

Abstract
Mitochondria have been shown to play an important role in cell death in mammalian cells. However, the importance of mitochondria in Drosophila apoptosis is still under investigation. Many proteins involved in the regulation of apoptosis in mammals act at mitochondria or are released from mitochondria, resulting in caspase activation. In addition, these organelles undergo significant ultrastructural changes during apoptosis. This review highlights similarities and differences in the roles of mitochondria and mitochondrial factors in apoptosis between Drosophila and mammals. In Drosophila, many key regulators of apoptosis also appear to localize to this organelle, which also undergoes ultrastructural changes during apoptosis. Although many of the proteins important for the control of apoptosis in mammalian cells are conserved in Drosophila, the role that mitochondria play in apoptosis in this model system remains an area of controversy and active research.
Many proteins important for apoptosis are highly conserved in organisms from C. elegans and Drosophila to mammals [1]. The core elements of the apoptotic machinery, including caspases, the adaptor proteins Apaf-1/Dark/Ced4, and the Bcl-2 family of proteins are present in all of these organisms. However, the mechanisms of caspase activation seem to show surprising distinctions. The role of the mitochondria in cell death is arguably the feature that is the most divergent between flies and mammals. In this review we will highlight both commonalities and unique aspects of mitochondrial participation in cell death, as well as new insights gained into the role of mitochondria in a variety of cell death paradigms.
Activation of caspases is important for most developmental cell death in the fly [2]. The Drosophila genome encodes seven caspases, including three long pro-domain containing “initiator” caspases and four effector caspases. The initiator caspase Dronc is required for most developmental apoptosis [3]. Dronc associates with the Apaf-1-like protein Dark, and becomes activated, in turn activating the effector caspases [4–6]. Drice and Dcp-1 are the major effector caspases in developmental apoptosis [7]. Current models suggest that Dronc activation is constitutive in most cells [2]. Caspase activity is held in check in living cells by DIAP1, a member of the Inhibitor of Apoptosis Protein family, which can inhibit caspase activity through direct binding and by ubiquitylation [8]. Developmental cell death in the fly is activated by the proteins Reaper (Rpr), Hid, Grim, and Sickle (Skl), which bind to DIAP1 and inhibit DIAP1’s caspase inhibitory function [1]. We discuss additional functions of these proteins below.
In contrast to the constitutive activation of caspases in the fly, caspase activation in mammalian cells is a major control point in cell death [9]. Caspase activation pathways can be simplistically divided into extrinsic and intrinsic mechanisms. Ligation of cell surface receptors activates the extrinsic pathway, while activation of the intrinsic pathway requires factors released from the mitochondria. The initiation of the intrinsic pathway involves Bax and Bak, proapoptotic members of the Bcl-2 family of apoptotic regulators. In the doomed cell, these proteins become localized to mitochondria, resulting in mitochondrial permeabilization. Cytochrome C, normally found in the intermembrane space of the mitochondria, is released to the cytosol where it can bind to the Apaf-1 adaptor protein. Apaf-1 binds Cytochrome C, dATP and Caspase-9, to form a large mulitmeric complex called the apoptosome [10]. Caspase-9 is activated in the apoptosome, and then activates effector Caspases 3, 6, and 7 to result in the apoptotic death of the cell.
At first glance, the differences between the Drosophila and mammalian cell death pathways are striking, especially with regard to the role of mitochondria. However, on closer examination there are considerable similarities in terms of the morphological changes in mitochondria, the role of the mitochondrial fission and fusion machinery in these changes, and in the proteins released from mitochondria during cell death. What remains to be further explored is how important these mitochondrial changes are to developmental apoptosis in the fly.
Structural changes in mitochondria during cell death
Mitochondria undergo characteristic changes during cell death. These include swelling and fragmentation of the mitochondrial network [11]. Swelling is likely a response to the permeabilization of the outer mitochondrial membrane, and can be observed in fly cells as well as in mammalian cells [12–14]. Mitochondrial membrane permeabilization occurs in a caspase independent fashion in mammals [15], although there is some controversy in this regard [16]. In contrast, ultrastructural changes, such as swelling, are caspase dependent in fly cells [12, 17]. It is interesting to note that alterations in mitochondrial structure are associated with spermatid individualization, a caspase dependent process that requires Cytochrome C (see below) [18]. Alterations in mitochondrial cristae were also reported in apoptotic flight muscles in aging or mutant Drosophila [19].
Mitochondria are highly dynamic organelles, undergoing continuous fission and fusion in living cells. This process is essential for normal mitochondrial function [20]. Several Dynamin-like GTPases are involved in mitochondrial dynamics, including Drp-1, which is important for mitochondrial fission, Opa-1, and the mitofusins, Marf and Fuzzy Onions in flies [21, 22] and MFN-1 and MFN-2 in mammals [11], which are involved in mitochondrial fusion (Fig 1). Increased mitochondrial fragmentation is seen during apoptosis in mammals, worms and flies [13, 23–25]. This fragmentation has been shown to involve Drp-1. Expression of a dominant negative Drp-1 or knock down of Drp-1 with RNAi or mutation results in decrease mitochondrial fragmentation after apoptosis induction in all three models [13, 23, 24, 26].
Figure 1. Drp-1 RNAi induces mitochondrial fusion in S2 cells.
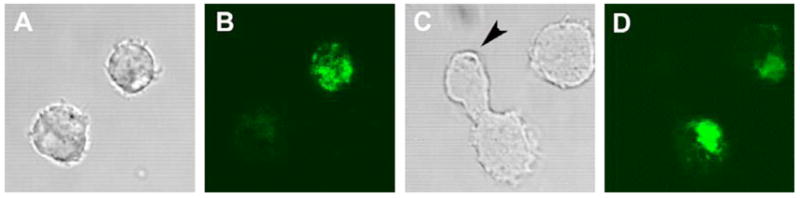
S2 cells were transfected with double-stranded RNA for Drp-1 or a control and mitochondrial localized YFP, incubated for 3 days, and imaged by confocal microscopy. A,B) mitochondrial localized GFP shows a reticular distribution of mitochondria throughout the cell. C,D) Knock down of Drp-1 results in mitochondrial clumping. It is interesting to note that mitochondria do not seem to segregate properly in diving cells (arrow).
The easily visualized alterations in mitochondrial morphology and dynamics in dying cells suggest that these processes could be involved in cell death. Alternatively, these changes could be secondary to other changes in the dying cell, and not play a critical role. One argument that mitochondrial fragmentation is not secondary to other changes in the dying cell is that it occurs prior to caspase activation in fly cells [13]. In addition, inhibition of caspase activity does not block mitochondrial fragmentation in worms, flies or mammalian cells [13, 23, 24].
The best evidence that mitochondrial dynamics play a role in cell death is the finding that inhibition of Drp-1, either by expression of a dominant negative Drp-1, RNAi knock down, or by mutation, results in an inhibition of cell death in flies as well as worms and mammalian cells [12, 13, 23, 24, 27]. However, these experiments do not rule out a second function for Drp-1 in apoptosis, independent of its effects on mitochondrial fission. Recent data suggest that inhibition of mitochondrial fission might have no effect or only delay cell death in mammalian cells [28, 29]. Modulation of mitochondrial dynamics by genetic manipulation of Drp-1 and other genes involved in mitochondrial fusion and fission in the worm suggests that Drp-1 and Fis-2 can inhibit cell death downstream of activated caspase [26]. In the fly, inhibition of Drp-1 clearly inhibits caspase activation [12, 13]. Although not necessarily contradictory, these data demonstrate the need for further investigation into how mitochondrial fission contributes to cell death.
There are several models for how mitochondrial fission might facilitate cell death. The fission machinery may contribute to the release of proapoptotic molecules, such as Cytochrome C, from mitochondria, as discussed below [11]. Alternatively, mitochondrial fission might contribute to mitochondrial membrane permeabilization leading to disruption of mitochondrial function. A third possibility is that sites of mitochondrial fission might serve as scaffolding for the localization of other proteins important in apoptosis induction, as is seen with the Bcl-2 family member Bax in mammalian systems [30].
Do Drosophila Bcl-2 proteins, Buffy and Debcl act at the mitochondria to regulate cell death?
In mammalian systems, the Bcl-2 family of proteins has been shown to play a role in mitochondrial dynamics as well as regulation of apoptosis. Members of the Bcl-2 family of apoptosis regulators co-localize with mitochondria, and Bax and Bak promote mitochondrial fission [31]. Bax rapidly translocates to mitochondria on apoptosis induction, where it co-localizes with Drp-1 [30]. However, recent work suggests that the role for Bax and Bak in mitochondrial dynamics may be independent of their role in induction of apoptosis [29].
Two members of the Bcl-2 family of apoptotic regulators have been identified in Drosophila, Buffy and Debcl. Both of these proteins have been shown to contain domains that can anchor them to membranes [32]. Interestingly, Debcl has been shown to localize to mitochondria, while Buffy localizes to endoplasmic reticulum [32]. Studies based on overexpression and knockdown by RNAi suggest that Debcl has a proapoptotic function [25, 33–36], while Buffy is antiapoptotic [37, 38], although one study has found that Debcl is protective against poly-glutamine induced death in the nervous system, which is antagonized by Buffy expression [39].
Recent genetic studies have clarified the roles of the fly Bcl2 genes in apoptosis [25, 40]. Mutant analysis confirms a proapoptotic function for Debcl in developmental apoptosis [25]. After irradiation, one study has shown that Debcl mutants show reduced cell death in the embryo [40], while others have shown no effect on post-irradiation cell death in the wing, although morphological defects were increased [25]. Interestingly debcl buffy double mutants show increased cell death after irradiation, suggesting that Debcl is required to inhibit the antiapoptotic function of Buffy [40]. Apoptosis induced by expression of mammalian Bax is inhibited by deletion of Debcl, suggesting that the fly proteins share functions with the mammalian proteins [25]. However, debcl mutants do not show any defects in mitochondrial organization in living cells or in mitochondrial fragmentation in dying cells. In contrast, Buffy expression was shown to protect cells from mitochondrial changes and apoptosis caused by loss of function of Drosophila PTEN-induced kinase (Pink1), as discussed below [38]. Although further studies may demonstrate a role for the Drosophila Bcl-2 proteins in regulating mitochondrial changes in apoptosis, it is also possible that other proteins such as Rpr, Grim, and Hid have taken over this function in flies (see below).
Are molecules released from the mitochondria important for apoptosis?
A variety of molecules are released from mitochondria during apoptosis in mammalian systems [41]. Cytochrome C, Apoptosis Inducing Factor (AIF), ARTS, SMAC/Diablo and Omi/HTRA2 are among the released mitochondrial proteins implicated in cell death [9]. Below we examine the evidence for a role for mitochondrial factors in Drosophila cell death in general, and these factors specifically.
Several observations support a role for mitochondrial components in Drosophila cell death. Early studies indicated that caspases in Drosophila S2 cell extracts could be activated by mitochondria from apoptotic S2 cells, suggesting that mitochondrial factors contribute to caspase activation [42]. However, others have found that mitochondrial lysates from untreated cells, or from apoptotic cells, are not able to induce or accelerate apoptosis in S2 extracts [43]. In the first study, apoptosis was induced by expression of Rpr or Grim, while in the second it was induced by UV. More recent data suggests that these two treatments can have different effects on mitochondria in Drosophila cells, which could explain these contradictory results [12].
The proapoptotic proteins Rpr, Grim, and Hid have been shown to localize to mitochondria [12, 44, 45]. Mitochondrial localization of these proteins is important for permeabilization of the mitochondrial membrane, and for efficient activation of apoptosis. This again suggests a role for mitochondria in Drosophila cell death. However, it is still unclear whether proteins released from the mitochondria play a role in cell death, or if mitochondrial disruption itself serves as a failsafe for insuring the elimination of doomed cells. Another intriguing possibility is that mitochondrial localization, perhaps at sites of mitochondrial fission, is required for the proapoptotic activity of these proteins independently of mitochondrial changes [45].
Cytochrome C
The mammalian apoptosome is formed when Cytochrome C and dATP bind to Apaf-1 resulting in Caspase 9 recruitment and activation [10]. The CARD and WD40 domains of Apaf-1 are required for this activation. One Drosophila isoform of the Apaf-1 homolog Dark also contains both WD40 and CARD domains, suggesting that this isoform could indeed be activated by Cytochrome C released from mitochondria. Dark has been reported to bind to Dronc and Cytochrome C, and the addition of Cytochrome C to a cytosolic S2 cell fraction can enhance the formation of a high molecular weight complex between Dark, Dronc, and Cytochrome C [4, 5, 46]. This suggests that Cytochrome C might play a role in cell death in flies.
The demonstration of Cytochrome C release in Drosophila cell death is an area of considerable controversy. Original studies by Varkey demonstrated that an epitope of Cytochrome C was exposed in apoptotic cells and not detected in living cells [42]. However, this epitope remained in a punctate, presumably mitochondrial, localization. This contrasts with the appearance of diffuse Cytochrome C in dying cells reported by Abdelwahid [12]. These disparities could reflect differences in antibody staining protocols. Fractionation studies done by many labs have failed to find detectable cytoplasmic release of Cytochrome C [12, 42, 46, 47]. However, cytosolic Cytochrome C has been seen in at least two studies using fractionation [4, 48]. Again, technical differences may explain these apparent contradictions, although it is clear that Cytochrome C release is not the easily detectable indicator of apoptosis initiation in flies that it is in mammalian cells.
A variety of strategies have been used to examine the role of Cytochrome C in Drosophila cell death. In 2003, Arama et al reported that disruption of one of the two Cytochrome C genes in flies, cyt-c-d, resulted in a defect in a spermatid individualization [49]. In this process, a cyst containing 64 connected spermatids separate into individual spermatozoa, with the removal of the majority of the cytoplasm of the cells. This process was shown to require caspase activation [7, 49, 50]. Loss of function of dark also results in a spermatid individualization defect that resembles loss of cyt-c-d, suggesting that the two proteins might act together in caspase activation in these cells, similar to the prevailing models for Apaf-1 and Cytochrome C in mammals [18]. Interestingly, Cyt-c-d appears to be the major form of Cytochrome C expressed in the germline where it is important for caspase activation, whereas Cyt-c-p is expressed in somatic cells and important for respiration. However either isoform can compensate functionally for the other [18]. These results indicate that Cytochrome C has a role in activating caspases in a non-apoptotic process.
There is also evidence that Cytochrome C might function in developmental apoptosis in certain tissues. The death of interommatidial cells in the developing eye is inhibited in animals lacking cyt-c-d, and in flies mutant for dark or dronc [51]. In addition, adult flies lacking cyt-c-d also show an extra scutellar bristle, another phenotype that is observed in dark mutants. above results suggest that Cytochrome C is important for caspase activation by the Drosophila apoptosome in some cases., the reported 3D structure of the Drosophila apoptosome shows that although Dark contains the WD40 region important for Cytochrome C binding, Drosophila apoptosome formation did not require Cytochrome C [52]. In addition numerous studies have failed to demonstrate a role for Cytochrome C in apoptosis of tissue culture cells [12, 47, 53]. Interestingly, apoptosis in Drosophila S2 cells, which does not require Cytochrome C release, is inhibited by Drp-1 knock down, showing that mitochondrial fission has a role separate from release of this factor [12].
IAP inhibitors
Three of the proteins released from mitochondria in mammalian apoptosis, ARTS, SMAC/Diablo and Omi/HTRA2, can act as IAP inhibitors. SMAC/Diablo is considered to be a functionally homologous to the Rpr, Grim and Hid IAP inhibitors. However, activation of SMAC/Diablo activity by release from mitochondria in doomed cells is quite different from the mechanism of activation of the Drosophila IAP inhibitors, which are for the most part transcriptionally upregulated in doomed cells [54]. ARTS is a splice variant of a septin that is localized to mitochondria, which can bind to and inhibit mammalian XIAP [55]. Genetic evidence suggests a role for the Drosophila ARTS homolog in apoptosis.
Mammalian HtrA2/Omi is released from mitochondria during apoptosis, and cleaved HtrA2/Omi can bind IAPs [56–58]. The Drosophila homolog of this protein, dOmi, was recently described [48, 59, 60]. dOmi was shown to be mitochondrially localized and is released, along with Cytochrome C, from the mitochondria upon activation of apoptosis [48, 59]. Cleaved dOmi can bind DIAP1 and promote DIAP1 cleavage and degradation. Expression of dOmi in the eye caused a rough eye phenotype that was at least partially dependent on the serine protease activity of dOmi [48, 60]. This suggests that dOmi has at least two pro-apoptotic activities. Importantly, reducing dOmi expression in the fly by RNAi was found to inhibit stress induced apoptosis [59]. This is in contrast to HtrA2/Omi knock outs in mouse, where stress induced apoptosis and neurodegeneration were increased [61]. It is possible that HtrA2/Omi has a proapoptotic function when released from mitochondria, and an anti-apoptotic function in the mitochondria. Both Cytochrome C and AIF also share dual pro-survival and pro-apoptotic functions.
Other mitochondrial proteins
Another protein found to be released from mitochondria in mammalian cells is apoptosis inducing factor (AIF) [62]. AIF is a flavoprotein that translocates from the mitochondria to the nucleus on death stimulation, triggering DNA fragmentation. is also required for proper mitochondrial function in living cells [63]. of the Drosophila homolog of AIF suggests both pro-survival and proapoptotic functions for this protein [64]. AIF knockouts are lethal at early larval stages, with reduced ATP. Survival through embryonic stages may be supported by maternal expression. These mutants also show reduced apoptosis in the developing embryo. Interestingly, expression of a truncated, cytoplasmic form of AIF induced apoptosis in the eye. This rough eye was not modified by mutations in Dark, or by the caspase inhibitor p35. The rough eye was suppressed, however, by expression of DIAP1 or DIAP2. Further examination of these phenotypes will help to clarify the role of AIF in cell death.
Are familial Parkinson’s associated genes involved in mitochondrial changes in apoptosis?
Mutations in PINK1, a mitochondrial Ser/Thr kinase, and Parkin, an E3 ubiquitin ligase, are associated with familial Parkinson’s Disease. Pink1 mutants in Drosophila exhibit defects in spermatid individualization (as was observed in cyt-c-d and dark mutants) and a progressive apoptotic death of flight muscles [38, 65, 66]. Loss of dopaminergic neurons was also reported [38, 66]. Interestingly, these phenotypes were associated with mitochondrial changes including swelling and cristae fragmentation. Double mutant analysis of parkin and Pink1 suggests that the two genes act in the same pathway. Overexpression of Parkin rescued the defects seen in Pink1 mutants, suggesting that Parkin functions downstream of Pink1. Inhibiting the Jun Kinase pathway also suppressed some Pink1 mutant phenotypes, including apoptosis, however it did not suppress the appearance of swollen mitochondria [38]. Intriguingly, expression of the BCL-2 homolog Buffy also suppressed Pink1 phenotypes.
Several reports have now looked for interactions between Pink1 or parkin mutants and regulators of mitochondrial fission and fusion in Drosophila [67–69]. Expression of Drp-1, promoting mitochondrial fission, or knockdown of Marf or Opa1, inhibiting mitochondrial fusion, suppresses Pink1 and parkin phenotypes. Loss of one copy of Drp-1 is lethal in Pink1 and parkin mutant backgrounds [67, 68]. Additionally, the mitofusin fuzzy onions interacts genetically with Pink1 in spermatogenesis. Interestingly, these results suggest that increased fission can protect PINK1 neurons from death. This contradicts other data indicating a proapoptotic effect of increased fission. One interpretation is that the role of fission in apoptosis could be context-dependent., the defects in mitochondrial fission seen in Pink1 mutations could cause cell death due to improper distribution of mitochondria within cells, as is seen in drp-1 mutants [70].
Interestingly, a substrate was recently identified for mammalian PINK1 [71]. HtrA2/Omi was phosphorylated in a PINK1-dependent manner after activation of the p38 stress pathway. Point mutations in HtrA2/Omi have also been associated with Parkinson’s disease, and the phosphorylation of HtrA2/Omi was reduced in patients with a PINK1 mutation. interaction between dOmi and PINK1 in Drosophila has been reported [72]. of dOmi partially suppressed PINK1 overexpression phenotypes. similar interaction was not found between dOmi and Parkin., loss of both dOmi and Parkin was reported to completely suppress the rough eye phenotype caused by PINK1 overexpression. The PINK1/Parkin/dOmi pathway showed both physical and genetic interactions with Rhomboid-7, a mitochondrial intermembrane protease that exhibits similar mutant phenotypes to PINK1 and Parkin. These observations are in contrast to a report that showed that loss of dOmi did not result in mitochondrial defects and did not suppress Pink1 overexpression phenotypes [73]. contradictions remain to be resolved.
Looking ahead
Though it remains controversial, increasing evidence points to a role for mitochondria and mitochondrial factors in the regulation of apoptosis in Drosophila. role of the “canonical” mitochondrial pathway in developmental apoptosis remains to be clarified., cell death defects seen in the absence of the Bcl-2 family member Debcl suggest that this pathway may have a more important role in developmental apoptosis than previously recognized [25]. It is also not yet clear whether Cytochrome C and other factors released from mitochondria only play a role in cell death in specific tissues, or if more general roles have been missed due to the dual survival and cell death roles of these proteins.
Several observations suggest interesting new functions for mitochondrial proteins implicated in apoptosis and their cellular targets that merit further study. Mutations in Drosophila optic atrophy 1-like protein OPA1 also lead to mitochondrial defects similar to those seen with rhomboid-7 [74]. Combined with the observations that lowering OPA1 and Marf suppressed phenotypes, including apoptosis, observed in Pink1 and parkin mutants in Drosophila, these results suggest that future studies be conducted to further elucidate the role that mitochondrial fusion proteins may play in regulating death.
A new role for Parkin was recently identified in mammalian cells [75]. Parkin appears to be necessary to recruit autophagosomes to damaged mitochondria. Mitochondrial damage due to uncoupling reagents results in recruitment of cytosolic Parkin to mitochondria. Significantly, defects in mitochondrial fusion also lead to depolarization and recruitment of Parkin to fragmented mitochondria. In Parkin deficient cells, damaged mitochondria fail to recruit autophagosomes, and this defect is rescued by expression of Parkin. Further studies in Drosophila may help elucidate a role for components of the autophagocytic machinery in apoptosis.
It is perhaps surprising that the role of mitochondria in Drosophila apoptosis is still unclear. Some of this lack of clarity may represent truly unique features of the cell death pathway in flies. For example, the central role of IAP inhibitors in Drosophila apoptosis may be a particular adaptation to the need for rapid induction of cell death during development, where caspase activation downstream of mitochondrial pathways may be too slow. Alternatively, some of the apparent differences between mammals and flies may reflect exchange of protein functions. For example the Rpr, Grim, and Hid proteins may have taken over some of the functions of the Bcl-2 family proteins, particularly with regard to regulating mitochondrial integrity. Finally, differences in the gene functions found to be important in flies and mammals may reflect more about differences between the study of cell death in an intact animal using loss of function strategies and the examination of cells adapted to culture conditions.
Acknowledgments
K.W. is supported by grants GM55568 and GM69541 from N.I.H.
References
- 1.Kornbluth S, White K. Apoptosis in Drosophila: neither fish nor fowl (nor man, nor worm) J Cell Sci. 2005;118:1779–1787. doi: 10.1242/jcs.02377. [DOI] [PubMed] [Google Scholar]
- 2.Hay BA, Guo M. Caspase-dependent cell death in Drosophila. Annu Rev Cell Dev Biol. 2006;22:623–650. doi: 10.1146/annurev.cellbio.21.012804.093845. [DOI] [PubMed] [Google Scholar]
- 3.Xu D, Li Y, Arcaro M, Lackey M, Bergmann A. The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development. 2005;132:2125–2134. doi: 10.1242/dev.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanuka H, Sawamoto K, Inohara N, Matsuno K, Okano H, Miura M. Control of the cell death pathway by Dapaf-1, a Drosophila Apaf-1/CED-4-related caspase activator. Mol Cell. 1999;4:757–769. doi: 10.1016/s1097-2765(00)80386-x. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez A, Oliver H, Zou H, Chen P, Wang X, Abrams J. DARK, a Drosophila homolog of Apaf-1/ced-4, functions in an evolutionarily conserved death pathway. Nature Cell Biology. 1999;1:272–279. doi: 10.1038/12984. [DOI] [PubMed] [Google Scholar]
- 6.Zhou L, Song Z, Tittel J, Steller H. HAC-1, a Drosophila homolog of APAF-1 and CED-4, functions in developmental and radiation-induced apoptosis. Mol Cell. 1999;4:745–755. doi: 10.1016/s1097-2765(00)80385-8. [DOI] [PubMed] [Google Scholar]
- 7.Muro I, Berry DL, Huh JR, Chen CH, Huang H, Yoo SJ, Guo M, Baehrecke EH, Hay BA. The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development. 2006;133:3305–3315. doi: 10.1242/dev.02495. [DOI] [PubMed] [Google Scholar]
- 8.O’Riordan MX, Bauler LD, Scott FL, Duckett CS. Inhibitor of apoptosis proteins in eukaryotic evolution and development: a model of thematic conservation. Dev Cell. 2008;15:497–508. doi: 10.1016/j.devcel.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 10.Bao Q, Shi Y. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ. 2007;14:56–65. doi: 10.1038/sj.cdd.4402028. [DOI] [PubMed] [Google Scholar]
- 11.Martinou JC, Youle RJ. Which came first, the cytochrome c release or the mitochondrial fission? Cell Death Differ. 2006;13:1291–1295. doi: 10.1038/sj.cdd.4401985. [DOI] [PubMed] [Google Scholar]
- 12.Abdelwahid E, Yokokura T, Krieser RJ, Balasundaram S, Fowle WH, White K. Mitochondrial disruption in Drosophila apoptosis. Dev Cell. 2007;12:793–806. doi: 10.1016/j.devcel.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Goyal G, Fell B, Sarin A, Youle RJ, Sriram V. Role of mitochondrial remodeling in programmed cell death in Drosophila melanogaster. Dev Cell. 2007;12:807–816. doi: 10.1016/j.devcel.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Means JC, Hays R. Mitochondrial membrane depolarization in Drosophila apoptosis. Cell Death Differ. 2007;14:383–385. doi: 10.1038/sj.cdd.4402036. [DOI] [PubMed] [Google Scholar]
- 15.Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 16.Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun MG, Williams J, Munoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, Green DR, Frey TG. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol. 2007;9:1057–1065. doi: 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]
- 18.Arama E, Bader M, Srivastava M, Bergmann A, Steller H. The two Drosophila cytochrome C proteins can function in both respiration and caspase activation. Embo J. 2006;25:232–243. doi: 10.1038/sj.emboj.7600920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker DW, Benzer S. Mitochondrial “swirls” induced by oxygen stress and in the Drosophila mutant hyperswirl. Proc Natl Acad Sci U S A. 2004;101:10290–10295. doi: 10.1073/pnas.0403767101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 21.Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–129. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- 22.Hwa JJ, Hiller MA, Fuller MT, Santel A. Differential expression of the Drosophila mitofusin genes fuzzy onions (fzo) and dmfn. Mech Dev. 2002;116:213–216. doi: 10.1016/s0925-4773(02)00141-7. [DOI] [PubMed] [Google Scholar]
- 23.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 24.Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 25.Galindo KA, Lu WJ, Park JH, Abrams JM. The Bax/Bak ortholog in Drosophila, Debcl, exerts limited control over programmed cell death. Development. 2009;136:275–283. doi: 10.1242/dev.019042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Breckenridge DG, Kang BH, Kokel D, Mitani S, Staehelin LA, Xue D. Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol Cell. 2008;31:586–597. doi: 10.1016/j.molcel.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–1094. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- 29.Sheridan C, Delivani P, Cullen SP, Martin SJ. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell. 2008;31:570–585. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- 32.Doumanis J, Dorstyn L, Kumar S. Molecular determinants of the subcellular localization of the Drosophila Bcl-2 homologues DEBCL and BUFFY. Cell Death Differ. 2007;14:907–915. doi: 10.1038/sj.cdd.4402082. [DOI] [PubMed] [Google Scholar]
- 33.Brachmann CB, Jassim OW, Wachsmuth BD, Cagan RL. The Drosophila bcl–2 family member dBorg-1 functions in the apoptotic response to UV–irradiation. Curr Biol. 2000;10:547–550. doi: 10.1016/s0960-9822(00)00474-7. [DOI] [PubMed] [Google Scholar]
- 34.Colussi PA, Quinn LM, Huang DC, Coombe M, Read SH, Richardson H, Kumar S. Debcl, a proapoptotic Bcl-2 homologue, is a component of the Drosophila melanogaster cell death machinery. J Cell Biol. 2000;148:703–714. doi: 10.1083/jcb.148.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Igaki T, Kanuka H, Inohara N, Sawamoto K, Nunez G, Okano H, Miura M. Drob-1, a Drosophila member of the Bcl-2/CED-9 family that promotes cell death. Proc Natl Acad Sci U S A. 2000;97:662–667. doi: 10.1073/pnas.97.2.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Huang Q, Ke N, Matsuyama S, Hammock B, Godzik A, Reed JC. Drosophila pro-apoptotic Bcl-2/Bax homologue reveals evolutionary conservation of cell death mechanisms. J Biol Chem. 2000;275:27303–27306. doi: 10.1074/jbc.M002846200. [DOI] [PubMed] [Google Scholar]
- 37.Quinn L, Coombe M, Mills K, Daish T, Colussi P, Kumar S, Richardson H. Buffy, a Drosophila Bcl-2 protein, has anti-apoptotic and cell cycle inhibitory functions. Embo J. 2003;22:3568–3579. doi: 10.1093/emboj/cdg355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 39.Senoo-Matsuda N, Igaki T, Miura M. Bax-like protein Drob-1 protects neurons from expanded polyglutamine-induced toxicity in Drosophila. Embo J. 2005;24:2700–2713. doi: 10.1038/sj.emboj.7600721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sevrioukov EA, Burr J, Huang EW, Assi HH, Monserrate JP, Purves DC, Wu JN, Song EJ, Brachmann CB. Drosophila Bcl-2 proteins participate in stress-induced apoptosis, but are not required for normal development. Genesis. 2007;45:184–193. doi: 10.1002/dvg.20279. [DOI] [PubMed] [Google Scholar]
- 41.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 42.Varkey J, Chen P, Jemmerson R, Abrams JM. Altered cytochrome C display precedes apoptotic cell death in Drosophila. J Cell Biol. 1999;144:701–710. doi: 10.1083/jcb.144.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Means JC, Muro I, Clem RJ. Lack of involvement of mitochondrial factors in caspase activation in a Drosophila cell-free system. Cell Death Differ. 2006;13:1222–1234. doi: 10.1038/sj.cdd.4401821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Claveria C, Caminero E, Martinez AC, Campuzano S, Torres M. GH3, a novel proapoptotic domain in Drosophila Grim, promotes a mitochondrial death pathway. Embo J. 2002;21:3327–3336. doi: 10.1093/emboj/cdf354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olson MR, Holley CL, Gan EC, Colon-Ramos DA, Kaplan B, Kornbluth S. A GH3-like domain in reaper is required for mitochondrial localization and induction of IAP degradation. J Biol Chem. 2003;278:44758–44768. doi: 10.1074/jbc.M308055200. [DOI] [PubMed] [Google Scholar]
- 46.Dorstyn L, Read S, Cakouros D, Huh JR, Hay BA, Kumar S. The role of cytochrome c in caspase activation in Drosophila melanogaster cells. JCell Biol. 2002;156:1089–1098. doi: 10.1083/jcb.200111107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zimmermann KC, Ricci JE, Droin NM, Green DR. The role of ARK in stress-induced apoptosis in Drosophila cells. J Cell Biol. 2002;156:1077–1087. doi: 10.1083/jcb.20112068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Challa M, Malladi S, Pellock BJ, Dresnek D, Varadarajan S, Yin YW, White K, Bratton SB. Drosophila Omi, a mitochondrial-localized IAP antagonist and proapoptotic serine protease. Embo J. 2007;26:3144–3156. doi: 10.1038/sj.emboj.7601745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arama E, Agapite J, Steller H. Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila. Dev Cell. 2003;4:687–697. doi: 10.1016/s1534-5807(03)00120-5. [DOI] [PubMed] [Google Scholar]
- 50.Huh JR, Vernooy SY, Yu H, Yan N, Shi Y, Guo M, Hay BA. Multiple apoptotic caspase cascades are required in nonapoptotic roles for Drosophila spermatid individualization. PLoS Biol. 2004;2:E15. doi: 10.1371/journal.pbio.0020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mendes CS, Arama E, Brown S, Scherr H, Srivastava M, Bergmann A, Steller H, Mollereau B. Cytochrome c-d regulates developmental apoptosis in the Drosophila retina. EMBO Rep. 2006;7:933–939. doi: 10.1038/sj.embor.7400773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu X, Wang L, Acehan D, Wang X, Akey CW. Three-dimensional structure of a double apoptosome formed by the Drosophila Apaf-1 related killer. J Mol Biol. 2006;355:577–589. doi: 10.1016/j.jmb.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 53.Dorstyn L, Mills K, Lazebnik Y, Kumar S. The two cytochrome c species, DC3 and DC4, are not required for caspase activation and apoptosis in Drosophila cells. J Cell Biol. 2004;167:405–410. doi: 10.1083/jcb.200408054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oberst A, Bender C, Green DR. Living with death: the evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 2008;15:1139–1146. doi: 10.1038/cdd.2008.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. Embo J. 2004;23:1627–1635. doi: 10.1038/sj.emboj.7600155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, Alnemri ES. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem. 2002;277:432–438. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- 57.Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–444. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi RA. Serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–621. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 59.Igaki T, Suzuki Y, Tokushige N, Aonuma H, Takahashi R, Miura M. Evolution of mitochondrial cell death pathway: Proapoptotic role of HtrA2/Omi in Drosophila. Biochem Biophys Res Commun. 2007;356:993–997. doi: 10.1016/j.bbrc.2007.03.079. [DOI] [PubMed] [Google Scholar]
- 60.Khan FS, Fujioka M, Datta P, Fernandes-Alnemri T, Jaynes JB, Alnemri ES. The interaction of DIAP1 with dOmi/HtrA2 regulates cell death in Drosophila. Cell Death Differ. 2008;15:1073–1083. doi: 10.1038/cdd.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alnemri ES. HtrA2 and Parkinson’s disease: think PINK? Nat Cell Biol. 2007;9:1227–1229. doi: 10.1038/ncb1107-1227. [DOI] [PubMed] [Google Scholar]
- 62.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 63.Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 64.Joza N, Galindo K, Pospisilik JA, Benit P, Rangachari M, Kanitz EE, Nakashima Y, Neely GG, Rustin P, Abrams JM, Kroemer G, Penninger JM. The molecular archaeology of a mitochondrial death effector: AIF in Drosophila. Cell Death Differ. 2008;15:1009–1018. doi: 10.1038/cdd.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 66.Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deng H, Dodson MW, Huang H, Guo M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 71.Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- 72.Whitworth AJ, Lee JR, Ho VM, Flick R, Chowdhury R, McQuibban GA. Rhomboid-7 and HtrA2/Omi act in a common pathway with the Parkinson’s disease factors Pink1 and Parkin. Dis Model Mech. 2008;1:168–174. doi: 10.1242/dmm.000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yun J, Cao JH, Dodson MW, Clark IE, Kapahi P, Chowdhury RB, Guo M. Loss-of-function analysis suggests that Omi/HtrA2 is not an essential component of the PINK1/PARKIN pathway in vivo. J Neurosci. 2008;28:14500–14510. doi: 10.1523/JNEUROSCI.5141-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McQuibban GA, Lee JR, Zheng L, Juusola M, Freeman M. Normal mitochondrial dynamics requires rhomboid-7 and affects Drosophila lifespan and neuronal function. Curr Biol. 2006;16:982–989. doi: 10.1016/j.cub.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 75.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]