UC2. The preliminary results indicated that these drugs could be useful as templates for designing SARS-protease inhibitors [32].
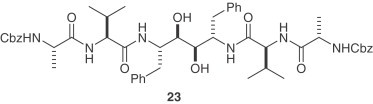
A collection of nearly 10,000 synthetic compounds and natural products was screened in an assay using SARS-CoV and Vero E6 cells [6], [33]. For the SARS-CoV 3CLpro inhibiton assay, a C2-symmetric anti-HIV agent, compound 23 was found to inhibit SARS-CoV 3CLpro with a K i value of 0.6 μM and showed a protective effect in the viral replication assay at a concentration of 10 μM [34]. The docking simulation of 23 showed that it is folded into a ring-like structure in the active site. Along similar lines, a compound library consisting of 960 commercially available drugs and biologically active substances was screened for inhibition of SARS-CoV 3CLpro [35]. Potent inhibition was achieved with the mercury-containing compounds thimerosal, phenylmercuric acetate and hexachlorophene in 1–10 μM range. Each compound inhibited viral replication in Vero E6 cell culture. Detailed mechanistic studies using a fluorescence-based protease assay demonstrated that the three compounds acted as competitive inhibitors (K i=0.7, 2.4 and 13.7 μM for phenylmercuric acetate, thimerosal and hexachlorophene, respectively). However, mercury-containing compounds pose toxicity problems. A panel of other metal ions including Zn2+ and its conjugates were also evaluated for their anti-SARS-CoV 3CLpro activities. Among these, 1-hydroxypyridine-2-thione zinc was shown to be the most potent competitive inhibitor (K i=0.17 μM). The addition of zinc-containing compounds such as (e.g. zinc acetate) as a supplement to the drug for Wilson's disease [36], suggests that zinc ion may be safe for human use.
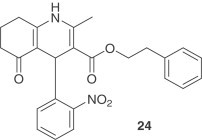
Based on the concept of chemical genetics, 50,240 structurally diverse small molecules were screened yielding 104 compounds with anti-SARS-CoV activities [37]. Compound 24, targeting SARS-CoV 3CLpro showed potent inhibitory activity with an IC50=2.5 μM and an EC50 of 7 μM in the Vero cell-based SARS-CoV plaque reduction assay. Another group of researchers, using a quenched FRET assay with a fully automated system screened 50,000 drug-like molecules, resulting in 572 hits [38]. After applying a series of virtual and experimental filters, five structurally novel molecules were identified which showed potent inhibitory activity (IC50=0.5–7 μM) against SARS-CoV 3CLpro. The inhibitory activities of the five compounds against four different proteases (HAV 3Cpro, NS3pro, chymotrypsin and papain) were also examined with the result that two compounds (25 and 26) showed apparent selectivity for SARS-CoV 3CLpro.
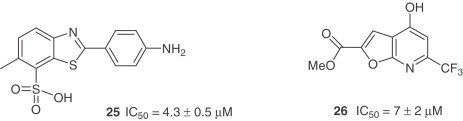
The screening of a database containing structural information for more than 8000 existing drugs identified the serotonin antagonist, cinanserin (SQ 10,643) [39], [40]. Both a homology model and the crystallographic structure of the binding pocket of the SARS-CoV 3CLpro were utilized in these docking studies. Follow up experiments showed that both cinanserin and its hydrochloride bind to SARS-CoV 3CLpro (IC50=4.92 and 5.05 μM, respectively).
Forty compounds emerged from a virtual docking screen after postdock screening filters, including pharmacophore model, consensus scoring and “drug-like” filters were applied. Among the three compounds found to inhibit SARS-CoV 3CLpro was the calmodulin antagonist, calmidazolium (C3930) [41]. It showed a K i value of 61 μM, moreover, calmidazolium is a non-covalent inhibitor.
Some ethacrynic acid derivatives were also tested as non-peptidic covalent inhibitors of the SARS-CoV 3CLpro [42]. An ethacrynic acid amide showed a K i value of 35.3 μM in a fluorimetric assay using a novel FRET pair-labeled substrate.
Finally, three natural products contained in tea, tanic acid, 3-isotheaflavin-3-gallate (TF2B) and theaflavin-3,3′-digallate (TF3) also showed inhibitory properties against SARS-CoV 3CLpro with IC50 values <10 μM [43].
4. PL inhibitors
Numerous studies on the structural and mechanistic aspects of SARS-CoV 3CLpro have provided multiple avenues for structure-based design of antiviral compounds targeted against the 3CLpro active site [9], [16], [44], [45]. On the other hand, structure-based design against the membrane-associated PLpro enzyme, either from SARS-CoV or related coronaviruses, has remained elusive due to lack of structural information. Unlike many coronaviruses that encode two PLpro paralogs (PLP1 or PLP2), SARS-CoV has a single copy of PLpro that cleaves pp1a at three sites at the N terminus to release nsp1, nsp2 and nsp3, respectively [10], [46].
Interestingly, these cleavage sites bear strong resemblance to the C-terminal tail of ubiquitin (consensus sequence LXGG). As a result, it was hypothesized that SARS-CoV PLpro may have de-ubiquitinating activity [47]. Recently, the catalytic domain of PLpro was purified and it was shown that it efficiently disassembles di- and branched polyubiquitin chains, cleaves ubiquitin-7-amino-4-methylcoumarin substrates, and has de-ISGylating activity [48], [49]. It has been reported that SARS-CoV PLpro can be inhibited by the specific de-ubiquitinating enzyme inhibitor, ubiquitin aldehyde with an inhibition constant of 0.21 μM [49]. However, the role of these de-ubiquitinating and de-ISGylating activities in the virus replication cycle remains unclear.
SARS-CoV PLpro is considered an equally viable target to 3CLpro for drug design because both are essential for viral replication. However, PLpro has likely not been pursued because of the paucity of structural information. Recently, the catalytic core of SARS-CoV PLpro was crystallized and its X-ray structure was determined to 1.9 Å[50]. The structure of SARS-CoV PLpro is the first to be elucidated for any coronavirus PLpro. This information should provide significant insight into its de-ubiquitinating function in vitro and expand the available structural templates of SARS-CoV enzymes that can be targeted for the discovery of novel therapeutic compounds that will halt the replication of SARS-CoV.
5. Viral entry inhibitors
Since viral entry into a cell is the first step of viral infection, it is an attractive target for anti-SARS chemotherapy. Blocking the entry of a virus may effectively minimize the chance for the virus to evolve and acquire drug resistance [51], [52]. The S1 domain of the SARS-CoV spike glycoprotein (S) can efficiently bind with a metallopeptidase, angiotensin-converting enzyme 2 (ACE2), at the virus entry step [53], [54]. It was also proved in vivo that the binding of the S1 domain to ACE2 on host cell is responsible for SARS-CoV entry into the cells [55]. Anti-ACE2 antibody showed inhibitory ability toward viral replication on Vero E6 cells [56]. A human IgG1 form of 80R was found to bind the S1 domain of the SARS-CoV S protein (K d=1.59 nM) with a higher affinity comparable to that of ACE2 (K d=1.70 nM), which suggests that the 80R human monoclonal antibody is a useful viral entry inhibitor for SARS treatment [57].
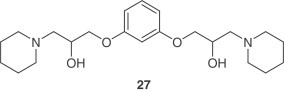
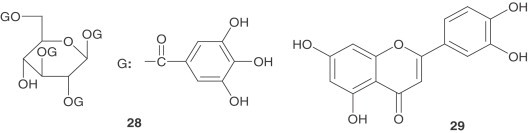
Virtual screening aided in identifying structural leads for viral entry inhibitors of SARS-CoV. Compound 27, which emerged from a 50,240 compound screen and inhibited pseudovirus entry and SARS-CoV plaque formation with EC50 values of 3 μM and 1.6 μM, respectively [37]. A two-step screening of Chinese herbal medicine-based, novel small molecules which bind avidly with the S protein was performed as well. Two virus entry inhibitors, tetra-O-galloyl-β- d-glucose (28) and luteolin ( 29 ) were identified and showed anti-SARS-CoV activities with EC50 values of 4.5 and 10.6 μM, respectively [58].
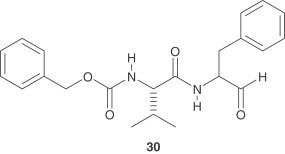
After binding with ACE2, SARS-CoV is taken up into a vesicle inside the cell. Special cellular enzymes (cathepsins) act in the acidic environment inside the vesicle, facilitating fusion of the viral membrane and the vesicle membrane, so that viral proteins and nucleic acids can enter the cell where viral replication occurs [59]. Thus, the cathepsin L inhibitor, MDL28170 ( 30 ) represents an attractive starting point for antiviral therapeutics targeting SARS-CoV entry.
6. Miscellaneous inhibitors
Several compounds have been identified that have shown inhibitory activity against SARS-CoV. However, no information regarding their mechanism of action or the corresponding target is known. Glycyrrhizin showed inhibitory activity for SARS-CoV replication with EC50=300 mg/l after virus absorption in Vero cells [60]. Some glycyrrhizin acid derivatives were found to inhibit SARS-CoV replication in vitro with EC50 values ranging from 5 to 50 μM. Unfortunately, these compounds show high cytotoxity [61]. The viral entry step was suspected to be inhibited by these derivatives. Nitric oxide (NO) has shown an inhibitory effect on some virus infections [62]. An organic NO donor, S-nitroso-N-acetylpenicillamine was shown to inhibit the replication cycle of SARS-CoV in a concentration-dependent manner, probably during the early steps of infection [63]. HIV protease inhibitor nelfinavir [64], antihelminthic drug niclosamide [65] and antimalarial agent chloroquine [66] all showed strong inhibitory activity (EC50=0.048 μM, EC50=1–3 μM, and IC50=8.8±1.2 μM, respectively) against SARS-CoV replication. However, no cytoprotective effect was found for nelfinavir in an independent study [67], [68]. None of the foregoing compounds showed inhibitory activity against SARS-CoV 3CLpro or viral entry. Ribavirin, a broad-spectrum inhibitor of RNA and DNA viruses, was used for treatment of SARS patients [69], but it did not inhibit viral growth at concentrations attainable in human serum. In contrast, interferon (IFN)-α showed an in vitro inhibitory effect starting at concentrations of 1000 IU/ml [70]. Interestingly, the combination of ribavirin and (IFN)-β synergistically inhibited SARS-CoV replication [71].
7. Future outlook
It is with unprecedented rapidity that a basic understanding of SARS-CoV life cycle has been achieved. Already, a number of targets including SARS-CoV 3CLpro and SARS-CoV PLpro appear very promising for anti-SARS-CoV chemotherapy. Since the global outbreak of SARS ended in 2003, only a small number of cases of SARS associated with laboratory exposures have been reported. However, with the identification of Chinese horseshoe bats as an animal reservoir for SARS-CoV, the potential danger of the transfer of this virus to humans still exists. To date, there is no effective therapy for the treatment of SARS in humans. While structure-based design and the screening of compounds have provided a number of promising structural leads for SARS-CoV 3CLpro, potent, low molecular weight inhibitors with less toxicity are needed for development. Interest in structure-based design and screening of SARS-CoV PLpro will increase since the X-ray structure of SARS-CoV PLpro was recently determined. Development of anti-SARS-CoV chemotherapy, based on the viral entry-step mechanism holds promise and requires further exploration. It will be of interest to determine if antivirals directed against SARS-CoV will be effective against the recently identified human coronaviruses NL-63 and HKU1, which cause respiratory infections and pneumonia in children and the elderly. Thus, it is likely that the quest for SARS-CoV chemotherapy will be of relevance to other coronavirus-related ailments.
Acknowledgments
Our research work was supported by a grant from the National Institutes of Health (NIAID) P01 AI060915. We thank Dr. Geoff Bilcer for helpful comments.
References
- 1.Lee N. N. Engl. J. Med. 2003;348:1986. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 2.Ksiazek T.G., Erdman D. N. Engl. J. Med. 2003;348:1953. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Drosten C., Gunther S. N. Engl. J. Med. 2003;348:1967. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 4.Kuiken T., Fouchier R.A. Lancet. 2003;362:263. [Google Scholar]
- 5.Peiris J.S., Lai S.T. Lancet. 2003;361:1319. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marra M.A., Jones S.J. Science. 2003;300:1399. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 7.Rota P.A., Oberste M.S. Science. 2003;300:1394. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 8.Snijder E.J., Bredenbeek P.J. J. Mol. Biol. 2003;331:991. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anand K., Ziebuhr J. Science. 2003;300:1763. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 10.Thiel V., Ivanov K.A. J. Gen. Virol. 2003;84:2305. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 11.Anand K., Palm G.J. EMBO J. 2002;21:3213. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matthews D.A. Proc. Natl. Acad. Sci. USA. 1999;96:11000. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh A.K., Xi K. J. Med. Chem. 2005;48:6767. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 14.Rao Z.H., Yang H.T. Proc. Natl. Acad. Sci. USA. 2003;100:13190. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seipelt J., Guarne A. Virus Res. 1999;62:159. doi: 10.1016/s0168-1702(99)00043-x. [DOI] [PubMed] [Google Scholar]
- 16.James M.N.G., Lee T.W. J. Mol. Biol. 2005;353:1137. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asgian J.L., James K.E. J. Med. Chem. 2002;45:4958. doi: 10.1021/jm025581c. [DOI] [PubMed] [Google Scholar]
- 18.Fang J.M., Liang P.H. Bioorg. Med. Chem. 2005;13:5240. doi: 10.1016/j.bmc.2005.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKeage M.J. Drug Saf. 1995;13:228. doi: 10.2165/00002018-199513040-00003. [DOI] [PubMed] [Google Scholar]
- 20.Woessner K.M. Clin. Rev. Allergy Immunol. 2003;24:149. doi: 10.1385/CRIAI:24:2:149. [DOI] [PubMed] [Google Scholar]
- 21.Venderas J.C., Jain R.P. J. Med. Chem. 2004;47:6113. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 22.Venderas J.C., Jain R.P. Bioorg. Med. Chem. Lett. 2004;14:3655. doi: 10.1016/j.bmcl.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 23.Ramtohul Y., Vederas J.C. J. Org. Chem. 2002;67:3169. doi: 10.1021/jo0157831. [DOI] [PubMed] [Google Scholar]
- 24.Fang J.M., Liang P.H. J. Med. Chem. 2005;48:4469. doi: 10.1021/jm050184y. [DOI] [PubMed] [Google Scholar]
- 25.Liu L.T., Chen S.F., Juang S.H. Bioorg. Med. Chem. Lett. 2005;15:3058. doi: 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webber S.E., Tikhe J. J. Med. Chem. 1996;39:5072. doi: 10.1021/jm960603e. [DOI] [PubMed] [Google Scholar]
- 27.Kuo C.-J., Chi Y.-H. Biochem. Biophys. Res. Commun. 2004;318:862. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freire E., Bacha U. Biochemistry. 2004;43:4906. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 29.Cai S.X., Zhang H.Z. J. Med. Chem. 2006;49:1198. doi: 10.1021/jm0507678. [DOI] [PubMed] [Google Scholar]
- 30.Vastag B. JAMA. 2003;290:1695. doi: 10.1001/jama.290.13.1695. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X.W., Yap Y.L. Bioorg. Med. Chem. 2004;12:2517. doi: 10.1016/j.bmc.2004.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pattaribaman N., Rajnayaranan R.V. Biochem. Biophys. Res. Commun. 2004;321:370. [Google Scholar]
- 33.Wu C.Y., Jan J.T. Proc. Natl. Acad. Sci. USA. 2004;101:10012. [Google Scholar]
- 34.Brik A., Lin Y.C. Chem. Biol. 2002;9:891. doi: 10.1016/s1074-5521(02)00184-9. [DOI] [PubMed] [Google Scholar]
- 35.Liang P.H., Hsu J.T.A. FEBS Lett. 2004;574:116. doi: 10.1016/j.febslet.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brewer G.J., Johnson V.D. Hepatology. 2000;31:364. doi: 10.1002/hep.510310216. [DOI] [PubMed] [Google Scholar]
- 37.Kao R.Y., Tsui W.H.W. Chem. Biol. 2004;11:1293. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown E.D., Eltis L.D. Chem. Biol. 2004;11:1445. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gunther S., Shen X. J. Virol. 2005;79:7095. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubin B., Piala J.J. Arch. Int. Pharmacodyn. Ther. 1964;152:132. [PubMed] [Google Scholar]
- 41.Lai L., Liu Y. J. Chem. Inf. Model. 2005;45:10. doi: 10.1021/ci049809b. [DOI] [PubMed] [Google Scholar]
- 42.Schirmeister T., Kaeppler U. J. Med. Chem. 2005;48:6832. doi: 10.1021/jm0501782. [DOI] [PubMed] [Google Scholar]
- 43.Chen C.N., Lin K.P.C. eCAM. 2005;2:209. doi: 10.1093/ecam/neh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang H., Rao Z. Proc. Natl. Acad. Sci. USA. 2003;100:13190. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harcourt B.H., Baker S.C. J. Virol. 2004;78:13600. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sulea T., Menard R. J. Virol. 2005;79:4550. doi: 10.1128/JVI.79.7.4550-4551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barretto N., Baker S.C. J. Virol. 2005;79:15189. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindner H.A., Menard R. J. Virol. 2005;79:15199. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ratia K., Mesecar A.D. Proc. Natl. Acad. Sci. USA. 2006;103:5717. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore J.P., Doms R.W. Proc. Natl. Acad. Sci. USA. 2003;100:10598. doi: 10.1073/pnas.1932511100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Este J.A. Curr. Med. Chem. 2003;10:1617. doi: 10.2174/0929867033457098. [DOI] [PubMed] [Google Scholar]
- 53.Jenwitheesuk E., Samudrala R. BMC Struct. Biol. 2003;3:2. doi: 10.1186/1472-6807-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tipnis S.R. J. Biol. Chem. 2000;275:33238. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 55.Penninger J.M., Kuba K. Nat. Med. 2005;11:875. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li W., Moore M.J. Nature. 2003;426:450. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marasco W.A., Sui J. Proc. Natl. Acad. Sci. USA. 2004;101:2536. [Google Scholar]
- 58.Deng H.K., Xu X.J. J. Virol. 2004;78:11334. [Google Scholar]
- 59.Simmons G., Bates P. Proc. Natl. Acad. Sci. USA. 2005;102:11876. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cinati J., Jr., Morgenstern B. Lancet. 2003;361:2045. doi: 10.1016/S0140-6736(03)13615-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cinati J., Jr., Hoever G. J. Med. Chem. 2005;48:1256. doi: 10.1021/jm0493008. [DOI] [PubMed] [Google Scholar]
- 62.Lane T.E., Paoletti A.D. J. Virol. 1997;71:2202. doi: 10.1128/jvi.71.3.2202-2210.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mirazimi A., Leijon M. J. Virol. 2005;79:1966. doi: 10.1128/JVI.79.3.1966-1969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamamoto N., Yang R. Biochem. Biophys. Res. Commun. 2004;318:719. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hsu J.T.A., Wu C. Antimicrob. Agents Chemother. 2004;48:2693. doi: 10.1128/AAC.48.7.2693-2696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ranst M.V., Keyaerts E. Biochem. Biophys. Res. Commun. 2004;323:264. doi: 10.1016/j.bbrc.2004.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tan E.L., Stanton L.W. Emerg. Infect. Dis. 2004;10:581. doi: 10.3201/eid1004.030458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu Y. Biochem Biophys Res Commun. 2005;333:194. doi: 10.1016/j.bbrc.2005.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koren G., King S. CMAJ. 2003;168:1289. [PMC free article] [PubMed] [Google Scholar]
- 70.Feldmann H., Li Y. J. Infect. Dis. 2004;189:1164. doi: 10.1086/382597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cinati J., Jr., Morgenstern B. Biochem. Biophys. Res. Commun. 2005;326:905. doi: 10.1016/j.bbrc.2004.11.128. [DOI] [PMC free article] [PubMed] [Google Scholar]