

Abstract
Background
More than 90% of Congenital Adrenal Hyperplasia (CAH) cases are associated with mutations in the 21-hydroxylase gene (CYP21A2) in the HLA class III area on the short arm of chromosome 6p21.3. In this region, a 30 kb deletion produces a non functional chimeric gene with its 5' and 3' ends corresponding to CYP21A1P pseudogene and CYP21A2, respectively. To date, five different CYP21A1P/CYP21A2 chimeric genes have been found and characterized in recent studies. In this paper, we describe a new CYP21A1P/CYP21A2 chimera (CH-6) found in an Italian CAH patient.
Methods
Southern blot analysis and CYP21A2 sequencing were performed on the patient. In addition, in order to isolate the new CH-6 chimeric gene, two different strategies were used.
Results
The CYP21A2 sequencing analysis showed that the patient was homozygote for the g.655C/A>G mutation and heterozygote for the p.P30L missense mutation. In addition, the promoter sequence revealed the presence, in heterozygosis, of 13 SNPs generally produced by microconversion events between gene and pseudogene. Southern blot analysis showed that the woman was heterozygote for the classic 30-kb deletion producing a new CYP21A1P/CYP21A2 chimeric gene (CH-6). The hybrid junction site was located between the end of intron 2 pseudogene, after the g.656C/A>G mutation, and the beginning of exon 3, before the 8 bp deletion. Consequently, CH-6 carries three mutations: the weak pseudogene promoter region, the p.P30L and the g.655C/A>G splice mutation.
Conclusion
We describe a new CYP21A1P/CYP21A2 chimera (CH-6), associated with the HLA-B15, DR13 haplotype, in a young Italian CAH patient.
Background
Congenital Adrenal Hyperplasia (CAH) is an autosomal recessive disorder mainly caused by defects in the steroid 21-hydroxylase gene (CYP21A2). Steroid 21-hydroxylase is a microsomal cytochrome P450 required for the synthesis of cortisol and aldosterone but not for the synthesis of sex steroids. The reduced synthesis of cortisol and aldosterone leads to excessive androgen production [1].
CAH includes a wide spectrum of clinical manifestations. Milder forms are referred to as Nonclassic (NC) or late-onset CAH [2]. Patients with the severe classic disease form are classified as either salt-wasting (SW) or simple-virilising (SV) depending on whether or not synthesis of the salt retaining hormone, aldosterone, is affected [3].
The gene encoding 21-hydroxylase, CYP21A2, is located in the HLA class III region on the short arm of chromosome 6p21.3 [4]. In this region, four tandemly arranged genes – serine/threonine Kinase RP, complement C4, steroid 21-hydroxylase CYP21, and tenascin TNX – are organized as a genetic unit designated as a RCCX module. In a RCCX bimodular haplotype, duplication of the RCCX module occurs and the orientation of genes, from telomere to centromere, is: RP1-C4A-CYP21A1P-TNXA-RP2-C4B-CYP21A2-TNXB. The three pseudogenes, CYP21A1P-TNXA and RP2, located between the two C4 loci, do not encode functional proteins [5,6].
In the Caucasian population, bimodular and monomodular RCCX organizations are present in about 69% and 17% of chromosome 6, respectively, while trimodular RCCX haplotypes have a frequency of about 14% [7]. The CYP21A2 gene and CYP21A1P pseudogene each contain 10 exons spaced over 3.1 Kb, their nucleotide sequences are 98% identical in exons and approximately 96% identical in introns [8]. Intergenic recombinations are responsible for 95% of the mutations associated with 21-hydroxylase deficiency; the remaining 5% of mutations do not appear to be the result of gene conversion events [9,10]. Among the intergenic recombinations, approximately 75% is represented by mutations normally present in the pseudogene and possibly transferred to the functional gene by microconversion events [11]. The remaining 20%–25% of mutations are CYP21A2 gene deletions or CYP21A1P/CYP21A2 chimeric genes. In fact, the 26 or 32 Kb deletion (depending on whether C4B is the long or short gene), involving the 3' end of CYP21A1P, all of the C4B gene, and the 5' end of CYP21A2, produces a single non functional chimeric gene with its 5' and 3' ends corresponding to CYP21A1P and CYP21A2, respectively [1,11]. To date, five different chimeric CYP21A1P/CYP21A2 genes have been found and characterized in recent studies [12-16]. In this paper, we describe a new CYP21A1P/CYP21A2 chimeric gene (CH-6) found in an Italian patient suffering from a severe form of CAH.
Methods
Patients
The patient is the first daughter of non consanguineous parents of Italian origin. No family history of CAH, of virilisation in female family members or of impaired fertility was reported. Pregnancy and delivery were uneventful and birth weight and length were normal (3400 g, 51 cm). Diagnosis of CAH was suspected due to genital virilisation (Prader IV). Thus, hormonal evaluation and kariotype were requested. Plasma levels of Na and K were 124 mEq/L and 6.2 mEq/L, respectively, while 17-OHP plasma value, on fourth day of life, was 223 nmol/L. Kariotype showed a normal female pattern (46, XX). Therapy with betamethasone and fluorohydrocortisone was started on the 11th day. No vomiting or other clinical signs of salt wasting were shown before onset of substitutive therapy.
Clitoromegaly was corrected at 4 yr of age and hydrocortisone therapy was started at 6 yr. The patient showed a normal pubertal development with menarche at 14 yr and she underwent vaginoplasty at 17 yr. She married at 26 yr with a healthy Italian male and she was pregnant at 29 yr.
Molecular analysis of CYP21A2 gene
Informed consent for genetic study was obtained, it was in compliance with the Helsinki Declaration and was approved by Catholic University Ethics Committee (Reference Number: P6242008).
Genomic DNA was isolated from peripheral blood samples using High Pure PCR Template Preparation Kits (Roche Diagnostic, USA), quantified by spectrophotometer at 260 nm and stored at -20°C until use. In addition, since the patient decided to undergo an amniocentesis at 18th week of gestation, a DNA sample was extracted from amniotic fluid cells.
Two genomic DNA fragments, spanning from the 5' region to exon 6 (FRAG 1) and from exon 6 to 3'region (FRAG 2) of the active CYP21A2 gene, were amplified using two sets of specific primers, as previously described [17]. In addition, the CYP21A2 promoter region was amplified using the primers: PROM-F 5'-gca ggg act gcc att ttc tc-3' (nucleotides 87034–87053; GeneBank accession AL049547) and PROM-R 5'-agc agg gag tag tct ccc aag-3'(nucleotides 85935–85955; GeneBank accession AL049547).
PCR-amplified fragments were directly sequenced using BigDye Terminator Cycle Sequencing kit v3.1(Applied Biosystems, USA) and ABI 3100 Avant Genetic Analyser (Applied Biosystems, USA) according to manufacturer's instructions.
The results were analysed using the SeqScape v2.5 software package (Applied Biosystems, USA). The CYP21A2 sequence references were: NCBI-AL049547 and NC-000006.
HLA typing
HLA-B and HLA-DRB1 genotypes were determined using a semi-automated, commercially available reverse dot-blot method (Inno-lipa, Innogenetics, Gent, Belgium), according to the manufacturer's instructions. Reaction patterns were interpreted using INNO-LiPA software.
CYP21/C4 haplotyping
Leukocyte DNA was digested with TaqI restriction enzyme and Southern blotting studies were conducted as previously described [18]. Blots were probed with a mixture of two fragments: a 500-bp BamHI-KpnI 5' fragment of C4 cDNA (pAT-A clone) and a 3.1-Kb genomic EcoRI-BamHI fragment of the 5.5-Kb BglII-BamHI fragment encompassing the entire CYP21A2 gene cloned in the BamHI site of bluscript SK+ plasmid [18,19].
Band ratios were measured by laser densitometry (Ultroscan XL laser densitometer; Pharmacia LKB).
Isolation of CYP21A1P/CYP21A2 chimeric gene
In order to isolate the chimeric CYP21A1P/CYP21A2 allele, two different strategies were used. The first was an allele specific PCR performed using the primers: PROMPS 5'-cca ggt cgg ggc gga cac cc-3' (nucleotides 433–452; GeneBank accession NC_000006.10 [region: 32.080.697...32.084.739]) and PROM-R (nucleotides 85935–85955; GeneBank accession AL049547). The PROMPS forward primer was specific for a CYP21A1P pseudogene 5' sequence while the reverse PROM-R primer was specific for exon 3 of the CYP21A2 active gene. The PCR fragment, spanning from the 5' region to exon 3 of the chimeric gene, was directly sequenced.
The second strategy was the amplification (Expand Long Template PCR System, Roche Diagnostic, USA), using CYP779f and Tena36F2 primers, of a 6.2Kb fragment encompassing the 5'-end of the CYP21A2 gene and exon 36 of the TNXB gene [20]. After amplification, 700 ng of the PCR product was incubated at 65°C for 2 h with 10 U of Taq I restriction enzyme. The completely digested PCR product was analyzed by electrophoresis on a 1.2% agarose gel to evaluate the presence of the 3.7- and 3.2-Kb fragments produced by the active CYP21A2 gene and the chimeric CYP21A1P/CYP21A2 gene, respectively [20]. The 3.2 Kb fragment including the whole CYP21A1P/CYP21A2 chimera [being generated by the Taq I cutting at 86870 position (GeneBank accession AL049547), corresponding to -209 C nucleotide in the CYP21A1P promoter region, and at 83662 position (GeneBank accession AL049547) within 5' region of TNXB gene] was isolated from agarose gel (QIAEX II Gel Extraction Kit, Qiagen, Hilden, Germany) and sequenced using internal primers (available on request). In order to confirm the obtained results the 6.2 kb fragment was cloned into the pGEM-T vector system (Promega, Madison, WI, USA) and the identified clone was directly used as template for DNA sequencing.
Results
Molecular analysis of CYP21A2 gene
The CYP21A2 sequencing analysis showed that the patient was homozygote for the g.655C/A>G mutation in intron 2 and heterozygote for the p.P30L missense mutation in exon 1. In addition, the promoter sequence revealed the presence, in heterozygosis, of 13 variants (g.-306G>C, g.-295T>C, g.-294A>C, g.-283A>G, g.-281T>G, g.-210T>C, g.-199C>T, g.-190insT, g.-126C>T, g.-113G>A, g.-110T>C, g.-103A>G and g.-4C>T) which are generally the result of microconversion events between gene and pseudogene. In this first sequencing analysis step, we were unable to perform the complete nucleotide analysis of intron 2 since the electropherogram showed various frameshift mutations resulting in two overlapping sequences.
The same problem was present when the fetus'CYP21A2 sequence was analyzed. However, the results showed that the fetus' DNA carried, in heterozigiosis, both the g.656C/A>G and p.P30L mutations in addition to the 13 variants in the promoter region.
Finally, the patient's husband resulted wild type for the CYP21A2 sequencing analysis.
HLA typing
The results of HLA-DRB and HLA-B genotypes were as follows: patient: HLA-B*35/44 HLA-DRB1*01/13; husband: HLA-B*15/51 HLA-DRB1*11/13; male fetus: HLA-B*15/44 HLA-DRB1*01/13. These results confirmed the correct segregation of HLA loci.
CYP21/C4 haplotyping
The Southern blot results are shown in Fig. 1 and were interpreted as follows: 1) the fetus' father (lane 1) carried two bimodular chromosomes with a total of two CYP21A2 genes (3.7 kb and 2.5 kb bands), two CYP21A1P pseudogenes (3.2 kb and 2.4 kb bands), two long telomeric C4 genes(7.0 kb band) and two short non-telomeric C4 genes (5.4 kb band); the patient (lane 2) carried a chromosome with a normal bimodular structure (one CYP21A2 gene, one CYP21A1P pseudogene, one long telomeric C4 gene and one long non-telomeric C4 gene corresponding to 6.4 kb band) and a chromosome with the classic 30 Kb deletion (as shown by decreased intensities of 6.4 kb band, corresponding to the C4B gene, and of 3.7 kb and 2.4 kb bands corresponding to the 5' section of the CYP21A2 active gene and to the 3' fragment of the CYP21A1P pseudogene, respectively); finally, the fetus (lane 3) inherited from the father a wild type bimodular chromosome with a short non-telomeric C4 gene and from the mother the chromosome with the 30 kb deletion (as shown by the absence of the 6.4 kb band and by the decreased intensities of 3.7 kb and 2.4 kb bands).
Figure 1.
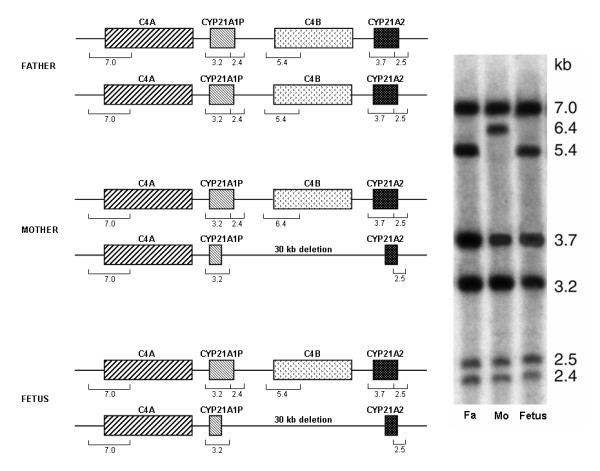
Southern blot analysis. Taq I restriction patterns of genomic DNA hybridized to a mixture of the CYP21A2 and C4 cDNA probes. Fa: father, Mo: mother. For pattern analysis see the text in results section.
Sequence of CYP21A1P/CYP21A2 chimeric gene
The 6.2 Kb long PCR digested with TaqI enzyme[20] combined with cloning and sequencing of 3.2 Kb fragment allowed to prove the existence of the new chimeric gene. In fact, the sequencing analysis showed that CH-6 gene have a CYP21A1P-like sequence up to 656 nucleotide of intron 2 while the remaining exons/intons have a wild type CYP21A2 sequence (Fig. 2).
Figure 2.

Sequence of the new chimeric gene. Sequence of CH-6 chimera from +1 nucleotide (exon 1) to +721 nucleotide (exon 3). Nucleotides were numbered starting from ATG. The lines showed: the amino acid sequence of CYP21A2 gene (M13936), the CYP21A2 nucleotide sequence (M13936), the CYP21A1P nucleotide sequence (M13935) and the CH-6 nucleotide sequence. Only nucleotide differences between CYP21A1P or CH-6 and CYP21A2 active gene are shown under corresponding nucleotides in CYP21A2 gene.
Discussion
The chimeric CYP21A1P/CYP21A2 gene is the consequence of the 26 or 32 Kb deletion including the complete XA, RP2, and C4B genes and the partial sequences of CYP21A1P and CYP21A2 genes [20].
The possible cause of the chimera formation is the presence of specific sequences, such as Chi-like and tandem-repetitive minisatellite consensus, which play a role in promoting genetic recombination [21]. In particular, it has been pointed out that the chi-like sequence GCTGGGG is present several times in the intronic regions of CYP21A2 and CYP21A1P genes [22]. However, the high degree of sequence homology of RP1-C4A-CYP21A1P-XA-RP2-C4B-CYP21A2-TNXB genes arranged in tandem, seems to be the most probable way to increase the chance of misalignment at meiosis to generate genetic recombination in the 6p21.3 chromosome area [23].
To date, five different chimeric CYP21A1P/CYP21A2 genes have been found and characterized [12-16] (Fig. 3). Three of them, found in ethnic Chinese (Taiwanese), are named CH-1, CH-2, and CH-3 [12-14]. In these molecules, the deletion g.707–724delGAGACTAC in exon 3 leads to a frameshift mutation which forms a TGA stop codon downstream 830 nucleotide producing a truncated protein.
Figure 3.

CYP21A1P/CYP21A2 chimeric genes. CH-1, CH-2, CH-3, CH-4, CH-5 and CH-6 represent six distinct chimeric CYP21A1P/CYP21A2 genes as described in the text. The black and white regions represent the non-functional CYP21A1P and the functional CYP21A2 sequences, respectively. The eight nucleotide deletion in exon 3 is marked by an asterisk.
CH-4 chimera was found in a Caucasian NC-CAH patient [15] and it is probably the same one earlier reported by Killeen et al. [24]. CH-4 junction site was located between the end of exon 1 and the beginning of intron 2, consequently, the resulting hybrid gene differs from the functional gene only for the presence of two deleterious mutations: the weak promoter region of the pseudogene and the p.P30L missense mutation in exon 1. Finally, CH-5 chimera, associated with the HLA-B47, DR7 haplotype, was described by White et al. [16] and results very common among CAH patients of Caucasian origin.
In this paper, we report a new CYP21A1P/CYP21A2 chimera (CH-6) found in an Italian woman suffering from salt-wasting form of CAH. The hybrid junction site was located between the end of intron 2 pseudogene, after the g.656C/A>G mutation, and the beginning of exon 3, before the 8 bp deletion. Consequently, CH-6 carries three mutations: the weak pseudogene promoter region, the p.P30L in exon 1 and the g.655C/A>G splice mutation in intron 2 (Fig. 3). Since the patient carried a second CYP21A2 allele with the g.656C/A>G mutation, her clinical severe phenotype was determined by the homozygote status for this mutation. Unfortunately, it was not possible to perform a more extensive familiar genetic study and therefore we can not determine if the patient inherited the new CH-6 chimeric gene from one of her parents.
However, our molecular CAH prenatal diagnosis showed that the fetus inherited the new chimeric gene from his mother and a wild type CYP21A2 allele from his father, as also confirmed by HLA typing.
Conclusion
In conclusion, we describe a new CYP21A1P/CYP21A2 chimera (CH-6), associated with the HLA-B15, DR13 haplotype, in a young Italian CAH patient.
Competing interests
The authors declare that they have no competing interests.
The study was carried out by means of funding of Institute of Biochemistry and Clinical Biochemistry, Catholic University of Rome.
Authors' contributions
PC carried out the molecular genetics studies, participated in the sequence alignment and drafted the manuscript.
EM participated in the molecular genetics study and in the sequence alignment.
AM participated in the molecular genetics studies and in the sequence alignment.
EG participated in the molecular genetic studies.
CZ participated in the design of the study and in its coordination.
VT performed clinical study and participated in the design of the study and in its coordination.
EC participated in the design of the study and drafted the manuscript.
All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
Written consent was obtained from the patients and their relative for publication of study.
Contributor Information
Paola Concolino, Email: paolaconcolino78@libero.it.
Enrica Mello, Email: enricamel@libero.it.
Angelo Minucci, Email: angelo.minucci@virgilio.it.
Emiliano Giardina, Email: emilianogiardina@uniroma2.it.
Cecilia Zuppi, Email: cecilia.zuppi@rm.unicatt.it.
Vincenzo Toscano, Email: v.toscano@uniroma1.it.
Ettore Capoluongo, Email: ecapoluongo@rm.unicatt.it.
References
- White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245–291. doi: 10.1210/er.21.3.245. [DOI] [PubMed] [Google Scholar]
- Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. 2009;10:77–82. doi: 10.1007/s11154-008-9097-x. [DOI] [PubMed] [Google Scholar]
- Bachelot A, Chakthoura Z, Rouxel A, Dulon J, Touraine P. Classical forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency in adults. Horm Res. 2008;69:203–11. doi: 10.1159/000113020. [DOI] [PubMed] [Google Scholar]
- White PC, Grossberger D, Onufer BJ, Chaplin DD, New MI, Dupont B, Strominger JL. Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci USA. 1985;82:1089–1093. doi: 10.1073/pnas.82.4.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular variations of the human major histocompatibility complex class III genes for serine/threonine kinase RP, complement component C4, steroid 21-hydroxylase CYP21, and tenascin TNX (the RCCX module). A mechanism for gene deletions and disease associations. J Biol Chem. 1999;274:12147–12156. doi: 10.1074/jbc.274.17.12147. [DOI] [PubMed] [Google Scholar]
- Haglund-Stengler B, Martin Ritzen E, Gustafsson J, Luthman H. Haplotypes of the steroid 21-hydroxylase gene region encoding mild steroid 21-hydroxylase deficiency. Proc Natl Acad Sci USA. 1991;88:8352–8356. doi: 10.1073/pnas.88.19.8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, Zipf WB, Rennebohm RM, Yung Yu C. Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. J Exp Med. 2000;191:2183–96. doi: 10.1084/jem.191.12.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA. 1986;83:5111–5. doi: 10.1073/pnas.83.14.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werkmeister JW, New MI, Dupont B, White PC. Frequent deletion and duplication of the steroid 21-hydroxylase genes. Am J Hum Genet. 1986;39:461–9. [PMC free article] [PubMed] [Google Scholar]
- Lee HH. CYP21 mutations and congenital adrenal hyperplasia. Clin Genet. 2001;59:293–301. doi: 10.1034/j.1399-0004.2001.590501.x. [DOI] [PubMed] [Google Scholar]
- Tusié-Luna MT, White PC. Gene conversions and unequal crossovers between CYP21 (steroid 21-hydroxylase gene) and CYP21P involve different mechanisms. Proc Natl Acad Sci USA. 1995;92:10796–800. doi: 10.1073/pnas.92.23.10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH. The chimeric CYP21P/CYP21 gene and 21-hydroxylase deficiency. J Hum Genet. 2004;49:65–72. doi: 10.1007/s10038-003-0115-2. [DOI] [PubMed] [Google Scholar]
- Lee HH, Lee YJ, Chan P, Lin CY. Use of PCR-based amplification analysis as a substitute for the southern blot method for CYP21 deletion detection in congenital adrenal hyperplasia. Clin Chem. 2005;51:480. doi: 10.1373/clinchem.2004.045872. [DOI] [PubMed] [Google Scholar]
- Lee HH, Chang SF, Lee YJ, Raskin S, Lin SJ, Chao MC, Lo FS, Lin CY. Deletion of the C4-CYP21 repeat module leading to the formation of a chimeric CYP21P/CYP21 gene in a 9.3-kb fragment as a cause of steroid 21-hydroxylase deficiency. Clin Chem. 2003;49:319–22. doi: 10.1373/49.2.319. [DOI] [PubMed] [Google Scholar]
- L'Allemand D, Tardy V, Grüters A, Schnabel D, Krude H, Morel Y. How a patient homozygous for a 30-kb deletion of the C4-CYP 21 genomic region can have a nonclassic form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2000;85:4562–7. doi: 10.1210/jc.85.12.4562. [DOI] [PubMed] [Google Scholar]
- White PC, New MI, Dupont B. HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proc Natl Acad Sci USA. 1984;81:7505–9. doi: 10.1073/pnas.81.23.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concolino P, Satta MA, Santonocito C, Carrozza C, Rocchetti S, Ameglio F, Giardina E, Zuppi C, Capoluongo E. Linkage between I172N mutation, a marker of 21-hydroxylase deficiency, and a single nucleotide polymorphism in Int6 of CYP21B gene: a genetic study of Sardinian family. Clin Chim Acta. 2006;364:298–302. doi: 10.1016/j.cca.2005.07.020. [DOI] [PubMed] [Google Scholar]
- Morel Y, Miller WL. Clinical and molecular genetics of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Adv Hum Genet. 1991;20:1–68. doi: 10.1007/978-1-4684-5958-6_1. [DOI] [PubMed] [Google Scholar]
- Belt KT, Carroll MC, Porter RR. The structural basis of the multiple forms of human complement component C4. Cell. 1984;36:907–14. doi: 10.1016/0092-8674(84)90040-0. [DOI] [PubMed] [Google Scholar]
- Lee HH, Tsai FJ, Lee YJ, Yang YC. Diversity of the CYP21A2 gene: A 6.2-Kb TaqI fragment and a 3.2-Kb TaqI fragment mistaken as CYP21A1P. Molecular Genetics and Metabolism. 2006;88:372–377. doi: 10.1016/j.ymgme.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Smith GR, Kunes SM, Schultz DW, Taylor A, Triman KL. Structure of chi hotspots of generalized recombination. Cell. 1981;24:429–436. doi: 10.1016/0092-8674(81)90333-0. [DOI] [PubMed] [Google Scholar]
- Chu X, Braun-Heimer L, Rittner C, Schneider PM. Identification of the recombination site within the steroid 21-hydroxylase gene (CYP21) of the HLA-B47, DR7 haplotype. Exp Clin Immunogenet. 1992;9:80–5. [PubMed] [Google Scholar]
- Koppens PF, Smeets HJ, de Wijs IJ, Degenhart HJ. Mapping of a de novo unequal crossover causing a deletion of the steroid 21-hydroxylase (CYP21A2) gene and a non-functional hybrid tenascin-X (TNXB) gene. J Med Genet. 2003;40:e53. doi: 10.1136/jmg.40.5.e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killeen AA, Sane KS, Orr HT. Molecular and endocrine characterization of a mutation involving a recombination between the steroid 21-hydroxylase functional gene and pseudogene. J Steroid Biochem Mol Biol. 1991;38:677–86. doi: 10.1016/0960-0760(91)90078-J. [DOI] [PubMed] [Google Scholar]